



120ADECUACIÓN DE LOS MOTIVOS DE SOLICITUD DE PROLACTINA EN PACIENTES REMITIDOS POR HIPERPROLACTINEMIA A UNA CONSULTA ESPECIALIZADA
O. Giménez Palop, C. Vilardell, A. Caixàs, J. González Clemente, M.J. Barahona, J. Arroyo, D. Subías y G. Giménez Pérez
Unidad de Diabetes, Endocrinología y Nutrición Hospital de Sabadell. Sabadell.
Introducción: La hiperprolactinemia es un motivo frecuente de consulta que comporta en muchos casos la realización de exploraciones complementarias para su diagnóstico diferencial por lo que su determinación en pacientes sin clínica atribuible a la misma puede dar lugar a exploraciones innecesarias.
Objetivo: Evaluar los motivos de solicitud de prolactina en los pacientes remitidos desde la atención primaria con el diagnóstico de hiperprolactinemia, las pruebas complementarias que se derivan y la orientación diagnóstica final.
Métodos: Estudio prospectivo en el que se recogen los datos relativos a las primeras visitas de endocrinología cuyo motivo de consulta fue "hiperprolactinemia" entre febrero de 04 y enero de 05. Las variables estudiadas fueron: edad, sexo, especialista que lo deriva, motivo por el que se determina la prolactina, cifra de prolactina, resultado de RMN hipofisaria (si existe), tratamientos farmacológicos y orientación diagnóstica. En todos los casos se determinaron hormonas tiroideas y se completó el estudio con las exploraciones complementarias necesarias (incluyendo la determinación de macroprolactina) para el diagnóstico diferencial a criterio médico.
Resultados: Se evaluaron un total de 93 pacientes (80 mujeres y 13 hombres) de edad media 32 años (15-77). La mayoria remitidos por su médico de familia (84%). En el 57% de los casos se solicitó la prolactina por síntomas relacionados con hiperprolactinemia (grupo 1): amenorrea/oligomenorrea (35,5%), galactorrea (8,6%), amenorrea+galactorrea (1,1%), disfunción eréctil (6,4%), infertilidad (5,4%). En el 43% restante, se solicitó por otros síntomas (grupo 2). Se realizó RMN hipofisaria en un 72% de los pacientes. No hubo diferencias en la cifra de prolactina entre los dos grupos (74,6 ± 66,6 vs 64,7 ± 42,4 ng/mL, P = 0,11) aunque el "pool" de prolactina fue mayor en el primero (53,7 ± 44,3 vs 35,5 ± 16,0 ng/mL, P = 0,03). Las orientación diagnóstica más frecuente en grupo 1 fue hiperprolactinemia idiopática (20%) y en el grupo 2 secundaria a fármacos (30%). El diagnóstico de micro o macroprolactinomas fue mayor en el primer grupo (8 pacientes) que en segundo (1 paciente) sin alcanzar significación estadística (P = 0,06). Tampoco hubo diferencias en la cifra de prolactina entre pacientes con RMN normal o no.
Conclusión: Prácticamente la mitad de los pacientes derivados por hiperprolactinemia no presentaban síntomas atribuibles a ésta. La magnitud de la hiperprolactinemia no ayudó al diagnóstico diferencial, sin embargo la presencia de prolactinomas fue mayor en aquellos pacientes que presentaron síntomas atribuibles a hiperprolactinemia.
121ADIPONECTINA Y FACTORES DE RIESGO CARDIOVASCULAR EN PACIENTES ADULTOS CON DÉFICIT DE GH TRATADOS CON GH
G. García Romero de Tejada, M. Lahera Vargas, F. Álvarez Blasco, C. Roa Llamazares, M. Peralta Watt y C. Varela da Costa
Endocrinología H.U. Ramón y Cajal. Madrid.
Estudiamos 18 pacientes adultos con déficit de GH antes y semestralmente tras iniciar tratamiento con GH durante 36 meses (3 pacientes) y 24 meses (15 pacientes). El tratamiento con GH se inició con 0,4-0,5 UI/día, incrementando la dosis hasta conseguir niveles séricos de IGF-1 adecuados para la edad y sexo de cada paciente. Se midieron niveles séricos de adiponectina, colesterol (Chol), LDL-c, HDL-c, triglicéridos, apoproteina B (apoB), apoproteina A (apoA), apoproteina E (apoE), lipoproteina (a) (Lp(a)), proteína C-reactive (PCR) y homocisteína. Se calcularon los cocientes Chol/HDL-c, LDL-c/HDL-c. En 16 pacientes se realizó un ecocardiograma transtorácico y se el midió el grosor de la pared del ventrículo izquierdo, las dimensiones auriculares y ventriculares y el volumen de inyección. También se les midió el grosor de la íntima media de la carótida interna. Los niveles de adiponectina (µg/ml) (mean ± SD) no se modificaron antes del tratamiento con GH (72,9 ± 14) (n = 18) vs a los 24 meses (64,9 ± 10,4) (n = 15) o a los 36 meses (77,3 ± 9,9) (n = 3). El Chol sérico se redujo a los 18 meses y se mantuvo a los 24 meses (basal: 268,7 ± 20 vs 18 meses: 215,4 ± 16,75 mg/dL, p < 0,001). El LDL-c sérico disminuyó al mes 12º, 18º y 24º (basal: 203,2 ± 21 vs 12º mes: 187 ± 22,6; p < 0,005, vs 18º mes: 180,5 ± 20,3, p = 0,002, vs 24º mes: 143,25 ± 19,2 mg/dL, p = 0,01). Los ratios COL/HDL-c y LDL-c/HDL-c disminuyeron a los 18 meses. Los niveles séricos de apoA, apoB y apoE no variaron, como tampoco hubo cambios en los niveles de TG o HDL-c. La Lp (a) sérica aumentó a partir del 6º mes (basal: 8,1 ± 2,1; 6º mes: 12,3 ± 2,9; 12º mes: 13,1 ± 2,9; 24º mes: 16,0 ± 3,4 mg/dL, p < 0,001). La PCR bajó de 0,6 ± 0,2 antes del tratamiento con GH hasta 0,3 ± 0,08 al 6º mes y 0,2 ± 0,1 al 12º mes (p < 0,05). Los niveles de homocisteína no se modificaron durante el seguimiento. No encontramos diferencias en el espesor de la íntima media carotídea ni en ninguno de los parámetros cardíacos estudiados. En contra de los datos obtenidos en acromegalia, el tratamiento sustitutivo con GH no modifica los niveles de adiponectina en adultos deficitarios de GH. Nuestros resultados concuerdan con tres estudios publicados realizados con adiponectina en este tipo de pacientes. El tratamiento con GH mejora los factores de riesgo cardiovascular tales como el perfil lipídico y los niveles de PCR y aumenta los niveles de Lp (a) hasta valores próximos a la normalidad.
122ALTERACIONES HIDROELECTROLÍTICAS SEVERAS TRAS CIRUGÍA HIPOFISARIA
M. González Boillos, J. Olivares Alcolea, S. Tofé Povedano, C. Sainz Álvarez y V. Pereg Macazaga
Endocrinología y Nutrición Hospital Universitario Son Dureta. Palma de Mallorca.
Paciente de 42 años remitida a nuestro hospital con el diagnóstico de craneofaringioma, ceguera por compresión quiasmática y panhipopituitarismo, para someterse a intervención quirúrgica. Se realiza abordaje transcraneal y en el postoperatorio la paciente inicia cuadro diabetes insípida central por sección de tallo hipofisario. En los días siguientes se produce un descenso paulatino de la natremia, reduciéndose la dosis de minurín. Posteriormente, inicia un cuadro brusco de poliuria (7000 ml en 24h) junto con disminución de conciencia y convulsiones con una concentración de Na en plasma de 175 mEq/dL, que obliga a reajustar el tratamiento. En los días posteriores se produce un descenso acusado de la natremia hasta alcanzar un valor de 116 mEq/dL. Se inició la determinación seriada de Na en orina observándose natriuresis elevadas (hasta 255 mEq/L) junto con hiponatremia y descenso persistente de la PVC. En una tomografía computerizada cerebral de control se aprecia hemorragia frontal bilateral, supraselar e intraventricular. Ante estos hallazgos, se llegó al diagnóstico combinado de diabetes insípida central + síndrome de pérdida de sal de origen cerebral. Se inició tratamiento combinado con desmopresina, fluidoterapia, ClNa hipertónico y fluorhidrocortisona, con lo que se consiguió mantener la natremia dentro de valores normales. No hemos encontrado un caso similar descrito en la literatura, en el que coexistan un déficit de ADH junto con la pérdida de sal de origen cerebral. Este último se debe a la elevación en sangre de péptidos natriuréticos de origen cerebral que disminuyen la reabsorción renal de sodio. El síndrome aparece con mayor frecuencia en niños y especialmente cuando se produce un sangrado parenquimatoso. Generalmente se trata de un cuadro autolimitado en el tiempo y suele ser suficiente el aporte extra de sodio, aunque en algunos casos, la administración de fluorhidrocortisona se ha mostrado útil para disminuir la natriuresis exagerada.
Conclusión: En pacientes con diabetes insípida central con difícil control de la natremia es preciso descartar la presencia de una natriuresis exagerada debida a la posible coexistencia de este cuadro.
123ANÁLOGOS DE LA SOMATOSTATINA EN ACROMEGALIA PERSISTENTE
M.T. Rivero Luis, I. Bernabeu Morón, I. Pinal Osorio, E. Fernández Rodriguez, J.M. Cabezas Agricola y J. Cabezas Cerrato
Endocrinología y Nutrición Hospital Clínico Universitario de Santiago. Santiago de Compostela, La Coruña.
Los análogos de la somatostatina (ASST) normalizan la IGF-1 en un 65% y la GH en un 70% de los casos aproximadamente. Sin embargo, estos datos proceden de estudios en los que con frecuencia ha existido una preselección de pacientes en base a su respuesta positiva al fármaco. En series sin preselección, su eficacia parece ser inferior y más acorde con nuestra impresión clínica previa. Para aclarar éste aspecto analizamos nuestros resultados en 21 pacientes no seleccionados con acromegalia persistente.
Material y métodos: 11 mujeres y 10 hombres de 45,57 ± 14,59 años de edad y con un seguimiento de 77,7 ± 56,45 meses.17 pacientes tenían un macroadenoma (1 intra, 6 supra, 8 con invasión de s. cavernoso y 2 ampliamente invasivos), 3 un microadenoma y 1 una silla turca vacía. El tratamiento primario fue quirúrgico en 17 (19 intervenciones: 14 TE y 5 TF) y con ASST en 4 (2 de ellos precisaron posteriormente 3 cirugías TE). Tras la cirugía, 5 presentaban restos intraselares, 4 en s. Cavernoso y 2 intra/supra/paraselares. 11 pacientes recibieron 12 tratamientos con radioterapia (10 convencional, 2 estereotáxica). 8 recibieron octreótido LAR y 13 lanreótido autogel, 10 recibieron simultáneamente cabergolina. La dosis y/o el intervalo de administración se ajustaron en función de la respuesta bioquímica.
Resultados: La GH media inicial fue 22,51 ± 13,61 ng/ml y la IGF-1 258,51 ± 125,79% (100% = limite superior del rango normal). En el primer control postquirúrgico la GH fue 12,5 ± 10,1 ng/mL y la IGF-1 243,85 ± 159,73 y al final del seguimiento de 3,06 ± 2,89 ng/ml y 121,78 ± 65,04% respectivamente. La IGF-1 disminuyó un 52,9% desde el diagnóstico y un 50% desde la cirugía y la GH un 86,4% y un 75,2%. El 24% consiguió buen control según consenso, el 62% una GH basal media < 2,5 ng/ml y el 43% una IGF-1 normal. En los pacientes con control inadecuado o malo, los ASST consiguieron un descenso de la IGF-1 de 37,52 ± 79% (en 3 no respondedores la IGF-1 aumentó un 86 ± 74%, en el resto disminuyó un 40,38 ± 26,47%) y de la GH de 54 ± 40%. Ninguno de los dos pacientes en tratamiento primario con ASST presentan buen control. Ningún paciente ha experimentado crecimiento tumoral durante el tratamiento.
Conclusiones: En nuestra serie, los ASST consiguen buen control en el 24% de los casos, GH segura en el 62% e IGF-1 normal en el 43%. Estos resultados son similares a otras series de pacientes no seleccionados. En los pacientes en los que no se alcanza un buen control se consigue un descenso promedio de la IGF-1 de un 37,52 ± 79% y de la GH de un 54 ± 40%.
124CARACTERIZACIÓN DE LOS EFECTOS DEL NEUROPÉPTIDO NEUROMEDINA U SOBRE LA SECRECIÓN DE GONADOTROPINAS EN LA RATA
E.M. Vigo Gago, J.M. Castellano Rodríguez, R. Fernández Fernández, V.M. Navarro Loro, J. Roa Rivas, A. Mayen Reyes, R. Pineda Reyes, E. Aguilar Benítez de Lugo, L. Pinilla Jurado y M. Tena Sempere
Dpto. Biol. Cel., Fisiología e inmunología Facultad de Medicina. Córdoba.
