



Los adenomas hipofisarios clínicamente relevantes son 3-5 veces más frecuentes de lo que inicialmente se pensaba. La mayoría son casos esporádicos, pero su presentación puede ser familiar dentro de síndromes conocidos: neoplasia endocrina múltiple (MEN) 1 y complejo de Carney. Cuando se expresan dos o más casos en la misma familia en ausencia de los síndromes anteriores, hablamos de adenomas hipofisarios familiares aislados (familial isolated pituitary adenomas [FIPA]), que suponen un 1-2% de todos los adenomas hipofisarios. Las mutaciones del gen AIP (aryl hydrocarbon receptor-interacting protein) pueden justificar el 15% de las familias con FIPA (el 50% de acromegalia familiar), pero su base genética continúa en estudio. Además, estas mutaciones de AIP se detectan en adenomas aislados en población joven (< 30 años). Se describen las características descritas en los FIPA detallando el estudio de una familia española, en este caso AIP negativa. También se detallan los hallazgos descritos en adenomas esporádicos en población joven con la presentación de una paciente de 19 años acromegálica con mutación de AIP intrónica. Los estudios multicéntricos han permitido conocer aspectos como las mutaciones de AIP, pero continúan siendo necesarios para conocer otros genes involucrados en los FIPA y los adenomas esporádicos que se presentan en edades tempranas.
Clinically relevant pituitary adenomas occur 3-5 times more frequently than previously thought. The majority are isolated cases, but their presentation can be familial in the setting of known syndromes such as multiple endocrine neoplasia (MEN)-1 and Carney complex. When 2 or more cases of pituitary adenomas occur in the same family in the absence of the above-mentioned syndromes, a diagnosis of FIPA (familial isolated pituitary adenomas) is made, which accounts for 1-2% of all pituitary adenomas. Mutations of the gene AIP (aryl hydrocarbon receptor-interacting protein) may account for 15% of FIPA families (50% of familial acromegaly), and as such the genetic causes continue to be studied. Also mutations in AIP can be detected in sporadic adenomas among young populations (< 30 years of age). We describe the characteristics of FIPA, detailing the study of a spanish family, in this case AIP mutation negative. Also, the reported findings in sporadic adenomas in the young population are detailed, accompanied by the description of a 19- year old patient with an intronic AIP mutation. Multicenter studies have provided understanding of aspects such as mutations in AIP; however, further studies are necessary to identify other genes involved in FIPA and sporadic pituitary adenomas occurring at a young age.
Clásicamente se ha considerado que los tumores hipofisarios suponen el 10-15% de todos los tumores cerebrales1,2. En un metaanálisis reciente se señala una prevalencia media de tumores hipofisarios incidentales del 14,4 al 22,5% en autopsias y series radiológicas respectivamente3. Se ha confirmado en estudios posteriores coordinados en Bélgica que los tumores hipofisarios diagnosticados clínicamente son más frecuentes en la población general, con una relación 1:1.0644. De este modo, los tumores hipofisarios son 3-5 veces más frecuentes de lo que inicialmente se pensaba, lo que incrementa la necesidad de conocer mejor sus mecanismos fisiopatológicos.
Un 2,5% de los adenomas hipofisarios tienen tinción positiva para somatotropina (GH) en los estudios histológicos. En cambio, la prevalencia de acromegalia diagnosticada es sólo 36-69 casos/millón de habitantes5. Se han puesto en marcha estudios que indican que la prevalencia actual de acromegalia debería ser mayor de lo que indican los datos epidemiológicos publicados6.
La etiología de los tumores hipofisarios es una cuestión que ha suscitado interés, ya que la investigación en este campo podría explicar aspectos como la diversa presentación clínica o los diferentes ritmos de crecimiento tumoral y ayudaría en el complejo abordaje de estos pacientes. Se han realizado diversos estudios de genética molecular en los adenomas hipofisarios con la finalidad de determinar su fisiopatología. La mayoría de los adenomas hipofisarios son esporádicos y hasta el 5% son casos familiares7,8. Entre las causas familiares de adenomas hipofisarios familiares, se diferencian:
- 1.
Neoplasia endocrina múltiple 1 (MEN-1): es una enfermedad autosómica dominante causada por mutaciones en el gen MEN1, localizado en el cromosoma 11q13, de las que se han descrito más de 500. Los adenomas hipofisarios aparecen en el 40% de los casos, y predominan los prolactinomas, con un comportamiento más agresivo y menor respuesta al tratamiento farmacológico9. En un 10-20% de los casos de pacientes con MEN-1 no se detectan mutaciones, por lo que es importante en estos casos el diagnóstico clínico, más frecuente en los casos de acromegalia. En algunos de los casos MEN-1 negativos, una mutación sin sentido (TGG>TAG en el codón 76) en el gen CDKN1B se ha identificado en una entidad MEN-4, que se caracteriza por macroadenoma hipofisario productor de GH e hiperparatiroidismo primario causado por hiperplasia o adenoma paratiroideo, que además se ha descrito concomitante a feocromocitoma, paraganglioma, tumor carcinoide, tumores renales o enfermedad de Cushing10. A pesar de estos hallazgos, todavía se conoce poco sobre los mecanismos moleculares por los que mutaciones en el gen MEN1 o en el gen CDKN1B conllevan el desarrollo de tumores hipofisarios y en otras localizaciones8.
- 2.
