


La obesidad abdominal (OA) constituye, junto con la resistencia insulínica, la base fisiopatológica del síndrome metabólico. El exceso de tejido adiposo visceral (TAV) desempeña un papel clave en las comorbilidades de la OA. La esteatosis multiorgánica promociona la resistencia insulínica, el estrés oxidativo y la inflamación, lo que da lugar a disfunción endotelial y arteriosclerosis. El exceso de TAV conduce a un perfil metabólico de riesgo independientemente de la cifra de índice de masa corporal. Los recientes estudios epidemiológicos defienden que es necesario medir sistemáticamente el perímetro de cintura (PC) en la valoración de la obesidad, así como incluir el valor del PC y los parámetros de síndrome metabólico en las escalas de valoración del riesgo cardiometabólico. La potencial capacidad patógena de la OA debe ser tenida en cuenta en la valoración de cualquier cuadro en que el riesgo cardiometabólico sea un objetivo de acción preventiva o terapéutica.
Abdominal obesity (AO), together with insulin resistance, forms the pathophysiological basis of metabolic syndrome. Excess visceral adipose tissue (VAT) plays a key role in the comorbidity associated with AO. Multiorgan steatosis promotes insulin resistance, oxidative stress and inflammation, giving rise to endothelial dysfunction and atherosclerosis. Excess VAT leads to a metabolic risk profile regardless of body mass index. Recent epidemiological studies confirm the need to measure waist circumference when evaluating obese patients and to include this value and metabolic syndrome parameters in scales to assess cardiometabolic risk. The pathogenic capacity of AO should be taken into account when evaluating any condition in which reducing cardiometabolic risk is a preventive or therapeutic goal.
Que la obesidad se acompaña de un aumento de la mortalidad y de la comorbilidad cardiovascular y metabólica es un hecho sobradamente demostrado que ha llevado a considerarla un factor de riesgo de gran trascendencia, tanto en sí mismo como en su papel promotor de enfermedades prevalentes generadoras de riesgo cardiometabólico1. Una de las causas de que la obesidad no haya sido reconocida desde el inicio como un factor de riesgo cardiovascular clásico es la heterogeneidad de su impacto clínico, en parte derivada de la participación de otros elementos diferentes del índice de masa corporal (IMC), como son el exceso de masa grasa y especialmente su distribución anatómica. En los últimos años se ha otorgado a la obesidad abdominal (OA) un papel trascendental en la fisiopatología del riesgo cardiovascular y metabólico. Conocer los factores implicados en su desarrollo y los mecanismos fisiopatológicos que de ella se derivan puede aportar claves esenciales en el control y el manejo del riesgo cardiometabólico (RCM).