La Neuromedina U (NMU) es un neuropéptido identificado inicialmente en médula espinal porcina y posteriormente descrito en numerosos tejidos (hipotálamo y CNS, tracto gastrointestional, tracto urogenital, hipófisis) de distintas especies animales. NMU actúa a través de dos receptores: NMU1R, abundante en tejidos periféricos, y NMU2R, localizado selectivamente en regiones específicas del cerebro. Las acciones biológicas de NMU incluyen la inducción de vasoconstricción y contracción uterina, así como la participación en respuestas neuroendocrinas a situaciones de estrés y el control de secreción de ACTH. Adicionalmente, se ha informado que NMU se expresa abundantemente en el área hipotalámica ventromedial, donde parece actuar como factor inductor de saciedad. Recientemente, diversas evidencias experimentales han demostrado que numerosas señales primariamente implicadas en el control de la ingesta (tales como leptina, orexinas y ghrelina) participan en la regulación del eje gonadotropo, contribuyendo de ese modo a los sistemas de control integrado de balance energético y la función reproductora. No obstante, el posible papel de NMU en la regulación de la secreción de gonadotropinas no ha sido suficientemente analizado hasta la fecha. En el presente trabajo, hemos evaluado los efectos de NMU sobre la liberación de LH en la rata, empleando diversos modelos experimentales in vivo. La administración intracerebroventricular de 1 nmol NMU estimuló la secreción basal de LH en ratas macho peripuberales y ratas cíclicas en diestro (bajos niveles de LH circulantes). Por el contrario, NMU indujo una respuesta bifásica en ratas hembra en la tarde de proestro (pico preovulatorio de gonadotropinas), con estimulación a los 15-min e inhibición a los 60-min tras la administración del neuropéptido. De modo análogo, NMU inhibió, 60-min tras su administración, los niveles estimulados de LH en ratas macho orquidectomizadas y tras pre-tratamiento con el neuropéptido KiSS-1. En resumen, nuestros resultados demuestran que, en función de los niveles prevalentes de gonadotropinas, NMU ejerce un efecto dual (estimulador sobre niveles basales; predominantemente inhibidor sobre niveles estimulados) sobre la secreción de LH y sugieren la participación de este neuropéptido en el complejo sistema de control integrado del balance energético y la función reproductora. Trabajo financiado por proyecto BFI 2002-00176 y Beca de la Dirección General de Investigación y Desarrollo, Xunta de Galicia.
125DÉFICIT DE GH Y OTRAS HORMONAS HIPOFISARIAS DESPUÉS DE TRAUMATISMOS CRANEOENCEFÁLICOS
C. Aragón Valera*, G. García Romero de Tejada*, M. Peralta Watt*, C. Roa Llamazares*, P.J. Pinés Corrales*, T. Antón Bravo*, R. Sanchón Rodriguez*, M.A. Valero** y C. Varela da Costa*
*Endocrinología y Nutrición Hospital Ramón y Cajal. Madrid, **Endocrinología y Nutrición Hospital Nuestra Señora del Prado. Talavera de La Reina.
Estudios recientes sugieren que el déficit de hormonas hipofisarias no es infrecuente entre los supervivientes a un TCE. Aunque la diabetes insípida se reconoce fácilmente en la fase aguda tras el traumatismo, la ausencia de otras hormonas puede estar presente de manera sutil. El DGH en adultos, caracterizado por sensación de debilidad y pérdida del bienestar, suele pasarse por alto o atribuirse a otras causas. El episodio traumático puede ser difícil de recordar bien porque fue pequeño o bien porque ocurrió años antes. El objetivo de este estudio es evaluar la secreción de GH y de otras hormonas hipofisarias después de un TCE. Se seleccionó a 35 pacientes que sufrieron TCE entre 4 y 60 meses antes. Se recogieron los siguientes datos: tipo de traumatismo, puntuación en la Escala de Glasgow (EG) al ingreso, fracturas craneales y pérdida de conocimiento. Además se hicieron determinaciones de IMC, sodio, potasio, cortisol 9 a. m., LH, FSH, testosterona o estradiol, TSH, T4, prolactina e IGF-1. En los pacientes con sospecha de hipopituitarismo postraumático además se realizó un test de hipoglucemia insulínica o un test de estimulación con glucagón intramuscular para valorar el DGH. También se realizaron test de LHRH y TRH. Todos los pacientes contestaron dos tests para evaluar la calidad de vida: SF-36 y QOL-AGHDA. Los datos se expresaron como media ± DE y rangos. De los 35 pacientes, 25 fueron varones (71,4%) y 10 mujeres (28,6%), la edad media en el traumatismo fue de 34 ± 14,6 años (rango 18-61). En el 63% de los casos el traumatismo se debió a un accidente de tráfico, el 28,6% a precipitación y el 8,5% a otras causas. El 51% tuvo fracturas craneales en el momento de la admisión y el 17,15% pérdida de conocimiento. La puntuación en la EG fue 12,8 ± 3,1. El lapso de tiempo entre el traumatismo y el estudio (media ± DE) fue de 23,1 ± 17,3 meses. Los niveles basales de IGF-1 fueron bajos en 19 pacientes (54,3%) y normales en 16 (45,7%). 6 de los 18 pacientes a los que se practicó el test de hipoglucemia insulínica o el test de estimulación con glucagón tuvieron una respuesta de GH pobre. En tres pacientes se diagnosticó DGH severo con un pico de GH≤3 µg/L. En otros tres pacientes se demostró déficit parcial con GH≤10 µg/L. Todos estos pacientes con respuesta anormal tuvieron bajos niveles de IGF-1. Tres de los 35 pacientes (8,7%) tuvieron déficit de gonadotropinas después de LHRH. No se observaron deficiencias en el eje tiroideo ni adrenal. Muchos de los apartados de los tests de calidad de vida estuvieron en correlación con los niveles de IGF-1 y/o la respuesta de GH a la estimulación. Más del 50% de los pacientes que sufrieron TCE mostraron alteraciones en la función de la hipófisis anterior, y la mayoría de ellas afectan al eje GH-IGF-1. En los pacientes afectados se encontró correlación entre muchos aspectos del test de calidad de vida y la función del eje GH-IGF-1.
126EFECTOS DEL PÉPTIDO KISS-1 SOBRE LA SECRECIÓN DE GONADOTROPINAS EN LA RATA HEMBRA EN DISTINTOS PERÍODOS DEL DESARROLLO Y ESTADOS FUNCIONALES DEL EJE REPRODUCTOR
J. Roa Rivas, E. Vigo Gago, V.M. Navarro Loro, R. Fernández Fernández, J.M. Castellano Rodriguez, A. Mayen Reyes, R. Pineda Reyes, E. Aguilar Benítez de Lugo, L. Pinilla Jurado y M. Tena Sempere
Dpto. Biol. Cel., Fisiología e Inmunología Facultad de Medicina. Córdoba.
La hormona luteinizante (LH) y la hormona folículo-estimulante (FSH) juegan un papel central en el control de la función gonadal. El regulador primario de la liberación hipofisaria de ambas gonadotropinas es el decapéptido hipotalámico GnRH, cuya secreción pulsátil se encuentra, a su vez, bajo el control de una gran variedad de señales centrales (neurotransmisores y neuropéptidos) y periféricas (esteroides sexuales y otros). Recientemente, se ha demostrado un papel muy relevante del sistema ligando/receptor KiSS-1/GPR54 en el control hipotalámico del eje reproductor. No obstante, numerosos aspectos de la función reproductora de este sistema permanecen aún desconocidos. En este contexto, datos recientes de nuestro grupo han demostrado que el péptido KiSS-1 estimula potentemente la secreción de gonadotropinas y adelanta la llegada de la pubertad en ratas hembra prepúberes. Considerando que la función del eje reproductor y los niveles de gonadotropinas cambian considerablemente a lo largo del desarrollo, en el presente trabajo nos propusimos caracterizar la respuesta, en términos de secreción de LH, a la administración central (i.c.v.) del péptido KiSS-1 en la rata hembra, en diversas etapas del desarrollo post-natal (períodos neonatal, infantil, juvenil, puberal y adulto) y en diversos estados fisiológicos del sistema reproductor (ciclo estral, gestación y lactancia).Nuestros resultados indican que, a pesar de la ausencia de activación del eje gonadotropo antes de la pubertad, la administración de 1 nmol i.c.v. del péptido KiSS-1 estimula muy potentemente la secreción de LH en ratas neonatales (5-d), infantiles (15-d) y juveniles (23-d); respuestas similares a las obtenidas en ratas peripuberales. Más aun, la inyección central de KiSS-1 indujo una elevación muy significativa de los niveles de LH en todas las fases del ciclo estral (estro, diestro, proestro), así como en la preñez y la lactancia. No obstante, la magnitud y el curso temporal de estas respuestas estimuladoras varió en función del estado funcional del eje reproductor, siendo máximas en estro y diestro y mínimas durante la lactancia. En conclusión, nuestros datos contribuyen a la caracterización del papel relevante del sistema KiSS-1 en el control del eje gonadotropo, y evidencian que el péptido KiSS-1 es un muy potente estimulador de la secreción de LH a lo largo del desarrollo y en diversas condiciones funcionales del sistema reproductor en la rata hembra.
Financiado por una beca del Instituto de Salud Carlos III (Red de Centros RCMN C03/08 y proyecto PI042082)
127EFECTOS DEL PYY3-36 SOBRE LA LIBERACIÓN DE GNRH Y GONADOTROPINAS EN RATAS PREPÚBERES
R. Fernández Fernández, E. Vigo Gago, M. Tena Sempere, E. Aguilar Benítez de Lugo y L. Pinilla Jurado
Departamento de Biología Celular, Fisiología e Inmunología Facultad de Medicina. Córdoba.
En los últimos años se ha avanzado significativamente en la identificación de las señales implicadas en el control integrado de peso corporal y la función reproductora. Entre estas señales, el neuropéptido Y (NPY), la leptina, la ghrelina, las orexinas y la resistina parecen ejercer un importante papel integrativo. El polipéptido YY 3-36 (PYY3-36) es un péptido de origen gastrointestinal estructuralmente relacionado con el NPY y que opera a través de los receptores NPY Y2 e Y5. Recientemente, ha sido destacada su participación en el control del peso corporal como señal saciante (anorexigénica). A pesar de la identificación de numerosos efectos del NPY en el control endocrino de la reproducción, las acciones del PYY3-36 en este ámbito han sido escasamente analizadas impidiendo evaluar su posible participación como integrador neuroendocrino en el control coordinado del balance energético y la función reproductora. En el presente trabajo hemos analizado la expresión de los genes que codifican los receptores Y2 e Y5 en hipotálamo e hipófisis y los efectos "in vivo" e "in vitro" del PYY3-36 sobre la secreción de GnRH y gonadotropinas en ratas prepúberes. Los resultados obtenidos demostraron la expresión de los genes que codifican ambos receptores (aunque a nivel bajo) en hipotálamo e hipófisis. Por otra parte, el PYY3-36 estimuló "in vitro" la secreción de LH y FSH por hipófisis obtenidas de hembras y machos prepúberes, permaneciendo el efecto en ausencia de calcio extracelular. La administración intraperitoneal de PYY3-36 (3, 10, 30 µg/kg) no modificó de forma significativa las concentraciones plasmáticas de LH y FSH, mientras que su administración intracerebroventricular (3 nmol/rata) inhibió la secreción de LH solamente en ratas macho. El PYY3-36 (10-6 M) inhibió la secreción de GnRH "in vitro" exclusivamente en hipotálamos de ratas macho. En resumen, nuestros resultados evidencian que el PYY3-36 (al igual que la leptina, ghrelina y orexinas) participa en el control de la función reproductora a través de acciones a nivel hipotalámico e hipofisario. Igualmente parece existir un dimorfismo sexual en la respuesta al PYY3-36.
Este trabajo ha sido financiado por el proyecto BFI2002-00176 (DGESIC).
128EFECTOS DEL TRATAMIENTO SUSTITUTIVO CON GH EN PACIENTES ADULTOS DEFICITARIOS SOBRE LAS CONCENTRACIONES SÉRICAS DE ADIPONECTINA, LEPTINA, INSULINA Y PAPP-A
E. Aguilera*, C. Joaquín*, C. Pastor**, I. Salinas*, A. Lucas*, A. Cantón* y A. Sanmartí*
*Endocrinología y Nutrición, **Laboratorio de Bioquímica Hospital Universitari Germans Trias i Pujol. Badalona.
El déficit de hormona de crecimiento (DGH) en la edad adulta se asocia a dislipemia, obesidad central, riesgo de enfermedad cardiovascular y a resistencia a la insulina. La concentración de PAPP-A (pregnancy associated plasma protein A) se ha hallado aumentada en el síndrome coronario agudo y en diabéticos tipo 2 con dislipemia y se considera un marcador de inestabilidad de la placa de ateroma.
Objetivo: Evaluar los efectos del tratamiento sustitutivo con GH sobre la adiponectina (ADP), leptina (LEP), PAPP-A y la resistencia a la insulina en enfermos con DGH.
Material y métodos: 14 enfermos con DGH (edad 40,9 ± 14,4 anos, 10 mujeres) fueron evaluados al inicio del tratamiento con GH, al ano y 10 de ellos también a los 4 anos. Se compararon estos pacientes con 14 controles apareados por edad, sexo e índice de masa corporal (IMC). Se determinaron: ADP, LEP, PAPP-A, insulinemia, glucemia, HbA1c, HOMA, perfil lipídico, IMC, índice cintura-cadera, masa magra y grasa.
Resultados: Al inicio no se objetivaron diferencias entre los pacientes con DGH y los controles respecto a la ADP, insulinemia, glucemia, perfil lipídico, HOMA o PAPP-A. La LEP fue significativamente mayor en los pacientes con DGH (24,69 ± 16,12 vs 13,69 ± 12,01 ng/ml, p < 0,04). Tras el tratamiento con GH, los enfermos con DGH mostraron un incremento significativo del IMC tanto al ano (26,4 ± 3,9 vs 27,3 ± 3,6 kg/m2 p < 0,02) como a los 4 anos (26,3 ± 4,1 vs 28,4 ± 4,3 kg/m2 p < 0,005) con tendencia hacia el aumento de la masa magra y la disminución de la masa grasa. No hubo diferencias en la ADP, LEP, glucemia o perfil lipídico, pero la insulinemia y el HOMA fueron significativamente más altos al ano (6,59 ± 3,74 vs 10,47 ± 7,01 mU/l p < 0,004 y 13,2 ± 6,9 vs 19,6 ± 11,9 mU/l p < 0,02 respectivamente) y a los 4 anos (1,43 ± 0,88 vs 2,47 ± 1,74 y 3,4 ± 1,5 vs 4,6 ± 2,7 p < 0,02) y la PAPP-A fue más baja al ano (1,27 ± 0,85 vs 0,99 ± 0,66 mcUI/ml p < 0,037).
Conclusiones: A pesar del pequeno número de enfermos estudiados no parece que ADP ni LEP jueguen un papel importante en la resistencia a la insulina en los enfermos con DGH dada la ausencia de cambios tras el tratamiento con GH. Dicho tratamiento aumenta la insulinemia y la resistencia a la insulina medida por HOMA, así como el IMC. La disminución de PAPP-A al ano de tratamiento con GH sugeriría una posible disminución del riesgo de síndrome coronario agudo en los enfermos con DGH.