Complejo de Carney: se asocian a mixomas mucocutáneos y cardíacos, hiperpigmentación cutánea, schwannomas e hiperplasia suprarrenal. En el 21% de los casos se asocia a adenomas hipofisarios, que suelen ser productores de GH, y dan lugar a acromegalias. No suelen tener un comportamiento agresivo y se diagnostican en la tercera década de la vida. En el 73% de los casos se detectan mutaciones en el gen PRKAR1A (protein kinasa A 1a regulatory subunit gene)11,12.
El síndrome de McCune-Albright está causado por mosaicismo debido a una mutación en el gen GNAS, localizado en el cromosoma 20q13, que codifica la proteína Gsa y está asociada con el mosaicismo, es decir, que el gen anormal está presente en una fracción y no en todas las células del paciente. Se caracteriza por displasia fibrosa, manchas café con leche e hiperfunciones de glándulas endocrinas que pueden dar lugar a pubertad precoz, tirotoxicosis, gigantismo-acromegalia y síndrome de Cushing. El exceso de secreción de GH se presenta en el 20% de los casos, pero una imagen tumoral hipofisaria sólo se detecta en pocos pacientes. La concomitancia de acromegalia empeora el cuadro de displasia fibrosa e incluso puede desembocar en una transformación sarcomatosa. En este síndrome, al igual que en el complejo de Carney, el defecto tiene relación con alteraciones genéticas que alteran la función del AMPc8.
ADENOMAS HIPOFISARIOS FAMILIARES AISLADOS (FIPA)Una nueva entidad denominada familial isolated pituitary adenomas (FIPA) se describió por primera vez hace 7 años y supone un 1-2% de todos los adenomas hipofisarios. Se define como la presencia de 2 o más casos de adenoma hipofisario en la misma familia en ausencia de MEN-1 o complejo de Carney. La mayoría de los casos son prolactinomas y acromegalias familiares. Los FIPA representan una nueva entidad de neoplasia endocrina cuya base genética está en fase de investigación13.
Así, la acromegalia familiar, ya reconocida como entidad desde hace años y de la que se han descrito 50 familias, quedaría incluida dentro de los FIPA. Su estudio genético inicialmente se relacionó con un locus de menos de 10Mb localizado en el cromosoma 11q13.1-13.3, que está cercano a MEN1, y que la pérdida de función de un gen supresor tumoral a este nivel sería la causa. Inicialmente más de 100 genes eran candidatos a este nivel, incluyendo genes que codifican proteínas de transducción, reguladores del ciclo celular y de la diferenciación, reparadores de ADN, apoptosis y factores de transcripción. También se excluyeron posteriormente mutaciones en el receptor de la somatoliberina (GHRH) y en los genes Gsa. Incluso en algunas familias se ha estudiado la posible relación de haplotipos HLA14,15.
Posteriormente se han puesto en marcha estudios multicéntricos para reducir la región candidata hasta poder localizar el gen causal. En el estudio coordinado desde la Universidad de Lieja, de 27 pacientes locales identificados en 200013, se aumentó la casuística, en colaboración internacional con otros centros, a 140 pacientes en 200416. Se recogieron datos clínicos, radiológicos, bioquímicos y de análisis anatomopatológicos17-19 hasta reunir finalmente, en 2007, datos de pacientes procedentes de 90 familias. Se realizó cribado genético de MEN1 al menos a un miembro de cada familia; la historia familiar no resultó compatible y las concentraciones de calcio, paratirina (PTH) y calcitonina fueron normales. Además se determinaron otros parámetros relacionados con MEN1 (gastrina, péptido intestinal vasoactivo [VIP] y polipéptido pancreático) en la mayoría de ellos. Estos datos clínicos y analíticos son importantes para descartar MEN1 porque al menos el 10% de los pacientes con MEN1 pueden tener un cribado genético negativo. La secuenciación del gen PRKAR1A permitió descartar la presencia del complejo de Carney en al menos un miembro por familia afecta de acromegalia. Ése es el estudio más amplio sobre FIPA y a continuación se detallan sus hallazgos.
CARACTERÍSTICAS CLÍNICAS Y CLASIFICACIÓN DE LOS FIPA19Podría establecerse una regla del 75%, ya que las acromegalias (30%) y los prolactinomas (41%) suponen casi el 75% de los FIPA, con predominio femenino, probablemente relacionado con la mayor preponderancia de prolactinomas, y el 75% de los pacientes tienen relación de primer grado entre ellos19. La expresión clínica dentro de la misma familia puede ser homogénea, cuando todos los miembros afectados de la misma familia presentan el mismo tipo de adenoma hipofisario, o heterogénea, cuando en la misma familia se expresan distintos tipos de adenomas hipofisarios. En los casos de FIPA heterogéneos, puede ocurrir cualquier tipo de adenoma, aunque al menos se presenta un caso de prolactinoma o de acromegalia por familia. La edad al diagnóstico es menor que en los pacientes con adenomas esporádicos, y la edad es menor en los casos de FIPA homogéneos (probablemente porque en los heterogéneos la existencia de adenomas no funcionantes retrase el diagnóstico). La edad es menor en los miembros más jóvenes de la misma familia (29 frente a 50 años; p < 0,001). No se sabe si se debe a un diagnóstico más temprano o a alguna forma de anticipación genética.
El tamaño tumoral o el grado de agresividad no son diferentes que en los casos esporádicos, la mayoría macroadenomas, sobre todo en los casos heterogéneos (casi el 75%) por el predominio de adenomas no funcionantes (que en estos casos sí que son más agresivos) y la mayor frecuencia de microprolactinomas en los casos homogéneos. Además, los prolactinomas en las familias con FIPA heterogéneas son de mayor tamaño y con mayor extensión supraselar que los de los casos esporádicos.