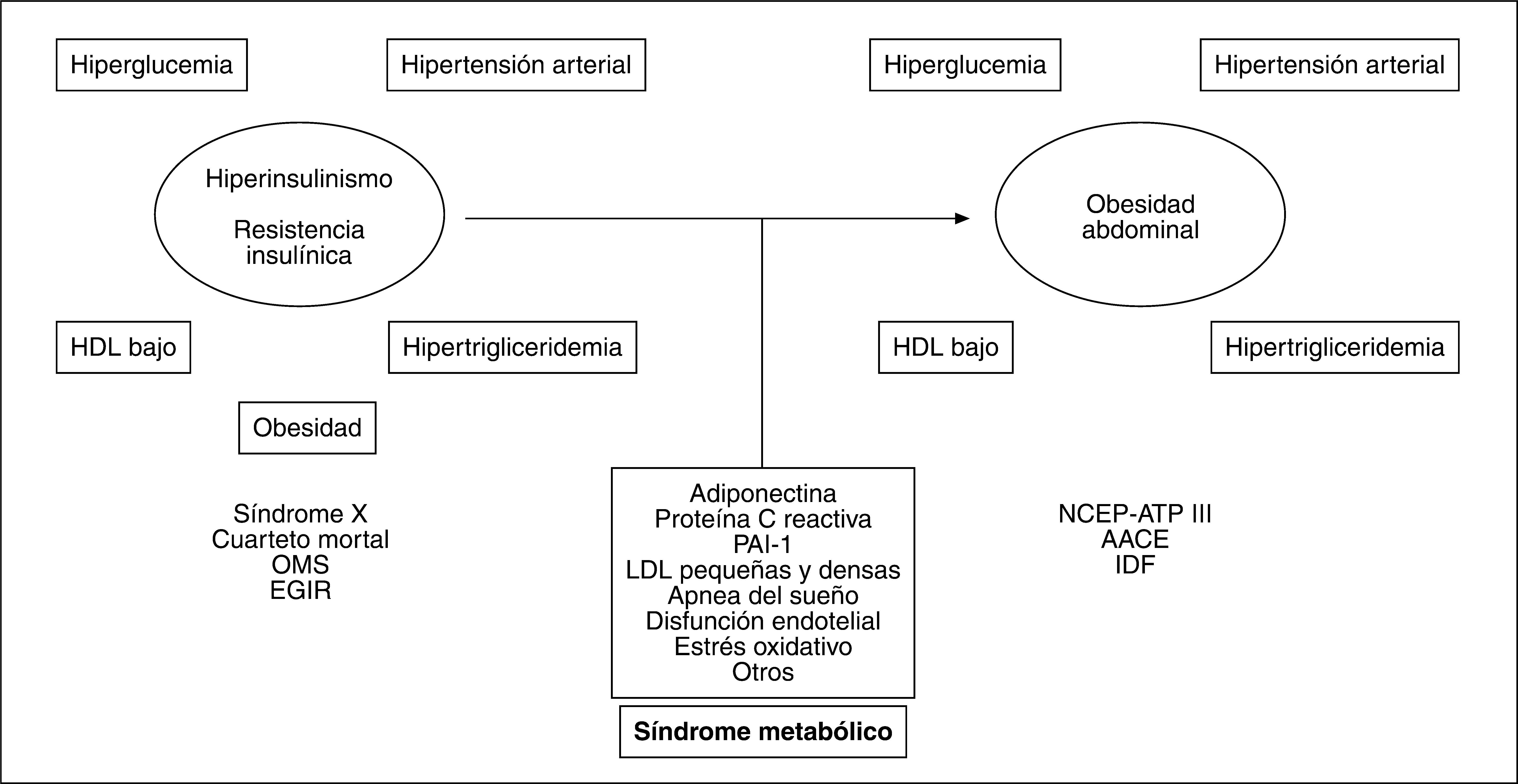
EVOLUCIÓN CRONOLÓGICA DE LA OBESIDAD ABDOMINAL HACIA EL NÚCLEO DEL SÍNDROME METABÓLICOLa capacidad de elevar el riesgo cardiovascular que tiene la relación entre algunos hallazgos clinicoanalíticos sentó las bases para la propuesta del concepto de síndrome metabólico. Desde la propuesta inicial de este síndrome llevada a cabo por Reaven2 en 1988, basada en el papel central de la resistencia insulínica (IR), se ha evolucionado hacia la adopción de nuevos criterios, pasando por el del Grupo Europeo de Estudio de la Resistencia Insulínica (EGIR) y el del Programa Nacional de Educación para el colesterol (NCEP-ATP III)3 hasta el más moderno propuesto por la Federación Internacional de Diabetes (IDF)4. El consenso NCEPATP III, que es de momento el más empleado en la práctica clínica, alude a la presencia de obesidad central medida por el perímetro de la cintura (PC), mientras que el establecido por la IDF considera la OA como condición sine qua non para determinar que hay síndrome metabólico. Adicionalmente, la propuesta de la IDF reduce las medidas de PC y les otorga un umbral distinto según el origen racial4. Por lo tanto, con el paso del tiempo hemos asistido a un viraje conceptual que evoluciona desde la perspectiva insulinocéntrica inicial a la obesocéntrica actual como clave diagnóstica del síndrome metabólico5 (fig. 1).

Evolución de la hipótesis insulinocéntrica a la obesocéntrica del síndrome metabólico con la posible incorporación de otros elementos propios del cuadro. AACE: American Association of Clinical Endocrinologists; EGIR: Grupo Europeo de Estudio de la Resistencia Insulínica; HDL: lipoproteínas de alta densidad; IDF: Federación Internacional de Diabetes; LDL: lipoproteínas de baja densidad; NCEP-ATP III: Programa Nacional de Educación para el colesterol; OMS: Organización Mundial de la Salud; PAI-1: activador del plasminógeno tipo 1.
Aun cuando la oportunidad del término y su significado clínico son controvertidos6, la conexión OA-IR se propone como el eje central de la fisiopatología del síndrome metabólico y sus complicaciones7. En este contexto, la medida del PC como valoración indirecta de OA se presenta como un elemento esencial en la valoración clínica de la obesidad.