129EL HIPOTIROIDISMO ADULTO PROVOCA GRAVES ALTERACIONES DE LA NEUROGENESIS Y DESÓRDENES DEL COMPORTAMIENTO QUE SON REVERSIBLES TRAS EL REEMPLAZO HORMONAL
A. Montero Pedrazuela*, C. Venero Núnez**, R. Lavado Autric* y A. Guadaño Ferraz*
*Endocrinología Molecular Instituto de Investigaciones Biomédicas. Madrid, **Dpto. Psicobiología Facultad Psicología, UNED. Madrid.
Las alteraciones hormonales están implicadas en muchas patologías asociadas con la edad como enfermedades neurodegenerativas y trastornos psiquiátricos. En el adulto, las alteraciones del estado tiroideo cursan con frecuencia con cambios psicológicos y trastornos del estado del ánimo como la depresión. En los últimos años se ha demostrado que en el adulto continúan generándose neuronas a partir de células madre en el giro dentado del hipocampo. Estas neuronas generadas de novo se integran funcionalmente y podrían estar implicadas en procesos de memoria y aprendizaje, así como en el mantenimiento del estado de ánimo de un individuo. Se ha descrito que la disminución de la neurogenesis adulta induce trastornos depresivos y que la eficacia del tratamiento con antidepresivos depende de la generación de nuevas neuronas en el hipocampo. Por ello, el conocimiento del proceso de adquisición de nuevas neuronas en el adulto y su modulación puede suponer una herramienta importante para paliar las enfermedades neurodegenerativas y psiquiátricas. Entre los factores descritos que modulan este proceso se encuentran factores circulantes, incluyendo hormonas. En este trabajo estudiamos in vivo si la deficiencia de hormonas tiroideas (HT) durante un corto período en la rata adulta: 1) afecta la neurogénesis en el hipocampo, 2) induce alteraciones del comportamiento en un modelo experimental de depresión (test de Porsolt) y 3) si el reemplazo hormonal revierte las anormalidades observadas. Los resultados indican que el hipotiroidismo adulto disminuye significativamente la capacidad proliferativa del hipocampo al reducir tanto el número de precursores neuronales en proliferación como el número de unidades proliferativas. Además, la deficiencia de HT afecta gravemente a los neuroblastos inmaduros en su número y distribución, y provoca un desarrollo anormal de su árbol dendrítico. Estos cambios en la neurogénesis afectan a la función hipocampal, dado que los animales hipotiroideos mostraron un mayor comportamiento depresivo que los controles, mientras que el tratamiento con HT fue capaz de recuperar la tasa de neurogénesis en el hipocampo y normalizar el comportamiento de estos animales. Nuestros estudios demuestran por primera vez que la neurogenesis en el adulto es sensible a HT y son la primera caracterización de una alteración reversible debida a deficiencia de HT sobre la proliferación y maduración neuronal en mamíferos adultos in vivo. Además estos resultados sugieren que los desórdenes del comportamiento depresivo provocados por el hipotiroidismo adulto en humanos pueden estar relacionados, entre otros, con la disminución de la neurogenesis en el hipocampo. Todo lo anterior podría ser importante para futuras aplicaciones terapéuticas en la modulación de la neurogénesis endógena y desórdenes del comportamiento. AMP es becaria predoctoral y AGF y CV contratados por el programa Ramón y Cajal del MEC.
Trabajo financiado por el proyecto BFI2001-2412 del MCYT.
130EL TRATAMIENTO CON HORMONA DE CRECIMIENTOS (GH) INCREMENTA LOS NIVELES DE BCL2, BLOQUEANDO LA ACTIVACIÓN DE CASPASA 9 Y LOS NIVELES DE CASPASA 3 EN EL HIPOTÁLAMO DE ANIMALES VIEJOS, PERO NO EN ANIMALES CASTRADOS
V. Salazar*, A.M. Lechuga**, C. Castillo*, A.F. Tresguerres*, E. Vara***, C. Ariznavarreta*, J. Tresguerres* y J.A. Chowen**
*Dpto. Fisiología Fac. Medicina. UCM. Madrid, **Dpto. Endocrinología Pediátrica y Laboratorio de Investigación H. Infantil Universitario Niño Jesús. Madrid, ***Dpto. Bioquímica Fac. Medicina. UCM. Madrid.
La GH ejerce un efecto neuroprotector en animales viejos. Su administración incrementa el número total de neuronas, posiblemente disminuyendo la apoptosis, pues no estimula la neurogénesis. Las caspasas son una familia de proteínas apoptóticas, cuya activación se bloquea por el factor de supervivencia celular Bcl2. Nuestro objetivo es analizar los mecanismos moleculares de apoptosis que puedan ocurrir en el hipotálamo con el envejecimiento, y el posible efecto de la administración de GH. Se usaron 5 grupos (n = 8) de ratas hembras Wistar. Un grupo se sacrificó a los 2 meses de vida (m), los demás a los 24m: un segundo grupo no fue tratado, y un tercero recibió GH s.c. (2 mg/kg/d) las últimas 10 semanas. Los restantes grupos fueron castrados a los 12m. De éstos, un grupo se mantuvo sin tratamiento, otro grupo recibió GH GH s.c. (2 mg/kg/d) las últimas 10 semanas de vida, y un último grupo recibió valerianato de estradiol S.C. (150 µg/animal/S) el mismo tiempo. Los cerebros fueron extraídos e inmediatamente congelados a 70ºC. Los niveles de Bcl2 se determinaron en homogeneizado de hemicerebro mediante ELISA. Se disecaron los hipotálamos del hemicerebro contrario, y se determinaron los niveles de caspasa 9 y caspasa 3, mediante Wetern Blot empleando un anticuerpo monoclonal anticaspasa 9 (MBL®), y otro policlonal anticaspasa 3 (BD Pharmingen®).
Resultados: El tratamiento con GH logra un incremento significativo (p < 0,01) de Bcl2 en todos los animales, al igual que el estradiol en las hembras castradas. Dentro del grupo de animales intactos, las ratas viejas presentan un incremento significativo (p < 0,05) del fragmento activo de la caspasa 9 con respecto a los animales jóvenes. Dicho incremento se revierte con la administración de GH. La caspasa 3 también presenta una tendencia a incrementar con el envejecimiento, y los niveles de ambas formas se reducen significativamente con la administración de GH. Las ratas castradas, por el contrario, la administración de GH tiende a incrementar los niveles de caspasa 3, si bien no parece ejercer efecto significativo sobre la caspasa 9.La disminución de caspasa 9 activa y de ambas fracciones de caspasa 3 observada con la administración de GH, concuerda su efecto neuroprotector. Esto podría ocurrir a través de la acción de Bcl2 que bloquea la activación de la caspasa 9, iniciadora de la apoptosis. Dicho efecto podría precisar de la presencia de estrógenos, pues no ocurre en las hembras castradas.
Trabajo subvencionado por CAM 8.5/0062/2001 y proyecto GHESME2002 del IMSERSO.
131¿ES ÚTIL LA RESPUESTA A ANTAGONISTAS DOPAMINÉRGICOS EN EL DIAGNÓSTICO DIFERENCIAL DE LA HIPERPROLACTINEMIA?
C. Abreu Padín*, J. Galofré Ferrater*, E. Santos Mazo*, A. González Hernández**, J. Zubieta Zárraga*** y J. Salvador Rodríguez*
*Departamento de Endocrinología y Nutrición, **Departamento de Bioquímica, ***Departamento de Radiología Clínica Universitaria de Navarra. Pamplona.
La utilidad de los test dinámicos mediante el uso de antagonistas dopaminérgicos (AD) en el estudio de la hiperprolactinemia es controvertido. Su correcta interpretación puede ayudarnos a identificar qué pacientes son susceptibles de evaluación mediante resonancia magnética (RM) teniendo en cuenta que es una prueba costosa y estresante. Algunos grupos estudian la respuesta de TSH y PRL al antagonismo con domperidona en pacientes hiperprolactinémicos en su práctica habitual. Con el objeto de evaluar la utilidad del test con antagonistas dopaminérgicos (AD) determinamos la respuesta de PRL y TSH a los 30 y 60 minutos tras la administración intravenosa de 10 mg de metoclopramida en 51 pacientes con hiperprolactinemia (PRL> 24ng/mL). La respuesta se consideró normal si la prolactina alcanzaba un incremento superior al 100% (N: PRL30/PRL0 ≥ 2) y el incremento de TSH no excedía de 2,1 mU/L, siendo este dato útil solo en el diagnóstico diferencial de respuestas de PRL patológicas. Los pacientes fueron agrupados según el diagnóstico final en: macroprolactinoma (Mp, n = 7), microprolactinoma (mp, n = 9), compresión del tallo hipofisario (CT, n = 5), hiperprolactinemia secundaria a fármacos (SF, n = 3), hiperandrogenismo funcional ovárico (HFO, n = 6) e hiperprolactinemia idiopática (HI, n = 21). Los datos están presentados como mediana y amplitud intercuartil y analizados mediante estadística no paramétrica para comparación de muestras independientes (Kruskal-Wallis). Se observan diferencias estadísticamente significativas en el incremento relativo de la concentración de prolactina entre las distintas patologías (Mp: 34,89 [19,71; 54,59]; mp: 43 [9,10; 48,43]; CT: 47,75 [28,50; 122,35]; SF: 31,33 [2,36; --]; HFO: 466,55 [70,89; 1066,95] y HI: 533,79 [211,86; 877,77]). Apenas se advierte incremento en los pacientes con mp, Mp, CT y SF. Sin embargo, el grupo de HI y HFO mostró un notable aumento de prolactina tras administración de metoclopramida. La concentración de TSH basal fue similar en los distintos grupos. Tras la administración de metoclopramida se objetivo un incremento exagerado de TSH en los pacientes con mp (2,72 [2,10; 3,89]) y Mp (1,82 [1,18; 2,75]), mientras que no existía estimulación significativa en los grupos CT (0,53 [25,01; 1,11]) y SF (-0,04 [-0,55; --]). Estos resultados sugieren que el test de metoclopramida es útil en la valoración etiológica inicial de los pacientes con hiperprolactinemia, reduciendo la posibilidad de imagen patológica en la RM en aquellos pacientes que presentan una respuesta normal al test.
132¿ESTÁ JUSTIFICADO EL TRATAMIENTO EMPÍRICO CON CORTICOIDES ANTE LA SOSPECHA CLÍNICA DE HIPOFISITIS?
L. Choque Uño*, R. Hernaez Rodríguez*, M. Giménez Álvarez*, I. Halperin Rabinovich*, B. Gómez Ansón**, M. Puig Domingo* y G. Sesmilo León*
*Endocrinología y Nutrición, **Radiodiagnóstico, CDI Hospital Clínic, Universitat de Barcelona. Barcelona.
El diagnóstico de hipofisitis representa un reto debido a la dificultad del diagnóstico diferencial con otros procesos de la región selar. La presentación habitual es en forma de diabetes insípida y disfunción hormonal más o menos severa. Los pacientes pueden presentar una recuperación de la función hipofisaria espontánea o inducida por corticoterapia, así como reducción del tamaño de la lesión, lo que confirma el diagnóstico y evita exploraciones cruentas.
Objetivo: Discutir aspectos relevantes del manejo y evolución de un paciente con sospecha diagnóstica de hipofisitis.
Caso: Varón de 18 años que consulta por polidipsia-poliuria de un mes de evolución; refería antecedentes de traumatismo craneoencefálico leve 6 meses antes. La evolución hormonal inicial mostraba una ligera hiperprolactinemia (57 ng/ml) con preservación de la función adenohipofisaria y diabetes insípida central. La RMN mostró aumento global de la hipófisis con extensión supraselar y contacto quiasmático, hipointensidad central y captación periférica de contraste; ausencia de señal neurohipofisaria y tallo hipercaptante y engrosado con posible captación de contraste en hipotálamo. Dos semanas después, antes de iniciar algún tratamiento, se objectivó panhipopituitarismo clínico y analítico. No había ningún otro dato clínico asociado que sugiriera un diagnóstico etiológico. Las exploraciones complementarias, que incluyeron radiología torácica, ortopantomografía, seriada ósea, punción lumbar por marcadores (ADA, betaHCG, alfafetoproteína) y cultivos para bacterias y micobacterias, ECA en plasma, y estudio de TBC sistémica, fueron negativas. Con el diagnóstico de presunción de hipofisitis, se inició tratamiento empírico con prednisona a dosis de 1mg/kg. El tratamiento con 50 mg/d de prednisona se mantuvo durante 2 meses, con reducción muy progresiva durante 6 meses más y después dosis sustitutivas de 5 mg hasta la recuperación del eje propio. Durante el seguimiento, se evidenció recuperación de la función tiroidal al mes, gonadal a los 6 meses y corticotropa al año, con persistencia de la diabetes insípida, quedando pendiente de reevaluar el eje somatotropo. Radiológicamente se evidenció una reducción de la lesión selar del 30% al mes, del 70% a los 3 meses; actualmente el volumen selar es normal, persistiendo hipointensidad central y captación periférica de contraste.

Conclusión: El diagnóstico de hipofisitis requiere un elevado índice de sospecha en base a los datos clínicos, biológicos y radiológicos. Antes de realizar exploraciones cruentas puede estar justificada una prueba terapéutica con corticoides, ya que en caso de respuesta positiva, contribuye a confirmar el diagnóstico y facilita la recuperación funcional hipofisaria.
133ETIOLOGÍA DE LA HIPERPROLACTINEMIA EN UN HOSPITAL TERCIARIO: ANÁLISIS RETROSPECTIVO
T. Antón Bravo*, A. Sánchez Arias**, M. Peralta Watt*, P.J. Pinés Corrales*, C. Roa Llamazares*, R. Sanchón Rodríguez* y C. Varela da Costa*
*Endocrinología Ramón y Cajal. Madrid, **Medicina Familiar y Comunitaria Gregorio Marañón. Madrid.