En el estudio del árbol genealógico, se observa que los tumores hipofisarios ocurren en el 14% de los familiares estudiados (con una media de 15 individuos estudiados por familia), lo que indicaría que el modo de transmisión sería al menos parcialmente dominante. La transmisión materna es más frecuente entre los casos homogéneos, probablemente por la mayor frecuencia de asociación madre-hija con prolactinomas, mientras que la transmisión paterna es más frecuente en los casos heterogéneos con acromegalia. En los casos con acromegalia familiar, en el 65% se presenta en hermanos.
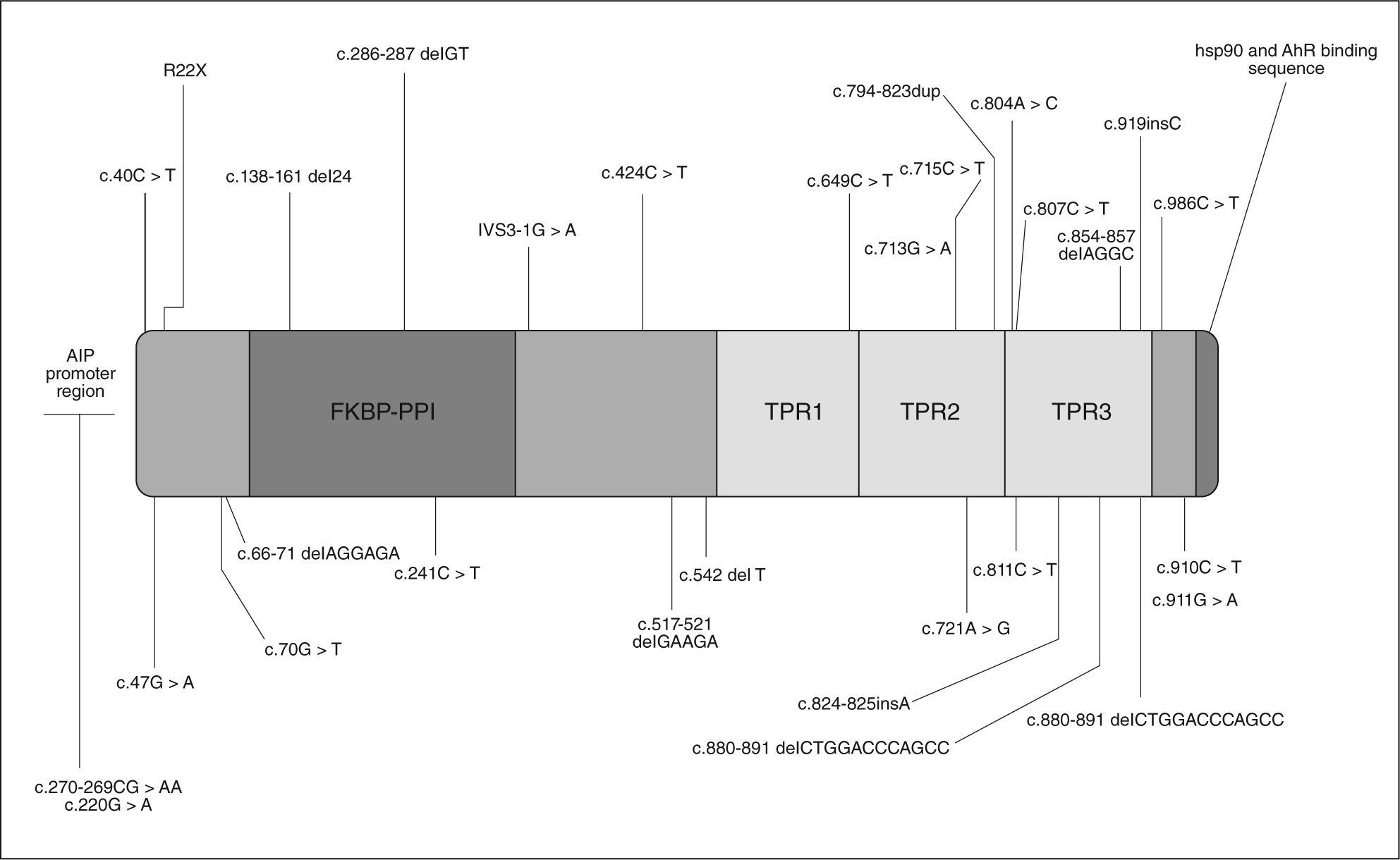
GENÉTICA DE LOS FIPAEn mayo de 200620 se comunicaron algunos casos de acromegalia y prolactinoma familiares relacionados con mutaciones de línea germinal en el gen de la aryl hydrocarbon receptor interacting protein (AIP), también conocida como ARA9 y XAP2, aunque AIP es su nomenclatura más reconocida, situada en el cromosoma 11q13.3. En la figura 1 se reflejan las mutaciones de AIP descritas.
.")
AIP actúa en la retención citoplásmica de la forma latente del receptor de aryl hydrocarbon y también tiene otras funciones. Contiene 330 aminoácidos y un número de regiones conservadas, entre ellas tres dominios de repetición TRP y un dominio de unión FKBPPPI. La mayoría de la información disponible sobre la relación estructura-función de AIP está en el tercer dominio TPR3 y en los aminoácidos carboxiterminales. El tercer dominio (TPR3) es necesario para la interacción de AIP con un dímero de la proteína Hsp90 y con el receptor AhR21.