Introducción: La hiperprolactinemia puede ser debida a multitud de causas: tanto fisiológicas como el embarazo, la lactancia, el ejercicio físico o el estrés, como patológicas, entre las que destacan por su frecuencia los adenomas hipofisarios y los fármacos. Otras causas menos frecuentes son el hipotiroidismo, la Insuficiencia renal crónica o el Síndrome de Ovario poliquístico.
Objetivo: Analizar, mediante un estudio retrospectivo, la etiología de las hiperprolactinemias en un hospital terciario durante un período de 12 meses, así como la distribución de dichas etiologías por servicios.
Material y métodos: Se valoraron 2352 determinaciones de prolactina realizadas en el laboratorio de Endocrinología y Nutrición del Hospital Ramón y Cajal de Madrid entre enero de 2003 y enero de 2004. Se obtuvieron 456 casos de hiperprolactinemia, considerándose como patológicas aquellas determinaciones mayores de 17 ng/mL en varones y 25 ng/mL en mujeres. De ellas se tuvo acceso a 153 historias clínicas, en las que se revisaron el servicio solicitante de la prueba, las características clínicas, etiológicas y el tratamiento recibido. Se consideró "sin diagnóstico" a aquellas hiperprolactinemias en las que no se llegó a solicitar RMN craneal, e "idiomáticas" a aquellos casos en los que no se encontró ninguna causa de la hiperprolactinemia a pesar de haberse completado el estudio.
Resultados: De los 153 pacientes, 18 eran varones y 135 mujeres. La edad media fue de 32,9 años. No hubo diferencias significativas en la distribución por edades de las etiologías salvo que los adenomas no funcionantes aparecían en sujetos mayores, con una edad media de 58,7 años. La etiología más frecuente fue "sin diagnóstico", con cifras de prolactina media de 40 ng/ml. De entre las diagnosticadas, lo más frecuente fueron las idiopáticas, seguidas de los microprolactinomas y los fármacos, siendo el resto de etiologías anecdóticas. Estudiando las etiologías por servicios, se encontró que en el servicio de Psiquiatría sólo se diagnosticaban las causas farmacológicas (15,40%) quedando el resto "sin diagnóstico". En el servicio de ginecología quedaban sin diagnosticar también un 60,7% de las hiperprolactinemias. En el servicio de Endocrinología, la distribución fue de 30,9% de microprolactinomas, 29% idiopática, 23% de otras causas infrecuentes, 10,9% "sin diagnóstico" y 3,6% fármacos.
Conclusiones: La hiperprolactinemia queda frecuentemente sin diagnóstico, especialmente cuando el servicio solicitante es psiquiatría o ginecología, o cuando las cifras de prolactina son bajas. En endocrinología suele progresarse más en el estudio. Por lo tanto disminuye la tasa de sin diagnóstico, y aumenta la hiperprolactinemia idiopática. Las causas más frecuentes entre las diagnosticadas son microadenomas y fármacos. La frecuencia elevada de microadenomas encontrados en nuestro servicio puede deberse al diagnóstico incorrecto como tales de incidentalomas hipofisarios acompañados de hiperprolactinemia de otro origen.
134EVALUACIÓN DE LA APLICACIÓN DEL PROTOCOLO DE RETIRADA DE TRATAMIENTO MÉDICO EN LOS MACROPROLACTINOMAS EN EL S. DE ENDOCRINOLOGÍA Y NUTRICIÓN DEL HOSPITAL CENTRAL DE ASTURIAS
C. García Delgado*, S. Valdés Hernández**, A. Urioste Fondo**, J. Aller Granda**, P. Boix Pallarés**, E. Delgado Álvarez**, M.J. Díaz Fernández**, F. Díaz Cadórniga**, A. Lavilla Corcovado** y A. Rabal Artal**
*Endocrinología y Nutrición Complejo Hospitalario Donostia. San Sebastián, **Endocrinología y Nutrición Hospital Central de Asturias. Oviedo.
En la mayoría de los pacientes con macroprolactinoma (MP), la cifra de prolactina (PRL) se normaliza y el tamaño tumoral disminuye significativamente durante el primer año de tratamiento. Sin embargo, el riesgo de recidiva hace que éste se mantenga de forma indefinida y que haya muy pocos estudios sobre qué ocurre tras la suspensión del mismo. Desde 1986, en el Servicio de Endocrinología y Nutrición del Hospital Central de Asturias, se realiza suspensión del fármaco en los MP que cumplen los siguientes criterios: tratamiento médico durante un mínimo de 5 años, cifra de PRL previa a la suspensión menor de 5 ng/dl en al menos 3 determinaciones consecutivas con dosis bajas (2,5 mg en el caso de la bromocriptina) y ausencia de restos o resto pequeño y con tamaño estable en las pruebas de imagen. Tras la retirada, el seguimiento se hace de la siguiente forma: determinación de cifra de PRL a los 1, 2, 4, 6 y 12 meses y después cada medio año; prueba de imagen durante el primer año y siempre que haya elevación significativa de la PRL. Los datos han sido recogidos de las historias clínicas de los MP diagnosticados entre 1980 y 1999.
Resultados: Se realizó retirada de tratamiento médico en 11 de los 23 MP diagnosticados (48%). El tiempo medio con tratamiento fue de 11,5 ± 5 años (5-20) y el tiempo medio de seguimiento tras la suspensión del fármaco de 6,6 ± 3,5 años (0,25-12). Hubo recidiva en 2 de los 11 pacientes (18%); en ambos casos se detectó a los 3 meses por elevación de la cifra de PRL, sin variación del tamaño tumoral ni clínica derivada de la hiperprolactinemia. En uno de ellos, no se cumplían los criterios previos a la retirada.
Conclusiones: La suspensión del tratamiento médico fue exitosa en el 82% de los pacientes, superior a lo recogido en la literatura, quizá en relación con el estricto protocolo de retirada. En los dos casos de recidiva, no hubo riesgo para los pacientes, ya que se detectó de forma temprana por elevación de PRL, sin modificación del tamaño tumoral ni clínica drivada de la hiperprolactinemia.
135FACTORES DE RIESGO CARDIOVASCULAR EN LA ACROMEGALIA. EFECTOS DEL TRATAMIENTO A LARGO PLAZO CON ANÁLOGOS DE SOMATOSTATINA
A. Abad, A. Aragón, M.C. Montáñez, J. Aller, A. Simal, P. Manzano y B. Oliván
Endocrinología y Nutrición Hospital Universitario Puerta de Hierro. Madrid.
La acromegalia se caracteriza por alta morbimortalidad que se produce, en un 60% de los casos, por patología cardiovascular (CV).
Objetivo: Analizar la prevalencia, en pacientes acromegálicos, de varios factores de riesgo CV (obesidad, hipertensión arterial, HDL-C, triglicéridos (Tg) y alteraciones del metabolismo glucémico), así como el efecto del tratamiento a largo plazo con análogos de somatostatina sobre estos factores. Se han estudiado de forma retrospectiva 32 pacientes (12 hombres y 20 mujeres, edad media 37 ± 13 años) intervenidos de acromegalia y tratados con análogos de somatostatina tras la cirugía. Se han determinado: test de tolerancia oral a la glucosa (TTOG), Tg, HDL-C, peso, talla, IMC y tensión arterial (TA). La valoración se realizó al diagnóstico, antes del inicio del tratamiento y durante la utilización de los análogos de somatostatina (tiempo de seguimiento entre 2 y 5 años). La diabetes se diagnosticó según los criterios de la Asociación de Diabetes Americana, la obesidad según los de la Sociedad Española para el Estudio de la Obesidad y el resto de parámetros según los del Panel de Tratamiento del Adulto III para el diagnóstico del síndrome metabólico (TA ≥ 130/80 mmHg; Tg ≥ 150 mg/dl y HDL-C < 40 mg/dL en hombres y < 50 mg/dL en mujeres). Al diagnóstico, el 53,8% eran hipertensos, el 27% obesos, y el 42% tenían sobrepeso; el 23% de los pacientes presentaron diabetes (DM), el 11,5% glucemia basal alterada (GBA), el 19% intolerancia a la glucosa (ITG); el 40% aumento de Tg y el 55,5% disminución de HDL-C. Tras la cirugía, y antes del inicio del tratamiento con análogos de somatostatina, 21 pacientes tenían glucemia normal, 2 GBA, 3 ITG, y 5 eran diabéticos. Se excluyó un paciente por no tener determinación de glucemia previa al inicio del tratamiento. De los 5 pacientes diabéticos, 1 empeoró su control glucémico, 3 mantuvieron el mismo nivel de control y 1 mejoró. De los 21 pacientes con normoglucemia antes del inicio del tratamiento 4 empeoraron: 3 desarrollaron GBA y 1 DM. Los 3 pacientes con ITG normalizaron la glucemia. El 11% de los pacientes mejoraron la TA, el 4% la empeoraron y en el resto no se modificó. El 70% mantuvo el mismo IMC, el 26% lo aumentó y un 4% lo disminuyó. El efecto sobre los Tg y el HDL-C no fue valorable por el escaso número de pacientes que lo presentaron alterado tras la cirugía.
Conclusiones: Los pacientes con acromegalia tienen un aumento de la prevalencia de HTA, obesidad y alteraciones del metabolismo glucémico y lipídico, factores de riesgo que contribuyen al aumento de la morbilidad y mortalidad cardiovascular de esta enfermedad. El tratamiento con análogos de somatostatina a largo plazo produce un efecto variable sobre el metabolismo de los carbohidratos en los pacientes acromegálicos. La mayoría de nuestros enfermos no mostraron cambios significativos, sin embargo, en un pequeño subgrupo, el control glucémico empeoró y en otro mejoró.
136GIGANTISMO HIPOFISARIO
A. Aragón*, A. Abad*, R. Domínguez*, M. Brito*, J. Alcañiz*, B. Lecumberri*, J. García-Uría** y T. Lucas*
*Endocrinología y Nutrición, **Neurocirugía Hospital Universitario Puerta de Hierro. Madrid.
El gigantismo hipofisario se produce por un exceso de secreción de GH (adenoma hipofisario en un 99% de los casos) antes del cierre epifisario. De nuestra serie de 509 pacientes acromegálicos, 15 (3%) son gigantes; 13 varones y 2 mujeres. La edad al diagnóstico fue de 21,9 ± 9,1 años pero retrospectivamente el inicio de la enfermedad se situó a los 12,7 ± 3,6 años (tiempo de evolución medio 7 ± 5 años). La talla media final fue de 193,5 ± 9,7 cm. Todos los pacientes tenían rasgos acromegálicos, 21% eran hipertensos, 15,3% diabéticos, ningún paciente presentaba galactorrea y el 35,7% tenía algún defecto campimétrico. La GH basal mediana era 34,7 µg/L (rango 2-162,4); la IGF-1 basal media, 1153 ± 269 m g/L; el 25% de los pacientes presentaban hiperprolactinemia y el 23% déficit de gonadotropinas. Los tumores eran invasores en un 70,9% (Grados III-IV) y sólo uno de ellos era un microadenoma. Todos los pacientes, a excepción de uno, fueron operados por vía transesfenoidal. El paciente que no se operó, fue tratado con radioterapia convencional en dos ocasiones controlando su enfermedad. Aunque se produjo un importante descenso de la GH basal postquirúrgica, mediana 6,2 µg/L (0,3-100), sólo dos pacientes se consideraron curados después de la cirugía. Dos pacientes precisaron una segunda intervención que no consiguió controlar el exceso de producción de GH. No hay seguimiento postquirúrgico de un paciente. Seis pacientes fueron tratados con diferentes técnicas de radioterapia a lo largo del seguimiento: 1 con radiocirugía (no controlado), 1 con radioterapia estereotáxica fraccionada (controlado) y 4 con radioterapia convencional (tres controlados). Al final del seguimiento: 7 de los pacientes están controlados, 6 no reúnen criterios de curación y están en tratamiento médico y una paciente falleció por la extensión tumoral.
Conclusiones: En nuestra serie, el gigantismo se presenta con comorbilidades (hipertensión arterial, diabetes mellitus) en el momento del diagnóstico en un número importante de pacientes. El crecimiento longitudinal se acompaña sistemáticamente de rasgos acromegálicos y el comienzo de la clínica no es tan agudo como se ha descrito en otras series de pacientes más jóvenes. Bioquímicamente es severo y se asocia a tumores grandes y agresivos lo que hace difícil el control y obliga a asociar varios métodos de tratamiento.
137LA ACROMEGALIA NO CONTROLADA SE ASOCIA CON UN INCREMENTO DE FACTORES DE RIESGO CARDIOVASCULARES, MORBILIDAD Y MORTALIDAD
N. Sucunza Alfonso*, M.J. Barahona Constanzo*, L. Sojo Vega*, T. Puig** y S. Webb*
*Endocrinología y Nutrición, **Epidemiología Hospital de la Santa Creu i Sant Pau. Barcelona.
Objetivo: Evaluar la prevalencia de los factores de riesgo cardiovasculares, morbilidad y mortalidad en una cohorte de pacientes con acromegalia seguidos en nuestro centro desde 1982.
Métodos: Se registraron retrospectivamente datos clínicos de 72 pacientes (de un total de 110) con acromegalia. Los datos de los pacientes que cumplían criterios de curación (GH < 2,0 µg/L 2 horas tras SOG e IGF-1 normalizada para edad y sexo) fueron comparados con los que no cumplían dichos criterios.
Resultados: La edad media al diagnóstico fue de 41 ± 3,2 años, IMC 27,7 ± 1,2 Kg/m2 y 64,3% eran mujeres (N = 110). De los pacientes revisados (N = 72), 77,8% presentaron macroadenomas, 82% recibieron cirugía transesfenoidal y 39,7/5,1% radioterapia convencional/esterotáxica. Hasta la fecha, 71% presentan remisión (75% microadenomas; 70% macroadenomas) y en 11,1% se observó recidiva de la enfermedad (100% macroadenomas) con un seguimiento medio de 15,5 años. Los factores de riesgo cardiovasculares observados fueron tabaquismo en 24,6%, hipertensión en 49,2%, 38,3% diabetes (1 tipo 1), 9,5% intolerancia hidrocarbonada y 35,3% dislipemia (20% mixta). La morbilidad observada fue 5 casos de enfermedad cardiovascular (6,9%) y 3 de cerebrovascular (4,2%), 2 arteriopatía periférica (2,8%), 3 apnea obstructiva del sueño (4,2%) y 11 neoplasias (15,3%; 5 mama, 2 colon, 1 páncreas, 1 tiroideo, 1 gástrico y 1 testicular). La mortalidad en nuestra serie fue 12,5% (4 cardiovascular, 3 cáncer, 1 cerebrovascular y 1 por broncoaspiración) a una edad media de 63,5 (30,1-85,5) años. De los pacientes que murieron 16,6% habían recibido radioterapia y 8,9% no (p < 0,05). La prevalencia de diabetes (64,7 vs 26,3%; p < 0,01) y de muerte (33,3 vs 4,7%; p < 0,005) fue mayor en pacientes que no cumplían criterios de curación.
Conclusiones: Los pacientes con acromegalia presentan una mayor prevalencia de factores de riesgo cardiovascular, morbilidad y mortalidad especialmente si persiste la hipersecreción de GH.
Financiado en parte por una beca de Pfizer, España
138LA ADIPOQUINA RESISTINA REGULA LA FUNCIÓN DE LAS CÉLULAS SOMATOTROPAS
F. Rodríguez-Pacheco*, A.J. Martínez-Fuentes*, S. Tovar**, C. Diéguez**, J.P. Castaño* y M.M. Malagón*
*Dpto. de Biología Celular, Fisiología e Inmunología Univ. de Córdoba. Córdoba, **Dpto. de Fisiología Univ. de Santiago de Compostela. Santiago de Compostela.
En los últimos años se han descubierto diversas proteínas de origen periférico que intervienen en el control neuroendocrino del metabolismo energético, entre las que destacan varias adipoquinas producidas por el tejido adiposo, que aparece como una fuente cada vez más importante de hormonas. Así, en 2001 se aisló una proteína expresada y secretada por adipocitos de ratón que se denominó resistina por su capacidad para reducir la tolerancia a glucosa en ratones y antagonizar la captación de glucosa inducida por insulina en células 3T3-L1. Recientemente, se ha descrito que el tratamiento con hormona del crecimiento (GH) aumenta los niveles de ARNm de resistina en el tejido adiposo, lo que ha llevado a proponer que la resistina podría mediar el efecto de GH en la sensibilidad a insulina. Puesto que se ha detectado la presencia de resistina en hipófisis de rata, en el presente estudio nos propusimos analizar si, de manera recíproca, la resistina puede regular la producción de GH por las somatotropas. Para ello, empleamos dispersiones celulares de hipófisis de rata que se mantuvieron en cultivo en presencia de resistina (1 microM, 4 h). Tras los tratamientos, los medios de cultivo se recogieron para la determinación de la secreción de GH mediante RIA específico. Igualmente, se extrajo el ARNm de las células tras el cultivo para un análisis de expresión génica por RT-PCR en tiempo real. Para determinar la posible acción directa de la resistina sobre las somatotropas, evaluamos mediante microfluorimetría la concentración intracelular de Ca2+ ([Ca2+] i) en células individuales en respuesta a resistina (1 microM). Los resultados muestran que, en los cultivos tratados con resistina, el nivel de secreción de GH fue el doble del observado en los correspondientes controles. Además, el tratamiento con resistina también indujo cambios significativos en la expresión de receptores reguladores clave de la actividad de las somatotropas, los receptores de ghrelina/secretagogos de GH (GHS-R). En concreto, el nivel de ARNm del GHS-R disminuyó en respuesta a resistina, un efecto que es similar al inducido por su ligando endógeno, la ghrelina. Por último, nuestros resultados demuestran que la resistina provoca aumentos de la [Ca2+] i en un elevado porcentaje de somatotropas, lo que apoya la idea de que las acciones descritas de esta proteína sobre secreción de GH y expresión de receptores son mediadas por una acción directa de la misma sobre este tipo celular. En conjunto, nuestros resultados demuestran, por primera vez, que la resistina regula la función de las somatotropas por un mecanismo doble: estimulando la secreción de GH y disminuyendo la sensibilidad de las somatotropas a ghrelina. Además, estos datos sugieren la existencia de una relación funcional entre GH y resistina que puede ser importante para el control del metabolismo y del balance energético.
Financiación: CVI-139(J. Andalucía); BFI 2001-2007, BFU2004-03883/BFI(MEC/FEDER).
139LA GHRELINA ACTIVA DIRECTAMENTE LAS CÉLULAS CORTICOTROPAS EN LA ENFERMEDAD DE CUSHING
J. Moreno-Fernandez1, A.J. Martínez-Fuentes2, R. Vázquez-Martínez2, M. Durán-Prado2, A. de la Riva3, M. Tena-Sempere2, C. Diéguez4, L. Jiménez-Reina5, P. Benito-López1, M.M. Malagón2 y J.P. Castaño2
1Serv. Endocrinología y Nutrición Hosp. Reina Sofía. Córdoba, 2Dpto. Biología Celular, Fisiología e Inmunol Univ. Córdoba. Córdoba, 3Serv. de Neurocirugía Hosp. Reina Sofía. Córdoba, 4Dpto. Fisiología Univ. Santiago de Compostela. Santiago de Compostela, 5Dpto. Ciencias Morfológicas Univ. Córdoba. Córdoba.
Los elevados niveles de ACTH y cortisol típicos de la enfermedad de Cushing se acompañan de una respuesta disminuida de GH. Se ha sugerido que la ghrelina, un nuevo estimulador de la secreción de GH, podría relacionar ambos fenómenos. En pacientes con enfermedad de Cushing, la secreción de ACTH estimulada por ghrelina aumenta notablemente mientras se reduce la de GH. La expresión del receptor de ghrelina GHS-R en tumores corticotropos ofrece una base para explicar esta observación, pero no hay pruebas a nivel celular que lo demuestren. Por ello, valoramos la acción directa de ghrelina sobre células de un adenoma corticotropo de una paciente con enfermedad de Cushing.
Pacientes y métodos: Mujer de 39 años remitida por sospecha clínica de síndrome de Cushing. El estudio hormonal confirmó hipercortisolismo dependiente de ACTH. En la RMN se detectó microadenoma hipofisario realizándose tumorectomía. Tras 6 meses se detectó recidiva obligando a hipofisectomía total, de la cual se tomaron muestras para: estudio ultraestructural; cultivo celular seguido de ensayo de la concentración intracelular de Ca2+, [Ca2+] i; y aislamiento de ARN para estudios de expresión por RT-PCR.
Resultados y discusión: Paciente con enfermedad de Cushing por adenoma hipofisario corticotropo densamente granulado. En el estudio microfluorimétrico la ghrelina (1 microM) aumentó la [Ca2+] i en un 89,5% de las células (n = 19). La inmunocitoquímica confirmó que el 99% de las células son corticotropos. El análisis por RT-PCR con cebadores específicos confirmó la expresión de POMC y reveló además la presencia de ARNm de las dos isoformas del receptor de ghrelina, GHS-R1a y 1b. Estos resultados demuestran que la ghrelina actúa directamente sobre las células adenomatosas corticotropas causantes de la enfermedad de Cushing, aumentando la [Ca2+] i. La demostrada correspondencia que existe entre dicho aumento y la subsiguiente secreción hormonal permite sugerir que la ghrelina puede aumentar directamente la secreción de ACTH por estas células, lo cual está apoyada por la presencia de receptores para ghrelina en este tejido. Estos datos ofrecen por primera vez una base celular para explicar la respuesta exagerada de ACTH a ghrelina en la enfermedad de Cushing y apoyan la posible implicación de este péptido en la regulación hormonal de dicha enfermedad. Estudios adicionales permitirán aclarar la contribución concreta de la ghrelina en la patogénesis tumoral.
Financ. CVI-139 (J.And.); BFI 2001-2007, BFU2004-03883/ BFI(MEC/FEDER).
140MONITORIZACIÓN DE INSULINRESISTENCIA Y SÍNDROME METABÓLICO EN ADULTOS CON DÉFICIT DE GH EN TRATAMIENTO SUSTITUTIVO A LARGO PLAZO
M.B. Molina, D. Peñalver, I. Pavón y S. Monereo
Endocrinología y Nutrición H.U. Getafe. Madrid.
Dado que los efectos del tratamiento sustitutivo con GH recombinante sobre el metabolismo hidrocarbonato son todavía objeto de debate, el presente estudio pretende evaluar el impacto del tratamiento sustitutivo con GH recombinante humana (rhGH) sobre la resistencia a la insulina y el posible desarrollo de síndrome metabólico (SM) en estos pacientes. Se utiliza un indicador simple de insulinresistencia (HOMA) en 20 pacientes sin alteración del metabolismo hidrocarbonado (6 hombres y 14 mujeres; edad media: 44 ± 13,9 años) con déficit de GH severo del adulto. Se evalúan medidas antropométricas como peso, talla, IMC, circunferencia de cintura, porcentaje de masa grasa (%MG), tensión arterial, glucemia e insulinemia basales, HbA1c y perfil lipídico, que se obtienen basalmente y tras la ultima visita, con un seguimiento de hasta 7 años (media: 5,1 ± 1,9 años; mediana: 5,5 años), iniciando el tratamiento con dosis media de rhGH de 0,2 ± 0,58 mg/día, con dosis finales de 0,4 ± 0,25 mg/día. Inicialmente todos los pacientes tenían niveles bajos de IGF-I, IMC elevado (28,8 ± 5,6 kg/m2) y un %MG por encima de lo normal (34,3 ± 9,5%). Las cifras basales de glucemia fueron normales (86 ± 11,3 mg/dl), insulinemias en rango alto del normal (12,3 ± 6,0 µU/ml), con HOMA de 2,7 ± 1,5 (media ± DS). Durante el tratamiento, se normaliza el IGF-I, y disminuye el perímetro abdominal y %MG sin alcanzar la significación estadística. La glucemia basal aumenta (86,4 ± 11,3 a 91 ± 15,3 mg/dl (p < 0,05) junto con un empeoramiento de la HbA1c (4,7 ± 0,73 a 5,14 ± 0,67% (p < 0,05), sin rangos patológicos. Asimismo, hay un incremento en la insulinemia basal (14,3 ± 6,5 µU/ml) y la resistencia insulínica medida por HOMA (3,1 ± 1,6) al final del seguimiento (p: ns.). Si bien los cambios en el metabolismo hidrocarbonato no fueron tan espectaculares, el perfil lipídico se vio modificado de forma favorable, sin variación en la cifra de colesterol total (p: ns) pero con aumento en HDL-col (51,2 ± 13,2 vs 67,5 ± 17,9 mg/dl; p < 0,05) y reducción de la fracción LDL-col (137,2 ± 55 vs 115,7 ± 30,8 mg/dl; p < 0,05), sin cambios en las cifras de triglicéridos (p: ns.). Ningún paciente desarrolló alteración del metabolismo glucémico. Del total de pacientes estudiados, inicialmente 4 cumplen criterios de síndrome metabólico con obesidad (IMC > 30) central, dislipemia y HOMA > 3,5, todas mujeres, añadiéndose 2 casos mas al final del seguimiento (5 a 7 años). En resumen, el tratamiento con rhGH origina aumento en la glucemia e insulinemia basales y discreto empeoramiento de la sensibilidad a la insulina, con posible aparición de SM en estos pacientes. Sin embargo, los cambios en la composición corporal y perfil lipídico, aunque exista mayor insulinresistencia, sugieren que los beneficios del tratamiento con rhGH superan la influencia negativa de la GH sobre el metabolismo hidrocarbonado.
141PANHIPOPITUITARISMO CONGÉNITO FAMILIAR Y ESPORÁDICO: CORRELACIÓN CLÍNICA, RADIOLÓGICA Y MOLECULAR
I. Alonso Troncoso*, C. Quinteiro García**, F. Barros Anguieira**, A. San Luis González***, V. Muñoz Leira*, P. Fernández Catalina*, A. Martís Sueiro*, J.R. Villar García* y S. Rodeiro Marta*
*Serv. de Endocrinología y Nutrición Complejo Hospitalario de Pontevedra. Pontevedra, **Unidad de Medicina Molecular H. Clínico Universitario. Santiago de Compostela, ***Serv. de Radiología Complejo Hospitalario de Pontevedra. Pontevedra.
Introducción: El Panhipopituitarismo se define por el déficit de GH asociado al déficit de al menos una de las otras cinco hormonas adenohipofisarias. Su presentación puede ser congénita (familiar ó esporádica) ó adquirida. Cualquier defecto en la organogénesis del eje Hipotálamo-Hipofisario puede provocar hipopituitarismo congénito. Se han descritos distintos tipos de mutaciones en los genes PROP1 y POU1F1 (PIT1) como responsables de hipopituitarismo congénito. Presentamos el cuadro clínico, analítico, radiológico y de estudio molecular de 14 casos con hipopituitarismo congénito.
Material y método: Presentamos 9 pacientes pertenecientes a 4 familias (dos de ellas consanguíneas) con cuadro de hipopituitarismo (déficit de GH, hipogonadismo completo e hipotiroidismo todos ellos y déficit de cortisol en 2 casos). En otros 5 casos la presentación fue esporádica, con déficit de GH y gonadotropo (total ó parcial) en todos ellos y déficit parcial de cortisol en 2. En 3 casos se verificó el antecedente de anoxia perinatal. Todos los casos presentaron agenesia ó hipoplasia de adenohipófisis con retracción de tallo y neurohipófisis normal en los casos familiares y agenesia de tallo y neurohipófisis ectópica en un caso esporádico. Se practicó análisis molecular de los genes PROP1 y POU1F1 por amplificación mediante PCR de toda la región codificante y posterior secuenciación cíclica bidireccional.
Resultados: En todos los casos familiares se objetivo delección 2bp(301-302 de lAG) en homocigosis en el exon 2 del gen PROP1. Dicha deleción provoca un desplazamiento en la pauta de lectura resultando en una terminación prematura en el codón 109 y consiguiente inactivación de la proteína codificada por PROP1. Se produce una proteína truncada que ha perdido el dominio de unión al ADN y el dominio transactivación C terminal. Adicionalmente también hemos encontrado distintos cambios nucleotídicos considerados polimorfismos poblacionales sin relevancia clínica conocida.