Se describieron dos mutaciones de AIP en población del norte de Finlandia en el 16% de los pacientes diagnosticados de acromegalia (Q14X e IVS3-1G-A) y en el 40% de los diagnosticados antes de los 35 años20. Así, se concluyó que AIP es un ejemplo de gen de susceptibilidad tumoral con baja penetrancia. Posteriormente se describió en una familia italiana una mutación R304X, mientras que estas mutaciones no se encontraron en familias turcas o alemanas.
Con la finalidad de conocer si las mutaciones de AIP contribuyen a la patogenia de los adenomas en los FIPA, se coordinó un estudio con la participación de 73 familias con FIPA procedentes de 9 países19. El 15,1% de los pacientes presentaron diez mutaciones diferentes de AIP: tres nuevas mutaciones que conducen a codones de stop prematuros (Q142X, Q217X y Q239X), una familia con tres miembros presentaba la mutación R304X ya descrita previamente en otra zona, se identificaron tres mutaciones de cambio de sentido (R16H, R271W y K241E), una mutación sin cambio de pauta de lectura (G47_R54del) y dos deleciones (E174frameshift y Q285frameshift) que conducen a un codón de stop tras aminoácidos incorrectos (fig. 1).
Solo el 50% de las familias con FIPA homogéneas con acromegalia presentaron mutaciones de AIP. Además, la ausencia de mutaciones de AIP en casos con evidente asociación familiar (3 o 4 miembros de la familia afectos) indicaría que en la fisiopatología genética de los FIPA estarían implicadas otras causas.
Los pacientes con FIPA y mutaciones de AIP se diagnosticaron a edades más tempranas que los que no tenían mutaciones de AIP (25,7 frente a 38,8 años; p = 0,0006) y además presentaron un tamaño tumoral significativamente mayor (24 frente a 10mm; p = 0,0005).
Georgitsi et al22 identificaron otras nueve mutaciones de línea germinal en 460 pacientes procedentes de Europa y Estados Unidos con adenomas hipofisarios de diverso origen (pacientes con acromegalia jóvenes, pacientes sin seleccionar con acromegalia, pacientes con adenomas hipofisarios esporádicos y pacientes con adenomas hipofisarios familiares sin mutaciones en MEN1).
En una de las familias con FIPA brasileñas, se estudió a 122 familiares de seis generaciones. Se identificó la mutación E174fs en 10 de ellos, 3 con adenoma hipofisario (dos acromegalias y un mixto GH-prolactina [PRL]) y en 7 portadores sanos. Los pacientes con acromegalia no tenían buena respuesta al tratamiento con octreotida. El estudio inmunohistoquímico de AIP en los pacientes intervenidos mostró una menor expresión que en tejido normal. En esta familia con amplio número de familiares estudiados, se encontró una penetrancia del 33,3% de los tumores hipofisarios en pacientes adultos con mutación de AIP23.
Se añade un grado de complejidad cuando se analizan los datos de inmunohistoquímica y los patrones de secreción hormonal. En la inmunohistoquímica los pacientes con acromegalia pueden dar positivo sólo para GH (59%), para GH y PRL (33%) o para GH y folitropina (FSH) (8%). Además, la misma mutación de AIP puede presentarse con varios fenotipos clínicos en diferentes familias; por ejemplo, en una familia la mutación R271W se manifestaba con una acromegalia y un prolactinoma, mientras que en otra sólo se presentaba acromegalia.
Además del estudio en sangre, se puede realizar el estudio inmunohistoquímico de AIP en los pacientes intervenidos. La mayoría de los adenomas hipofisarios sin mutación de AIP captan en núcleo y citoplasma, mientras que los adenomas con mutaciones tempranas de AIP (truncaciones) no tiñen para AIP ni en el núcleo ni en citoplasma. En éstos los leucocitos positivos sirven de control interno. El patrón de captación en la tinción de AIP es heterogéneo y depende del tipo de tumor y de la mutación (p. ej., Q14X tinción negativa)22.
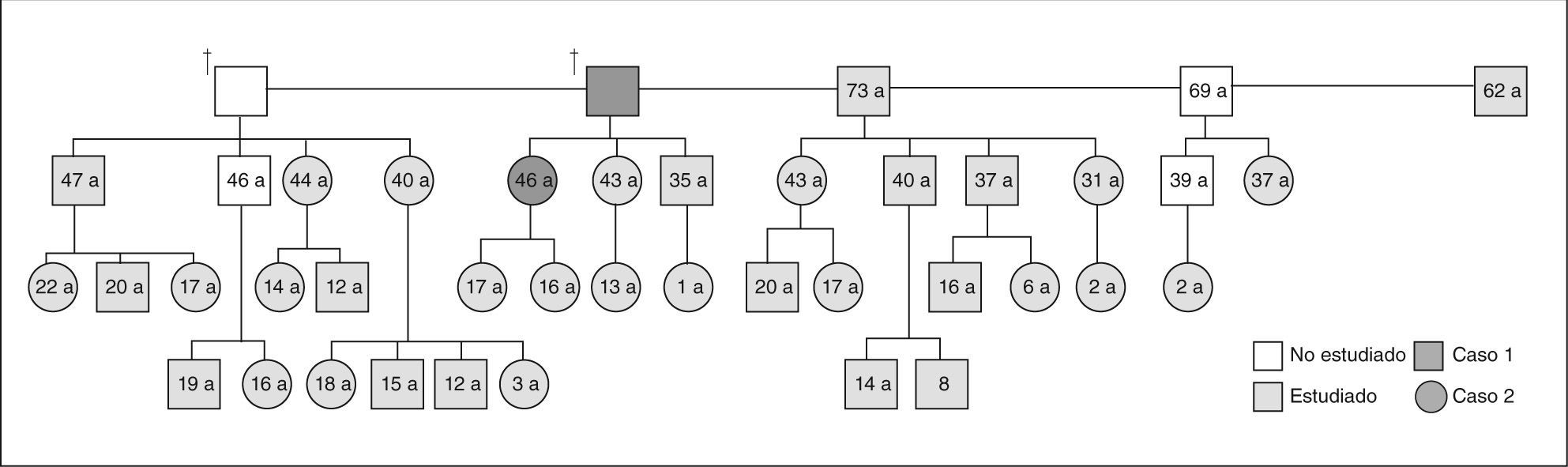
Acromegalia familiar: aportación de una familia española al estudio de FIPAUna de las 73 familias estudiadas por Daly et al19 procede de nuestro centro. Se presenta una familia afectada con dos miembros (padre e hija). El caso índice (caso 1) es una mujer de 45 años, sin antecedentes personales de interés, que consultó remitida desde ginecología por una concentración sérica de PRL de 83ng/ml (aparentemente secundaria a tratamiento farmacológico por cuadro vertiginoso) y un microadenoma hipofisario de 4mm (fig. 2). Refería trastornos menstruales sin baches amenorreicos de 4 años de evolución y sin galactorrea. El estudio hipofisario demostró que se trataba de una acromegalia con escasos síntomas, sin repercusión orgánica y sin cambios físicos objetivos (fig. 3). La concentración inicial de factor insulinoide de crecimiento 1 (IGF-1) fue de 849,84ng/ml (normal, 55-329ng/ml) y la concentración de GH tras sobrecarga oral de glucosa (SOG) para GH, medida a los tiempos basal y 15, 30, 60, 90 y 120min, fue de 21,4, 6,97, 2,11, 0,96, 0,47 y 0,47ng/ml, respectivamente, con el resto de los parámetros de función hipofisaria normales. El estudio genético de MEN1 resultó negativo, y tanto la evaluación del metabolismo fosfocálcico como el estudio de la función pancreática resultaron normales, lo que excluía en ese momento que se tratara de un caso clínico de MEN-1 no diagnosticado genéticamente. Fue intervenida en diciembre de 2004, y el estudio inmunohistoquímico fue positivo para GH, corticotropina (ACTH) y PRL. En la actualidad cumple criterios de curación. Su padre (caso 2) fue diagnosticado en 1995 de macroadenoma hipofisario (20 × 22mm) productor de GH con cambios fenotípicos evidentes en fotos seriadas. La concentración de GH basal fue de 20ng/ml y tras SOG, > 21ng/ml, sin llegar a determinarse el IGF-1, ya que el paciente falleció 1 mes después del diagnóstico por accidente cerebrovascular, por lo que no se llegó a tratar.


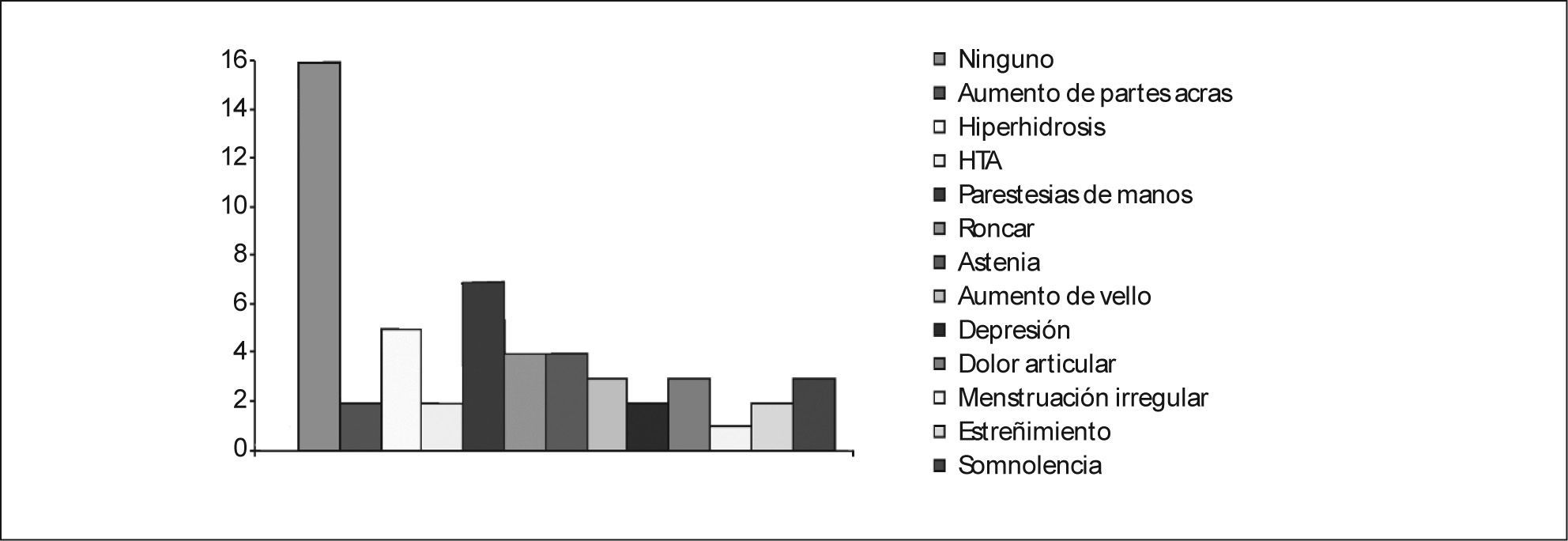
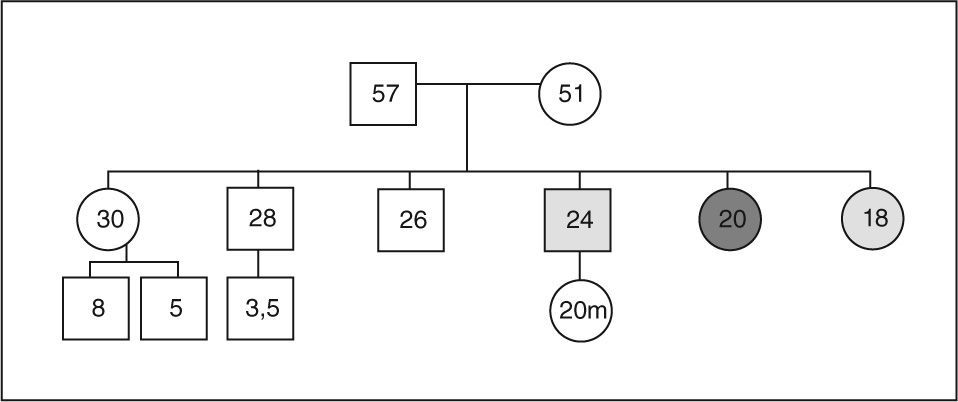
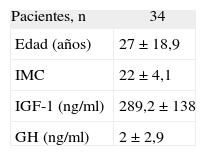