Conclusiones: El eje somatotropo y gonadal se afectan en todos los casos familiares o esporádicos siendo el eje suprarrenal el menos afecto. En la mayoría de los pacientes se demuestra un daño anatómico subyacente que en todos los casos familiares de nuestra área se justifica por una misma mutación en el gen PROP 1 y en los casos esporádicos probablemente por un daño perinatal. No se ha descrito hasta ahora ningún caso de neurohipófisis ectópica y mutaciones en PROP1. La alta prevalencia de polimorfismos concuerda con la de otras series.
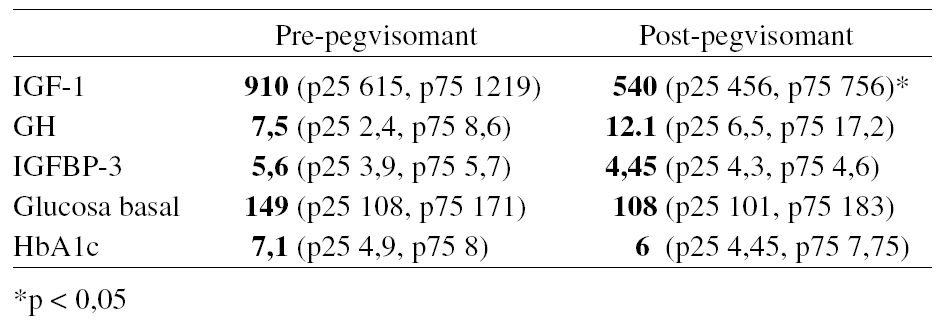
142PEGVISOMANT EN ACROMEGALIA RESISTENTE CON GRANDES RESTOS TUMORALES
I. Pinal Osorio, I. Bernabeu Morón, M.T. Rivero Luis, J.M. García López y J. Cabezas Cerrato
Endocrinología y Nutrición Hospital Clínico Universitario de Santiago. Santiago de Compostela, La Coruña.
El tratamiento crónico con pegvisomant normaliza la IGF-I en un alto porcentaje de pacientes acromegálicos. Existe preocupación respecto a la posibilidad de que induzca crecimiento tumoral a largo plazo y no existe suficiente experiencia en pacientes con acromegalia resistente y/o con grandes restos tumorales. Describimos nuestra experiencia en dos pacientes con éstas características.
Material y métodos:Caso 1: Varón de 42 años, diagnosticado en octubre de 1998 de acromegalia (GH basal media 27,2 ng/mL, Nadir GHxSOG 15,6 e IGF-1 996 ng/mL) por macroadenoma hipofisario (70 mm de diámetro y 132 cc de volumen calculado). Intervenido por vía transesfenoidal (Nov. 98) y transcraneal (Abr.99) y radiado (54 Gy Dic 99) con hipersecreción de GH resistente al tratamiento médico. En marzo 2004 con octreótido-lar 30 mg/15d y cabergolina 1 mg/sem: GH basal media 7,4, IGF-1: 748 y nadir GH: 5,4, con resto tumoral estable de 28,1 cc. Caso 2: Mujer de 30 años, diagnosticada en Julio 2001 de acromegalia (GH basal media 29,5 ng/ml, nadir GH*SOG 16,4 e IGF-1 593ng/mL) por macroadenoma hipofisario (4 cm diámetro máximo y 73 cc). Intervenida por vía transesfenoidal y transcraneal (Ag. 2001) y radiada (50 Gy en Nov. 2001). En marzo 2004, en tratamiento con octreótido-lar 30mg/25d. y cabergolina 1,5 mg/sem.: GH basal media 12.68ng/mL e IGF-1 de 556 ng/ml con resto tumoral estable de 10,1 cc. En ambos casos se sustituyó el tratamiento con análogos por 10 mg de pegvisomant, ajustando la dosis según respuesta bioquímica al mes, a los 3, 5 y 7 meses. Valoramos la respuesta de IGF-I, los efectos adversos, y los cambios del volumen tumoral.
Resultados: La IGF-1 disminuyó un 31% en el caso 1 y un 67% en el caso 2 al mes de tratamiento; un 59,9% y un 68,8% a los 3 meses; un 69,2% y 77,4% a los 5 meses y en el caso 1 un 70,3% a los 7 meses. El descenso promedio de la IGF-1 al final del seguimiento fue de un 73,85%. La IGF-1 se normalizó a los 5 meses en el caso 1 (con 40 mg de pegvisomant) y al mes en el caso 2 (con 10 mg de pegvisomant). Los estudios de imagen a los 3 meses en ambos casos y a los 6 en el caso 1, no mostraron cambios significativos del tamaño tumoral. La tolerancia al tratamiento fue inicialmente buena, si bien la paciente del caso 2 (polimedicada con varios fármacos potencialmente hepatotóxicos: paracetamol, atorvastatina, anticonceptivos e ibuprofeno) desarrolló una hepatitis tóxica que obligó a suspender el tratamiento a las 24 semanas.
Conclusión: El pegvisomant ha conseguido normalizar la IGF-1 en 2 pacientes con acromegalia resistente y con grandes restos tumorales. La sustitución de análogos de la somatostatina por pegvisomant no ha dado lugar a corto plazo a crecimiento tumoral.
143PREVALENCIA DE HIPOPITUITARISMO EN PACIENTES QUE HAN SUFRIDO UN TRAUMATISMO CRANEOENCEFÁLICO. ESTUDIO EPIDEMIOLÓGICO PILOTO
A.I. Castro-País, R. Peinó, M. Lage, M. Lorenzo-Solar y F.F. Casanueva
Unidad de Desórdenes Alimentarios - Endocrinología. Hospital Provincial de Conxo. Laboratorio de Endocrinología Molecular. Facultad de Medicina. Complejo Hospitalario Universitario de Santiago. Universidad de Santiago de Compostela. Santiago de Compostela.
Objetivos: El traumatismo craneoencefálico se ha asociado a hipopituitarismo y deficiencia de hormona de crecimiento (GH). Sin embargo, la prevalencia de hipopituitarismo como consecuencia de un traumatismo craneoencefálico es en gran medida desconocida. Este estudio epidemiológico, realizado en pacientes que han sufrido un TCE moderado o severo hace al menos un año antes del estudio, tiene dos objetivos: a) evaluar la prevalencia de hipopituitarismo (tanto sintomático como no sintomático) y b) en particular, evaluar la prevalencia de déficit de GH.
Material y métodos: A partir de un grupo no seleccionado de pacientes atendidos en la Unidad de Urgencias de nuestro Hospital, diagnosticados de TCE moderado o severo, e ingresados durante más de 24 horas por esa causa durante los últimos 7 años, 140 fueron seleccionados mediante randomización. De éstos, 112 contestaron a un cuestionario de síntomas de hipopituitarismo incluido en la carta de invitación para participar en el estudio. 84 sujetos (65 hombres y 19 mujeres) aceptaron participar en el estudio. A todos estos pacientes se les ha realizado un estudio bioquímico y hormonal basal. La secrección de GH se ha estudiado con el test GHRH+GRHP-6. Además de las hormonas hipofisarias se han analizado IGF-I, IGBP-3, fT4, estradiol o testosterona y cortisol.
Resultados: Partiendo de una muestra de 84 sujetos (100%), 12 (14,2%) presentaron déficit de TSH; 2 (2,5%) déficit de ACTH y 8 (9,5%) déficit de gonadotropinas. Se observó déficit de GH en 12 sujetos (14,3%) y de éstos 8 (9%) presentaron un déficit aislado de GH. Finalmente, 5 sujetos (5,9%) mostraron un déficit combinado de hormonas.
Conclusión: Tras un TCE moderado o severo, los déficits hipofisarios más comunes fueron el déficit de GH, TSH y Gonadotropinas. El déficit de ACTH supuso un 2,5% de la muestra. En conjunto, un 32,1% de los sujetos estudiados mostraron algún tipo de déficit hipofisario.
Este trabajo ha sido parcialmente financiado con una beca de colaboración de Pfizer y Fundación MMA Investigación Médica.
144PREVALENCIA DE SÍNDROME METABÓLICO Y FACTORES DE RIESGO CARDIOVASCULAR EN OBESIDAD MÓRBIDA INFANTIL. INDICADORES DE INSULINRESISTENCIA
D. Peñalver, M.B. Molina, S. Monereo y B. Vega
Endocrinología y Nutrición H.U. Getafe. Madrid.
El síndrome metabólico (SM) asocia la insulinresistencia (IR) con un conjunto de factores de alto riesgo cardiovascular (FRCV) siendo la obesidad la causa mas frecuente de IR en niños. Este estudio intenta relacionar la obesidad mórbida infantil (OMI), IMC > p97 para edad y sexo, con la prevalencia de SM y otros FRCV en la infancia, buscando índices que se correlacionen con IR en la edad pediátrica. Se realizó un estudio retrospectivo en niños y adolescentes con obesidad mórbida analizando edad, sexo, talla, peso, IMC, circunferencia de cintura, tensión arterial (TA), presencia de acantosis nigricans, glucemia basal y tras sobrecarga oral de glucosa (SOG), insulinemia basal, HOMA (HOMA > 3,5 para IR), colesterol total y fracciones, triglicéridos (TG) y HbA1c. Asimismo los índices calculados fueron cintura/talla (ICT) y TG/HDL. Un total de 35 pacientes obesos mórbidos (16 niños y 19 niñas) con edades comprendidas entre los 4,6 y 16 años (11,8 ± 3,1 años, 74% menores a 10 años) fueron estudiados utilizando el coeficiente de correlación de Pearson como herramienta estadística, obteniéndose los siguientes resultados (media ± DS). La glucemia e insulinemia basales fueron 90,6 ± 7,3 mg/dl y 26,3 ± 15,9 mU/ml respectivamente, con un 76,4 % de pacientes con HOMA > 3,5 (IR). El 11,4% presentó glucemia basal alterada en ayunas (GBA) y el 28,5% intolerancia a hidratos de carbono (IH) tras SOG, cumpliendo un paciente criterios de DM. Respecto al perfil lípidico, el 20% presentó hipertrigliceridemia, con colesterol total 167,7 ± 34,8 mg/dl, (HDL 49 ± 11,6 mg/dl, LDL 93,7 ± 28,3 mg/dl) teniendo 42,8% cifras bajas de HDLcol. De entre otras características del Síndrome Metabólico (SM) se encontró que 54,2% tenía acantosis nigricans, con obesidad central en 51,4% de los casos, siendo 34,7% hipertensos. El SM completo (3 o mas componentes) lo presentaban el 17,1% de los niños. Las correlaciones del IMC con ICT (r = 0,7) y con insulina (r = 0,56) respectivamente fueron muy positivas (p < 0,001). Asimismo se encontraron correlaciones positivas entre HOMA con ICT (p = 0,03; r = 0,38) y con TG/HDL (p = 0,013; r = 0,44) aunque de menor intensidad. El ICT se correlacionó positivamente y de forma significativa (p < 0,03) con parámetros del SM como TA sistólica (r = 0,51), HDL (r = 0,42) e insulinemia (r = 0,38), no asi con glucemia y cifras de TG.
Conclusiones: Más de dos tercios de los niños con obesidad mórbida tienen insulinresistencia, con una prevalencia de SM completo del 17% en estos pacientes. El IMC se correlaciona fuertemente con el ICT, y este a su vez es buen medidor de SM infantil, especialmente en relación con HTA y cifras de HDL bajas, así como con la presencia de hiperinsulinismo en la edad pediátrica. Son necesarios estudios posteriores para evaluar otros indicadores de insulinresistencia en niños con obesidades extremas.
145PRIMEROS RESULTADOS 2001-2005 DEL REGISTRO ESPAÑOL DE TUMORES GASTROENTEROPANCREÁTICOS (RETEGEP)
J.A. Díaz Pérez1, E. Villar2, I. Sevilla García3, J. Sastre Valera4, R. Salazar Soler5, C. Villabona Artero6, V. Alonso Orduña7, D. Castellano8, M. Marzuela Azpiroz9 y A. Abad Esteve10
1Endocrinología y Nutrición Hospital Clínico Universitario San Carlos. Madrid, 2Oncología Médica Hospital Vall d`Hebró. Barcelona, 3Oncología Médica Hospital Virgen de la Victoria. Málaga, 4Oncología Médica Hospital Clínico Universitario San Carlos. Madrid, 5Oncología Médica Hospital Duran i Reynals (ICO). Hospitalet (Barcelona), 6Endocrinología y Nutrición Hospital de Bellvitge. Hospitalet (Barcelona), 7Oncología Médica Hospital Miguel Servet. Zaragoza, 8Oncología Médica Hospital Doce de Octubre. Madrid, 9Endocrinología y Nutrición Hospital de la Princesa. Madrid, 10Oncología Médica Hospital Germans Trias i Pujol. Badalona. Grupo Español de Tumores Neuroendocrinos (GETNE).
Introducción: La incidencia de tumores gastroenteropancreáticos (TGEP) está aumentando en las últimas décadas, según datos actualizados del registro de tumores carcinoides norteamericano. La incidencia en España es desconocida, como así también su morbilidad y mortalidad. En el contexto de un grupo de trabajo multidisciplinario nacional recientemente creado, el grupo español de tumores neuroendocrinos (GETNE) hemos analizado los datos del Registro Español de TGEP (RETEGEP) procedentes de diversos hospitales de todo el país para conocer la prevalencia e incidencia de esta patología.
Material y métodos: se ha realizado un análisis descriptivo de los datos de RETEGEP, recogidos en 5 años a través de un registro en Internet (situado en http://www.retegep.net) mediante el programa estadístico SPSS V.11.5 (SPSS Inc.).