Se estudió en el caso índice la mutación del gen AIP en julio de 2006, como parte del estudio multicéntrico internacional de FIPA, y resultó negativo para las 10 mutaciones descritas (como en el 50% de los casos de acromegalia familiar y el 85% de los casos con FIPA). También se estudió a 34 familiares pertenecientes a cuatro generaciones de primero, segundo, tercero y cuarto grado, y en todos ellos el estudio de la mutación resultó negativo (tabla 1). El estudio genético se obtuvo con consentimiento informado en todos los casos. Se realizó el árbol genealógico familiar (fig. 4) incluyendo la recogida de fotografías de las tres generaciones ascendentes al caso 1 (fig. 5). Se recogieron datos demográficos, antropométricos y clínicos (antecedentes personales y síntomas mediante cuestionario dirigido de síntomas compatibles con afección hipofisaria) (fig. 6). Para el cribado de acromegalia, en todos los familiares se determinaron GH e IGF-1. Algunos de ellos están en estudio por concentraciones basales de GH e IGF-1 alteradas o por talla alta (> percentil 97), sin que se haya demostrado por el momento ningún nuevo caso clínico. La hermana del caso 1 es una mujer de 41 años que presentaba concentraciones de GH basales elevadas (5,43-27,1ng/ml) y cifras de IGF-1 reiteradamente normales (140-199ng/ml); se han realizado varias SOG y se alcanzaron concentraciones nadir de GH de hasta 0,4ng/ml, sin que se detectara imagen compatible en la resonancia magnética (RM) hipofisaria, y es objeto de especial seguimiento. El resto de los casos en estudio no son valorables de momento por estar en periodo peripuberal.
Descripción de la serie familiar
| Pacientes, n | 34 |
| Edad (años) | 27 ± 18,9 |
| IMC | 22 ± 4,1 |
| IGF-1 (ng/ml) | 289,2 ± 138 |
| GH (ng/ml) | 2 ± 2,9 |
GH: somatotropina; IGF-1: factor de crecimiento insulinoide 1; IMC: índice de masa corporal.
Valores expresados como media ± desviación estándar salvo otra indicación.
Valores de referencia. IGF-1 (inmunoanálisis): hasta 6 años, 52-297ng/ml; 6-9 años, 74-388ng/ml; 9-12 años, 143-693ng/ml; 12-15 años, 237-996ng/ml; 15-25 años, 116-358ng/ml; más de 25 años, 55-329ng/ml.


")
En Finlandia se han estudiado estas mutaciones en 79 pacientes con otros tipos de neoplasias endocrinas y en 32 adenomas hipofisarios esporádicos, y se ha encontrado la mutación Q14X en 2 pacientes con prolactinoma22. Esta mutación sería característica de una región geográfica concreta (norte de Finlandia) y no se encuentra en los estudios realizados en el resto de Europa, Japón y Estados Unidos. En Japón se estudiaron las mutaciones de AIP en 40 acromegalias esporádicas, y sólo se encontró una mutación de sentido erróneo V49M en la línea germinal, lo que indica que las mutaciones en pacientes con adenomas productores de GH esporádicos son raras24. En un estudio multicéntrico se estudió a 107 pacientes con adenomas esporádicos (49 prolactinomas, 29 adenomas no funcionantes, 26 acromegalias, 2 enfermedades de Cushing y 1 tirotropinoma) no se encontraron mutaciones de AIP en las muestras sanguíneas. Se estudiaron las muestras histológicas en otro grupo de 41 pacientes, y se encontró una nueva mutación (R22X), lo que confirma el escaso papel que tienen las mutaciones de AIP en la tumorogénesis de los casos esporádicos25.
Actualmente están en marcha estudios genéticos en pacientes con adenomas hipofisarios esporádicos con presentación clínica temprana (menores de 30 años), y en algunos casos se ha encontrado mutaciones de AIP, con una frecuencia del 17%26.