Resultados: Hasta el momento, han participado en RETEGEP 28 investigadores de 26 centros, que han incluido 188 pacientes (57,4% hombres, 42,6% mujeres, con una media de edad de 59,3 años) con 190 tumores registrados, procedentes de Cirugía General y Digestiva (4,2%), Endocrinología y Nutrición (26,8%) y Oncología Médica (69,0%). No se han incluido en los resultados los casos de tumores de pulmón y timo (no GEP). El registro incluye datos de 1979 a 2005 (86% desde 1995). Los tumores registrados son: carcinoides 49,5%; tumores pancreáticos no funcionantes 15,8%; insulinomas 10,5%; metástasis de primario desconocido 6,8%; gastrinomas 6,3%; otros tumores con menor incidencia (glucagonomas, t. pancreáticos con producción hormonal ectópica, vipomas, t. pancreáticos funcionantes mixtos y somatostatinoma) 11,1%. El motivo de diagnóstico correspondió mayoritariamente a clínica secretora (44,2%). La asociación a MEN1 se observó en 4,2%; 83,3% de los tumores fueron aislados. Estadiaje: 38,4% diseminado, 32,1% local, 8,9% locorregional y 20,5% no definido. Un 50,5% de los casos fueron unifocales, un 11,6% no localizados y un 7,4% multifocales. El tamaño tumoral (Ø mayor) más frecuente para los unifocales fue de 1-4 cm (38,1%). Los tumores se localizaron mayoritariamente en la cabeza del páncreas (18,4%), en cuerpo de páncreas (10,0%) y en íleon (8,4%). Los parámetros bioquímicos fueron estudiados en un 68,9% de los tumores y la prueba de imagen más utilizada fue el TAC (83,2%). Se realizó estudio anatomopatológico en el 91,6% de los tumores registrados, empleando mayoritariamente biopsia (75,9%). Los tratamientos aplicados a los pacientes fueron mayoritariamente el quirúrgico y farmacológico. Minoritariamente se empleó quimioembolización, radioterapia, tratamiento ablativo y embolizació tumoral. Se presentarán datos de supervivencia a los 5 y 10 años por tipo de tumor (carcinoide y no carcinoide) y por estadiaje (local, locorregional y diseminado).
146REEVALUACIÓN EN LA EDAD ADULTA DEL DÉFICIT DE GH DIAGNOSTICADO EN LA INFANCIA
R. Peinó1, L. Castro-Feijóo2, M. Lage3, J. Barreiro2, M. Lorenzo-Solar1, M. Pombo2 y F.F. Casanueva4
1Unidad de Desórdenes Alimentarios - Endocrinología. Hospital Provincial de Conxo. Complejo Hospitalario Universitario de Santiago. Santiago de Compostela., 2Endocrinología Pediátrica, Crecimiento y Adolescencia. Departamento de Pediatría. Complejo Hospitalario Universitario de Santiago. USC. Santiago de Compostela, 3Laboratorio de Endocrinología Molecular. Facultad de Medicina. Universidad de Santiago de Compostela. Santiago de Compostela., 4Laboratorio de Endocrinología Molecular. Departamento de Medicina. Facultad de Medicina. Complejo Hospitalario Universitario de Santiago. Santiago de Compostela.
Objetivo: antes de reconocer el déficit de hormona de crecimiento (GH) como una patología crónica, los pacientes deficitarios en edad pediátrica recibían tratamiento hasta alcanzar la talla adulta y abandonaban, posteriormente, el seguimiento y la terapia con GH. Actualmente, se acepta la existencia del síndrome del déficit de GH en el adulto (GHDA) existiendo claras evidencias sobre la mejoría que se produce con la terapia sustitutiva con GH. Se realizó una reevaluación clínica y hormonal en un grupo de pacientes (pac) diagnosticados de déficit de GHD en la infancia que habían alcanzado talla final y en los cuales se suprimió la GH.
Material y métodos: Se evaluaron 28 pac (18 varones y 10 mujeres), con una media de edad de 19,9 ± 2 años e IMC 23,7 ± 4,2. Se valoró GH tras hipoglucemia insulínica (-30 a 120 min) y GHRH+GHRP6 (0 a 60 min), IGF-I, estudio hormonal hipofisario basal y evaluación de la región hipotálamo-hipofisaria por RM.
Resultados: 7 de 28 pac presentaron GHDA (25%) según los criterios internacionales de respuesta de GH para ambos test, en 17 pac (61%) los estudios GH fueron normales y en 4 (14%) dudosos, con mala respuesta a la hipoglucemia insulínica, se necesitaron estudios ulteriores. En 6 de 7 pac con GHDA existían otros déficit hormonales, 3 con panhipopituitarismo y diabetes insípida y 3 con déficit parciales, todos ellos presentaban anomalías en la RM. En el resto de los pac se demostró un único déficit gonadotropo y 2 alteraciones hipotálamo-hipofisaria en la RM.
Conclusión:1. Se confirma la importancia de la reevaluación en los pac con déficit de GH diagnosticado en la infancia. 2. La utilidad del test GHRH+GHRP6 frente a la hipoglucemia insulínica: ausencia de falsos negativos, de efectos secundarios y rapidez. 3. GHDA grave se asocia a déficit hormonales múltiples y anomalías estructurales hipotálamo-hipofisarias en RM.
Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Red de Grupos RGTO (G03/028), Red de Centros RCMN (C03/08).
147REGISTRO ESPAÑOL DE ACROMEGALIA (REA): ¿QUÉ INFORMACIÓN NOS PERMITE OBTENER?
G. Sesmilo León
Endocrinología Hospital Clínic. Barcelona. Investigadores del REA.
El REA se concibió en 1997 como un estudio multicéntrico prospectivo y retrospectivo con el objetivo de obtener datos demográficos, características clínicas, datos de tratamiento, de evolución y de morbimortalidad en la acromegalia.
Objetivo: Analizar datos demográficos, de evolución y morbimortalidad en el REA a fecha de 9/2/2005. Evaluar las limitaciones del registro con el fin de diseñar estrategias que permitan una mejor explotación de los datos.
Método: Estadística descriptiva de la base de datos REA.
Resultados: El 9/2/2005 había 1277 pacientes incluidos (61% mujeres), con una media de edad al diagnóstico de 45 años (DE 14 a). La incidencia máxima se registró en 1997 (2,1 cpm/año). La prevalencia global es de 37 cpm, con una amplia variabilidad entre comunidades autónomas (de 16 a 93 cpm). La etiología fue tumoración hipofisaria en 1245 casos (70% macroadenomas) y secreción ectópica en 8 casos. Un 82% de pacientes recibieron tratamiento quirúrgico, el 92 por vía transesfenoidal, un 43% de pacientes recibieron radioterapia, estas modalidades se combinaron entre ellas y/o con tratamiento farmacológico en un 77% de los casos. Según criterios de normalidad de IGF-I + concentración de GH tras SOG < 2 ng/dl, un 45% de los pacientes fueron curados, un 55% no curados. Los datos de morbilidad reflejan HTA en un 33%, DM en un 30%, hipopituitarismo en un 15%, bocio en un 19%, síndrome del túnel carpiano en un 15%, dislipemia en un 11% y SAOS en un 9%. El 84% de pacientes fueron introducidos de forma transversal sin ningún seguimiento tras la introducción, mientras que en el 16% de los pacientes se reportaron datos de seguimiento posteriores a la inclusión, de los cuales, en el 35% hubo sólo un seguimiento, en el 20% dos y en el resto tres o más. Se registraron 58 fallecimientos a una edad media de 60 años (DE 14), la mayoría (36%) de causa cardiovascular. La tasa de mortalidad estándar (SMR) ajustada por edad fue inferior a 1, aunque este valor no es fiable dada la no obligatoriedad de actualización de los pacientes incluidos y que probablemente no se hayan registrado muchos fallecimientos dada la naturaleza del registro de acromegalia.
Conclusiones: El REA nos permite obtener datos aproximados de morbilidad y de tendencias de tratamiento en acromegalia. Los datos epidemiológicos: incidencia, prevalencia, datos de curación y de mortalidad son orientativos dado el planteamiento actual del registro. Para que lo sean, sería conveniente actualizar los datos de todos los pacientes incluidos.
148RESECCIÓN TRANSESFENOIDAL ENDOSCÓPICA FRENTE A CONVENCIONAL EN EL TRATAMIENTO DEL MACROADENOMA HIPOFISARIO
J.C. Ferrer García*, S. Escrivá Cerrudo*, J. Merino Peña**, J.M. Gallego Sánchez**, J.A. Barcia Albacar**, C. Barcia Moriño** y A. Herrera Ballester*
*Sección Endocrinología. Servicio Medicina Interna, **Servicio de Neurocirugía Hospital General Universitario. Valencia.
Objetivos: Comparar los resultados del tratamiento quirúrgico del macroadenoma hipofisario utilizando resección transesfenoidal endoscópica frente a convencional.
Material y métodos: Se incluyeron 29 pacientes diagnosticados de macroadenoma hipofisario. Según el tipo de cirugía transesfenoidal realizada se diferenciaron en 2 grupos: 11 casos mediante resección endoscópica y 18 casos mediante resección convencional. Se estudiaron como variables prequirúrgicas el tamaño tumoral, la extensión supraselar y la infiltración carotídea. Como variables de resultado post-cirugía se analizaron: estancia media hospitalaria, resto tumoral, complicaciones en el post-operatorio inmediato, crecimiento del tumor en la evolución y déficit hormonal residual. Los resultados se analizaron con el software SPSS versión 11.0.
Resultados: Los macroadenomas tenían un tamaño medio de 2.8 cm. Se diagnosticaron 2 prolactinomas y 1 acromegalia. El resto de adenomas se consideraron no funcionantes. No existieron diferencias en el estudio prequirúrgico al analizar tamaño tumoral, extensión supraselar e infiltración carotídea en ambos grupos de tratamiento. Al analizar los resultados post-cirugía se encontraron diferencias al comparar las estancias medias: 11,0 ± 4,3 días para la resección endoscópica vs 17,3 ± 7,3 días para la convencional; p < 0,05. En el resto de parámetros no se hallaron diferencias.
Conclusiones: La resección transesfenoidal endoscópica en casos de macroadenoma hipofisario es al menos tan eficaz como la convencional, y permite reducir la estancia media hospitalaria de estos pacientes.
149RESULTADOS DE LA CIRUGÍA TRANSESFENOIDAL EN LA ENFERMEDAD DE CUSHING
M. Vázquez Gutiérrez*, M.J. Amaya García*, R. Guerrero Vázquez*, M.A. Pomares*, A. Soto Moreno*, N. García Hernández*, A. Leal-Cerro*, M.A. Mangas Cruz*, J.M. Montero Elena** y F. Villamil Fernández*
*Endocrinología y Nutrición, **Neurocirugía Hospital Universitario Virgen del Rocío. Sevilla.
Introducción: Los resultados sobre la cirugía en la enfermedad de Cushing son muy dispares en las distintas series publicadas. En esta variabilidad influyen distintos factores, entre ellos, la falta de uniformidad en los criterios de curación, el tiempo de seguimiento evaluado o el grado de experiencia de los diferentes centros.
Objetivo: Determinar la tasa de curación de la enfermedad de Cushing intervenida en nuestro centro.
Pacientes y método: Hemos realizado un estudio retrospectivo sobre una serie de 40 pacientes remitidos para cirugía por enfermedad de Cushing, intervenidos entre el año 1985 y 2003, con una edad media al diagnóstico de 36±14 años. Veintiséis casos han completado un seguimiento mínimo de 12 meses en nuestro servicio, de éstos 20 han continuado seguimiento a largo plazo y 6 han sido remitidos a sus centros de origen.
Resultados: Se ha evaluado la situación de 26 pacientes a los 12 meses de la cirugía, de los cuales 22 (84,6%) se consideran curados en función de los siguientes criterios: la necesidad de tratamiento esteroideo sustitutivo (n = 18, 69,2%) y/o la desaparición clínica y bioquímica del hipercortisolismo (n = 4, 15,4%). En el subgrupo de pacientes sin tratamiento esteroideo se preservó la función hipofisaria integramente, salvo un caso de diabetes insípida permanente; mientras que entre los 18 casos con insuficiencia suprarrenal post-quirúrgica, 8 (44%) presentaron al menos otro déficit. En 4 pacientes (15,4%) se ha objetivado persistencia de enfermedad, ya sea por no curación o bien por recidiva. Han continuado seguimiento en nuestro servicio 20 de los 26 casos inicialmente evaluados, durante un período de 12 a 228 meses. Dieciséis pacientes (80%) se consideran curados, 11(55%) de ellos en tratamiento sustitutivo con esteroides. Los 4 casos restantes (20%) presentan datos de hipercortisolismo.
Conclusión: La tasa de curación inicial de la enfermedad de Cushing en nuestro centro es del 84% y a largo plazo del 80%, siendo estos datos concordantes con los recogidos en la bibliografía.
150RESULTADOS DE LA RADIOTERAPIA ESTEREOTÁXICA FRACCIONADA EN LA ENFERMEDAD DE CUSHING
M.C. Montáñez*, J. Aller*, I. Quiroga*, I. Zapata**, R. Magallón**, S. Díez*, L. Salto* y J. Estrada*
*Endocrinología y Nutrición, **Oncología Radioterápica Hospital Universitario Puerta de Hierro. Madrid.
La radioterapia hipofisaria convencional es un tratamiento eficaz en la Enfermedad de Cushing persistente o recidivante tras cirugía transesfenoidal (TSS). La radioterapia estereotáxica fraccionada (RTEF) permite una mayor precisión en la inmovilización y definición de la lesión, con reducción del volumen de tejido sano irradiado. Entre 1998 y 2004, 14 pacientes (11 mujeres y 3 hombres) con enfermedad de Cushing persistente (6) o recidivante (8) tras TSS fueron sometidos a RTEF, entre 7 y 140 meses (mediana 45) tras la cirugía. La edad media al diagnóstico fue de 29,6 años (19-41). Los niveles de cortisol libre urinario (CLU) previos al tratamiento con RTEF fueron de 130 a 571 µg/24h (media 289,1, valores normales hasta 120µg/24h). Se utilizó un sistema de inmovilización y marco estereotáxico de BrainLab, con estudios de fusión de TC y RM con contraste cada 2mm. El volumen de tratamiento incluyó la lesión visible (4 pacientes) o todo el contenido selar y ambos senos cavernosos cuando no existía evidencia de adenoma (10 pacientes), añadiendo 5mm de margen en ambos casos. El tratamiento se realizó con acelerador lineal, utilizando una mediana de 6 campos fijos no coplanares conformados con micromultiláminas, con una dosis por fracción de 1,8 Gy y una dosis total de 50,4 Gy, excepto en cuatro pacientes: tres irradiados en otro centro que recibieron 46 Gy (dos) y 54 Gy (uno), y uno previamente irradiado con radioterapia convencional (12 años antes de la RTEF) que recibió 45 Gy. Después de un seguimiento medio de 31,3 meses (6-79), 9 de los 14 pacientes (64,28%) están en remisión (normalización del CLU). El tiempo medio hasta la remisión fue de 17,8 meses (6-24). Las diferencias entre los pacientes que remitieron y los que no en el CLU pretratamiento (252,1 vs 372,2 µg/24 h) y en el tiempo de seguimiento (39,5 vs 16,4 meses) no alcanzaron significación estadística. Un nuevo déficit de gonadotropinas y tres nuevos déficits de GH fueron diagnosticados durante el seguimiento postradioterapia. No se presentaron casos de alteraciones visuales, necrosis cerebral y/o desarrollo de tumores radioinducidos.