Otros autores relacionan algunas mutaciones de AIP con tumorogénesis de otro origen (gastrointestinal, de mama, de próstata), pero esta relación no está demostrada y es anecdótica, sólo se ha demostrado en 2 casos de 373 tumores colorrectales, y no se ha encontrado en mama (0/82) o en próstata (0/44).
Acromegalia aislada con mutación de AIP: contribución de un casoSe presenta a una paciente de 19 años de edad, ecuatoriana, diagnosticada de acromegalia secundaria a un macroadenoma hipofisario. Refería clínica con aumento de partes acras y cefalea de aproximadamente 2 años de evolución, sin trastorno visual subjetivo. Menarquia a los 12-13 años, y desde entonces ciclos regulares hasta 18 meses antes de la consulta, con baches amenorreicos, por lo que inició tratamiento con anticonceptivos. Respecto a su crecimiento previo a la menarquia, refería haber sido siempre una “niña grande”, pero no destacaba en altura respecto a sus compañeros (teniendo en cuenta que en Ecuador las gráficas de talla son diferentes, con una media más baja). No refería ningún antecedente familiar de interés.
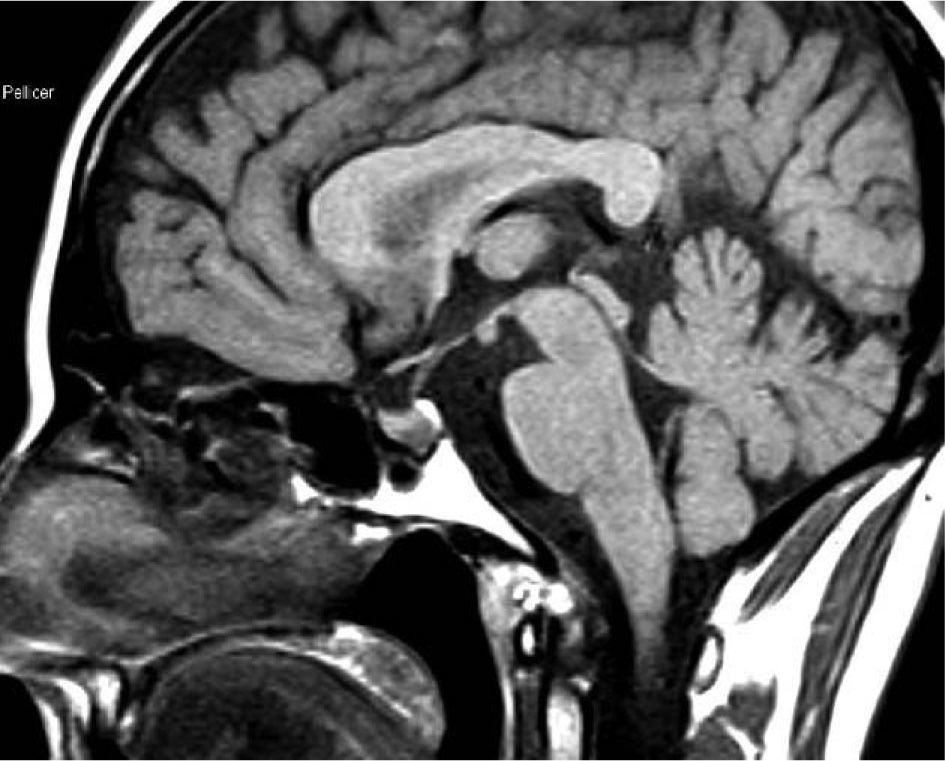
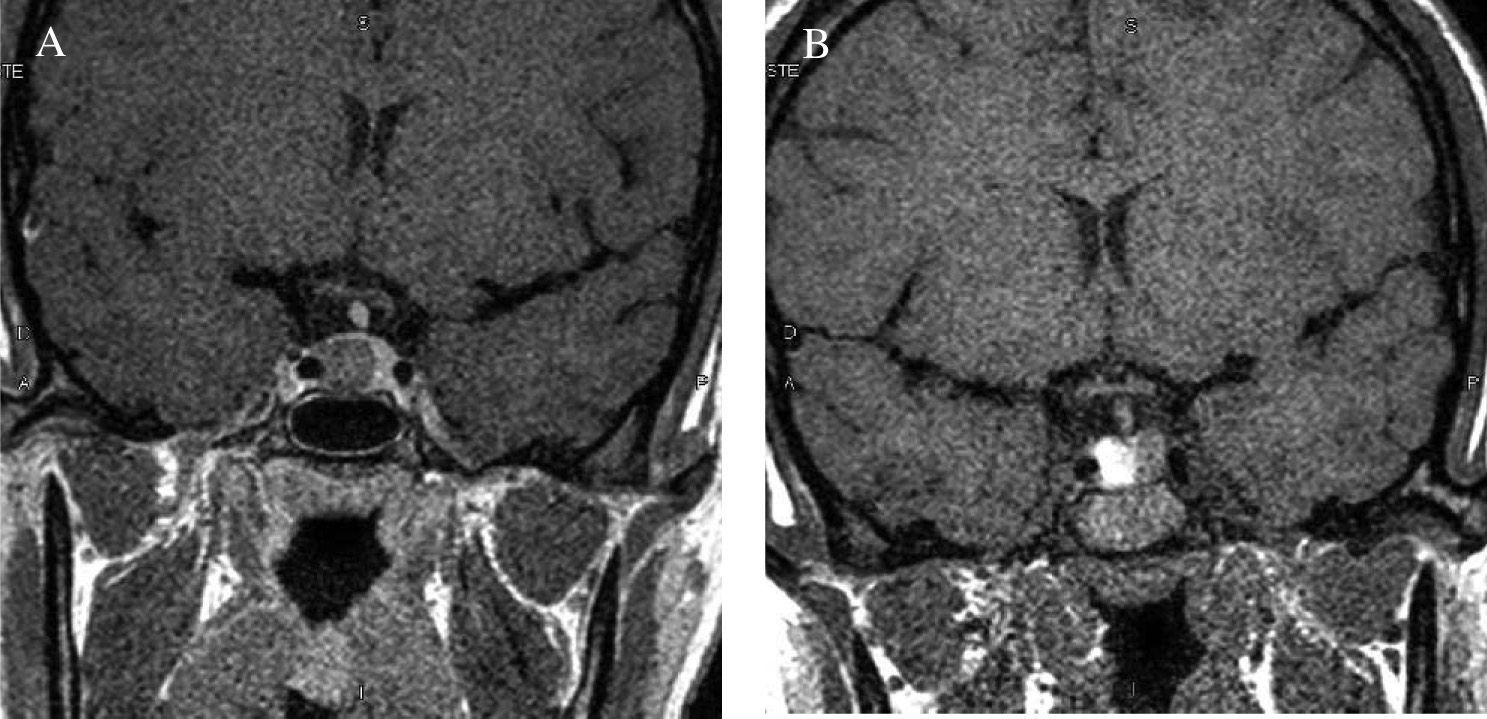
Se diagnosticó por la sospecha clínica de un familiar médico que la remitió a nuestro centro para tratamiento quirúrgico. En el estudio analítico inicial destacaban concentraciones plasmáticas de GH, 54,8ng/ml; PRL, 12,3ng/ml, e IGF-1, 635ng/ml (72-385). Las concentraciones de GH tras SOG para GH fueron de 54,8, 58,8, 76,6 y 11,6ng/ml, respectivamente; las concentraciones de tiroxina (T4) libre, 1,13ng/dl; las de TSH, 1,98mU/l, y las de cortisol, 12,54μg/dl. La resonancia magnética hipofisaria mostró un macroadenoma hipofisario de 13,4mm de diámetro, localizado en la vertiente derecha de la glándula. No se apreciaba invasión de seno cavernoso (fig. 7). A la exploración física el peso era de 91kg y la talla, de 165cm, y presentaba rasgos marcados de acromegalia, con macroglosia y aumento de pliegue cutáneo.

Se realizó intervención quirúrgica en septiembre de 2007 por vía transesfenoidal con resección completa de la tumoración (demostrada con resonancia magnética en el postoperatorio inmediato). El perfil inmunohistoquímico resultó positivo para lutropina (LH), folitropina (FSH), PRL y GH, positivo focal para ACTH y negativo para TSH.
La evolución en el postoperatorio fue favorable desde el punto de vista clínico, con disminución de una talla de calzado y del diámetro anular, menstruación espontánea y desaparición de la cefalea. Inició tratamiento sustitutivo con hidrocortisona a dosis sustitutivas de 10mg/5mg/5mg, corrigiéndose la diabetes insípida en 2 semanas.