Conclusiones: Los resultados preliminares de nuestra serie indican que la RTEF es un tratamiento eficaz y seguro en la enfermedad de Cushing persistente o recidivante tras TSS.Son necesarios estudios con mayor número de pacientes y seguimiento a largo plazo para determinar si la RTEF presenta ventajas en términos de eficacia y aparición de efectos secundarios respecto a la radioterapia convencional.
151RESULTADOS DEL TRATAMIENTO CON PEGVISOMANT EN CINCO PACIENTES ACROMEGÁLICOS
S. Aznar, P. Revert, O. Moreno, S. Martínez, A. López-Maciá y A. Picó
Sección de Endocrinología y Nutrición Hospital General Universitario Alicante. Alicante.
Introducción: Pegvisomant es un análogo humano de GH modificado genéticamente para actuar como antagonista del receptor del mismo. Recientemente, se ha aprobado su utilización en el tratamiento de pacientes acromegálicos con persistencia de enfermedad activa tras tratamiento quirúrgico, radioterápico y con análogos de somatostatina.
Objetivo: Evaluar el efecto clínico y analítico del tratamiento con pegvisomant en cinco pacientes acromegálicos.
Material y métodos: La edad mediana de los pacientes ha sido de 49 años (p25 46, p75 57) siendo 3 varones y 2 mujeres, con una mediana de 9 años de evolución de la acromegalia (p25 5, p75 14,5). Todos los pacientes habían recibido tratamiento quirúrgico inicial (cuatro de ellos por vía transesfenoidal y uno transcraneal) seguido de tratamiento médico con análogos de somatostatina. Cuatro de los pacientes recibieron tratamiento radioterápico (convencional o estereotáxico). Analíticamente, ninguno cumplía criterios de control, siendo la mediana de los valores de IGF-1 910 ng/ml (p25 615, p75 1219) y de GH 7,5 ng/ml (p25 2,4, p75 8,6) previo al inicio de tratamiento con Pegvisomant. Se inicia tratamiento con Pevisomant a dosis de 10 mgr diarios vía subcutánea incrementando a 15 y 20 mgr en función de la respuesta inicial. La duración mediana del seguimiento ha sido de 10 meses (p25 8, p75 10). Se analizan los valores de IGF-1, GH, IGFBP-3, perfil glucémico (glucosa, HbA1c) y los cambios en el volumen tumoral (RMN) antes y después del tratamiento.
Resultados:

La realización de RMN selar, no puso de manifiesto la existencia de crecimiento tumoral. En cuanto a los efectos secundarios, un paciente presentó cefalea y prurito sin condicionar modificación terapéutica.
Conclusiones: La administración de Pegvisomant en pacientes acromegálicos activos no controlados con análogos de somatostatina, consigue un descenso significativo de las concentraciones de IGF-1. Se evidencia una mejoría del perfil glucémico con descenso de los niveles de HbA1c y las cifras de glucemia basal. Los efectos secundarios atribuibles a la administración de Pegvisomant son transitorios y no han supuesto en ninguno de nuestros casos el abandono del tratamiento.
152
SISTEMA KISS-1 Y PUBERTAD: MECANISMOS DE ACCIÓN Y POSIBLE DISRUPCIÓN POR FACTORES NUTRICIONALES Y EXPOSICIÓN A COMPUESTOS ESTROGÉNICOS EN PERÍODOS CRÍTICOS DEL DESARROLLO
J.M. Castellano Rodríguez, V.M. Navarro Loro, R. Fernández Fernández, E. Vigo Gago, J. Roa Rivas, A. Mayen Reyes, R. Pineda Reyes, E. Aguilar Benítez de Lugo, L. Pinilla Jurado y M. Tena Sempere
Dpto. Biología Celular, Fisiología e Inmunología Facultad de Medicina. Córdoba.
La activación del eje reproductor en la pubertad es el punto final de una serie de fenómenos madurativos que conducen a la activación del generador hipotalámico de pulsos GnRH y la adquisición de la capacidad reproductora. Recientemente, se ha demostrado que mutaciones inactivantes del gen GPR54, que codifica el receptor putativo para los péptidos derivados del gen supresor de metástasis KiSS-1, se asocian a hipogonadismo hipogonadotropo en roedores y en humanos, lo que sugiere un papel relevante de este sistema en el control del desarrollo y/o la función del eje reproductor. No obstante, el papel fisiológico del sistema KiSS-1 en el control de la pubertad es aun desconocido. En este contexto, en el presente trabajo, hemos evaluado la expresión en hipotálamo de los genes KiSS-1 y GPR54 durante el desarrollo post-natal, y su alteración en condiciones de ayuno y tras la administración neonatal del estrógeno sintético benzoato de estradiol (EB). Además, hemos evaluado el efecto de la administración central y periférica de KiSS-1 sobre la secreción de gonadotropinas, su capacidad para regular la liberación de GnRH, y el efecto de la administración crónica de KiSS-1 sobre la llegada de la pubertad en condiciones de alimentación normal y subnutrición. Nuestros resultados muestran que los genes KiSS-1 y GPR54 se expresan en hipotálamo a lo largo del desarrollo post-natal, con máximos niveles en a la pubertad. La expresión hipotalámica del gen KiSS-1 disminuyó tras el ayuno y la exposición neonatal a EB. La administración tanto central como periférica del péptido KiSS-1 produjo una elevación significativa y dosis-dependiente de los niveles de gonadotropinas en ratas macho y hembra; respuestas que fueron observadas igualmente en ratas en ayuno y tras estrogenización neonatal. A su vez, el péptido KiSS-1 estimuló, en forma dosis-dependiente, la liberación de GnRH por fragmentos hipotalámicos de ratas control y en ayuno. De forma análoga, el KiSS-1 estimuló la secreción de GnRH por la línea neuronal murina GT1-7, donde se observó la expresión del gen GPR54. Finalmente, la inyección crónica de KiSS-1 en hembras inmaduras nutridas ad libitum, anticipó la llegada de la pubertad, mientras que en ratas subnutridas (con ausencia de pubertad) la administración repetida de KiSS-1 revirtió parcialmente la falta de apertura vaginal (60%) e indujo respuestas de LH en todos los animales estudiados. En conclusión, nuestros resultados demuestran que, a través de un mecanismo dependiente de GnRH, el sistema KiSS-1 participa en el control de la secreción de gonadotropinas y la llegada de la pubertad, y sugieren que la alteración de este sistema puede estar implicada en la disrupción del desarrollo puberal en situaciones de subnutrición y tras exposición a compuestos con actividad estrogénica.
Trabajo financiado por el Instituto de Salud Carlos III (Red de Centros 03/08 y PI042082) y el proyecto EDEN QLK4-CT-2002-00603.
153UTILIDAD DE LA DETERMINACIÓN DE LA SUBUNIDAD ALFA TRAS ESTÍMULO CON TRH COMO INDICADOR DE PERSISTENCIA Y/O RECURRENCIA TUMORAL EN GONADOTROPINOMAS
A. Estepa*, C. Villabona*, E. Mena*, D. Pérez Asensio*, M. Dastis**, M. Sahún*, E. Solano*, P. Rosel**, M.Á. Navarro** y J. Soler*
*Endocrinología y Nutrición, **Sección de Bioquímica Hormonal y Génica Hospital Universitario de Bellvitge. L'Hospitalet de Llobregat.
Introducción: Los adenomas secretores de gonadotropinas (gonadotropinomas) muestran una respuesta de la subunidad alfa de las gonadotropinas (LH y FSH) al estímulo con TRH. Ello permite diferenciar estos tumores de los no funcionantes y podría tener utilidad tras la cirugía dado que con frecuencia existen problemas en la identificación de restos tumorales respecto a cambios posquirúrgicos.
Objetivo: Analizar la respuesta de la subunidad alfa y gonadotropinas al estímulo con TRH en macroadenomas productores de gonadotropinas tras la intervención quirúrgica como indicador de persistencia y/o recurrencia tumoral.
Material y métodos: Se estudiaron 14 pacientes intervenidos de macroadenoma hipofisario productor de gonadotropinas comprobados histológicamente. La distribución según el sexo fue 10 hombres y 4 mujeres. La media de edad fue 52,2 años (34-80). Se estudió a 9 pacientes antes de la intervención quirúrgica y a 9 pacientes tras la intervención, en 4 pacientes se realizó el estudio pre y postquirúrgico. A todos se les practicó resonancia nuclear magnética (RNM) en los 6-12 meses posteriores a la intervención. Método: se determinaron en suero las gonadotropinas y la subunidad alfa en los tiempos: -90,-60,-30, basal, 30, 60 y 90 minutos; en el momento basal se administró 400 µg de TRH. Método analítico: radioinmunoanálisis con anticuerpos monoclonales anti subunidad alfa. Estándar internacional WHO 1er IRP 75/569. Se consideró respuesta normal de la subunidad alfa y gonadotropinas al incremento inferior al 33% del valor basal en varones y nulo en las mujeres. Como respuesta positiva un incremento en varones de la subunidad alfa ≥ 75%, FSH ≥ 25% y LH ≥ 60%; en mujeres (pre y postmenopáusicas) incremento de la subunidad alfa ≥ 50%, FSH ≥ 90% y LH ≥ 75% respecto al valor basal.
Resultados: En el grupo estudiado después de la intervención la respuesta de la subunidad alfa fue positiva en 5/9 pacientes (basal 0,06 ± 0,05; minuto 30: 0,14 ± 0,07 UI/L). Respecto a la LH y la FSH la respuesta fue positiva en 4/9 y en 1/9 de los gonadotropinomas. En el estudio prequirúrgico la respuesta de la subunidad alfa fue positiva en 5/9 (basal 0,18 ± 0,14; minuto 30: 0,34 ± 0,19 UI/L). La respuesta de la LH fue positiva en 4/9 y en 1/9 para la FSH. Por RNM 10/14 presentaban persistencia tumoral y/o cambios postquirúrgicos, de los cuales 5 presentaban respuesta positiva para la subunidad alfa.
Conclusiones. La determinación de la subunidad alfa al estímulo con TRH en gonadotropinomas puede ser útil como indicador de persistencia y/o recurrencia tumoral cuando las pruebas de imagen no son concluyentes.
154UTILIDAD DE LA SUBUNIDAD ÁCIDO-LÁBIL COMO PARÁMETRO DE ACTIVIDAD DE LA ACROMEGALIA
I. Halperin Rabinovich*, R. Casamitjana Abella**, R. Pagés Bayarri**, R. Hernáez Rodríguez*, M. Giménez Álvarez*, L. Choque Uño*, S. Rueda Alfaro*, G. Sesmilo León* y M. Puig Domingo*
*Endocrinología y Nutrición, **Lab. Hormonal, CDB Hospital Clínic, Universitat de Barcelona. Barcelona.
Introducción: En el seguimiento de la acromegalia se utilizan diversos parámetros de secreción de GH (GH basal y tras sobrecarga oral de glucosa, IGF1 y en menor medida su proteína de transporte IGFBP3). En algunas circunstancias se han observado discordancias entre GH e IGF1 (p.ej., tras radioterapia hipofisaria). Además, en pacientes tratados con pegvisomant el único parámetro establecido de seguimiento es IGF1. Se ha postulado la utilidad de la subunidad ácido-lábil (ALS) en la valoración de la actividad de la enfermedad.
Objetivo: Valorar ALS como parámetro de actividad de la acromegalia en comparación con otros marcadores de utilidad clínica establecida en un grupo de pacientes en tratamiento farmacológico.
Material y métodos: Se estudiaron 16 muestras de 12 pacientes (4 mujeres de 60-78 años, 8 varones de 30-70 años), todos con acromegalia activa, intervenidos y/o irradiados excepto en 1 caso, y con diversos grados de control bajo análogos de somatostatina (8 casos, 8 muestras) o pegvisomant (4 casos, 8 muestras). En todas ellas se determinó GH (quimioinmunoluminiscencia Nichols, límite de detección 0,1 ng/ml, coeficientes de variación intra e inter ensayo 5,4 y 8,7% respectivamente), IGF1 (IRMA InmunoTech, límite de detección 30 ng/ml, coeficientes de variación intra e inter ensayo 5,2 y 8,4% respectivamente), IGFBP3 (IRMA BioCode, límite de detección 50 ng/ml, coeficientes de variación intra e inter ensayo 10,2 y 12,4% respectivamente), y ALS (enzimoinmunoanálisis DSL, con límite de detección de 0,7 ng/ml y coeficientes de variación intra e interensayo son < 10%).
Resultados: Se observó una excelente correlación entre los niveles de IGF1, IGFBP3 y ALS (IGF1/IGFBP3 r = 0,950, p < 0,01; IGF1/ALS r = 0,953, p < 0,01; IGFBP3/ALS r = 0,943, p < 0,01). La correlación de GH con los demás parámetros fue muy inferior (0,60, 0,64 y 0,54 respectivamente), tal como era previsible dado que 10 de los 12 pacientes estaban irradiados, y 8/16 muestras procedían de pacientes tratados con pegvisomant.
Conclusión: Según los resultados de este estudio preliminar, ALS puede ser un parámetro útil como complemento de IGF1 en el seguimiento de pacientes acromegálicos bajo tratamiento farmacológico. No tenemos datos sobre su utilidad en el diagnóstico de la enfermedad, o para establecer su curación.