En la actualidad reside en Ecuador y se ha suspendido el tratamiento con hidrocortisona, por lo que el resto de la hipófisis está conservada. Las concentraciones de IGF-1 están dentro de la normalidad. En diciembre de 2007 presentaba concentraciones de IGF-1, 222ng/ml (prequirúrgica, 653ng/ml); IGFBP3, 7,8μg/ml (prequirúrgica, 9,33μg/ml), TSH, 1,45 μU/ml; T4L, 1,28ng/dl; FSH, 11 μU/ml; LH, 43 μU/ml (pico ovulatorio); PRL, 14,8ng/ml, cortisol, 6,1μg/dl, y ACTH, 8,4pg/ml, hematocrito del 39% y glucemia de 73mg/dl.
Al tratarse de una paciente joven, se decidió estudiar las mutaciones de AIP, y se indentificó una mutación intrónica (c.468+15C>T). Se ha realizado estudio familiar y dos de sus hermanos presentan la misma mutación, por lo que van a ser sometidos a control clínico y de imagen. El árbol genealógico se ve en la figura 8.
CONCLUSIONES
Se debería reclasificar los tumores hipofisarios incluyendo los FIPA como nueva entidad, que no sólo incluiría a los casos de acromegalia familiar, sino a familias con adenomas hipofisarios con presentación clínica homogénea (mismo tipo de adenoma en cada caso) o heterogénea (tipos distintos de adenoma en casos afectados). Esta clasificación se basa en datos clínicos y la historia familiar es fundamental en la anamnesis de los adenomas hipofisarios. Además, es un diagnóstico de exclusión. Su base genética está en estudio, ya que las mutaciones de AIP no explican todos los casos.
Los familiares asintomáticos con cribado positivo para mutaciones de AIP deben someterse a un estudio hormonal hipofisario basal y controlarse anualmente como mínimo con determinaciones de IGF-1 y PRL. Del mismo modo, es importante analizar las mutaciones de AIP en los familiares con historia de acromegalia, ya que en este grupo se identifica mayor número de mutaciones (50%), o con inicio temprano de la enfermedad. Parece recomendable estudiar mutaciones de AIP en pacientes jóvenes (menores de 30 años) con adenomas hipofisarios aislados. En los casos positivos a mutaciones de AIP debería realizarse el estudio familiar para la detección precoz de posibles casos.
Los estudios multicéntricos han permitido conocer aspectos como las mutaciones de AIP, pero continúan siendo necesarios para conocer otros genes involucrados en los FIPA y en los adenomas aislados que se presentan en edades tempranas.