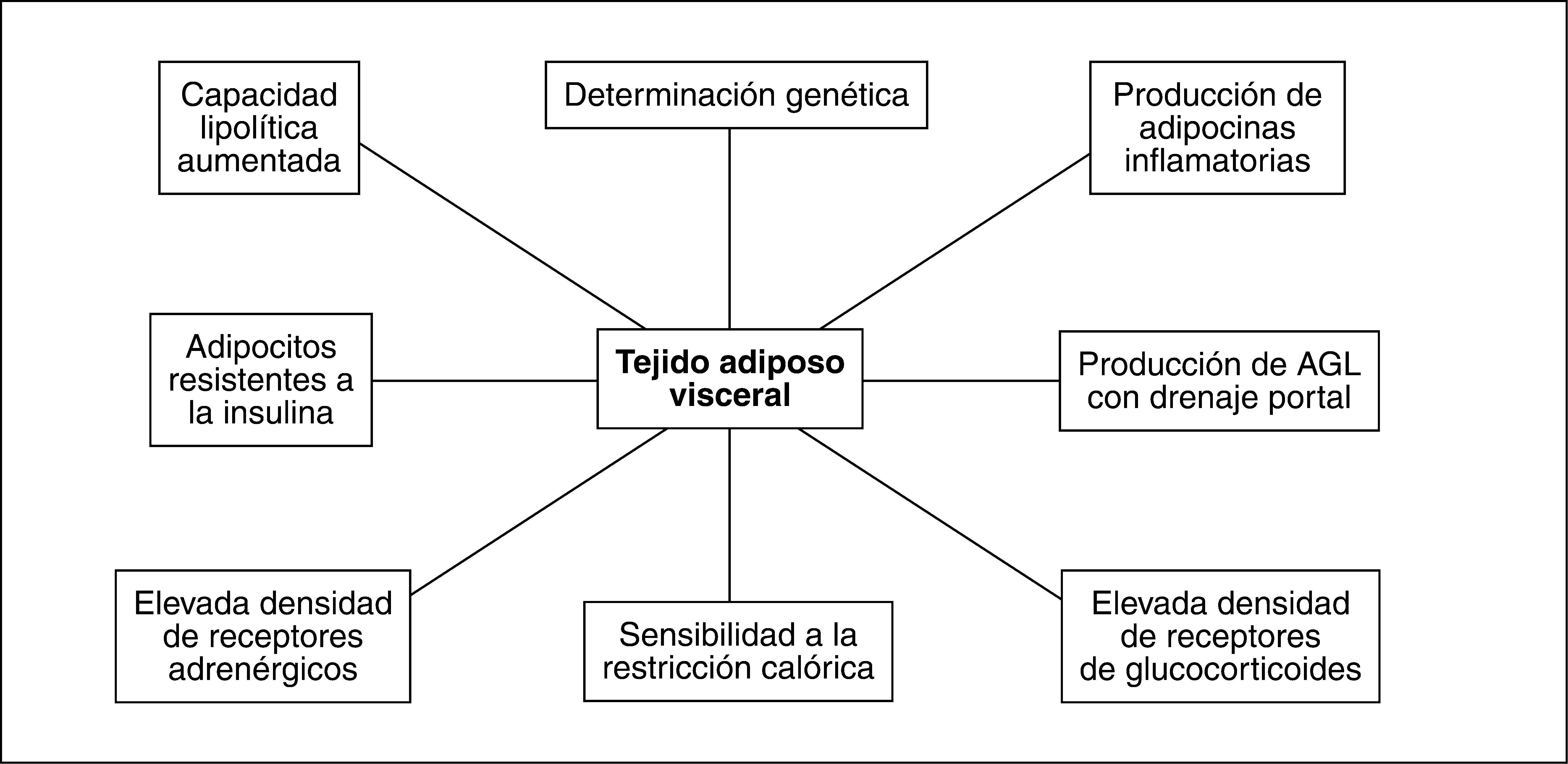
LOS COMPONENTES DE LA GRASA ABDOMINAL: DIFERENCIAS E INTERACCIONES ENTRE TEJIDO ADIPOSO SUBCUTÁNEO Y TEJIDO ADIPOSO VISCERALLa medida del PC se ve influida tanto por el tejido adiposo subcutáneo (TAS) como por el tejido adiposo visceral (TAV). Estos compartimentos poseen características biológicas muy diferenciadas, y aunque mantengan relaciones funcionales bilaterales, su papel en la fisiopatología de las complicaciones derivadas de la obesidad es muy distinto. El TAV comprende hasta el 20% del total del tejido graso en el varón y hasta el 8% en la mujer, que posee proporcionalmente mayor magnitud de TAS. El TAS se encuentra predominantemente localizado en las regiones femoral y glútea8, si bien se sitúa también en el compartimento subcutáneo abdominal.
Existen numerosas diferencias entre los adipocitos del TAV y los del TAS. Clásicamente se ha considerado que los adipocitos del TAV poseen mayor actividad lipolítica que los del TAS9. Sin embargo, observaciones recientes muestran que los adipocitos del TAS en términos absolutos tienen mayor actividad de lipoproteinlipasa y superior efecto lipolítico tras estimulación farmacológica10. No obstante, la capacidad de respuesta lipolítica relativa respecto al nivel basal es superior en los adipocitos del TAV. Estos hallazgos son compatibles con una mayor sensibilidad del TAV a estímulos lipolíticos10. En este sentido, el posible papel de una sensibilidad diferente de la regulación de la lipólisis por catecolaminas que explique las diferencias entre TAS y TAV es controvertido10,11. La producción de citocinas proinflamatorias y generadoras de IR como la interleucina (IL) 6 y el factor de necrosis tumoral alfa (TNFα), así como del inhibidor del activador del plasminógeno tipo 1 (PAI-1), es superior en el TAV que en el TAS12, mientras que éste genera más leptina y adiponectina. La producción de leptina aumenta conforme lo hace el tamaño del adipocito, lo que representa un mecanismo de autorregulación, ya que la hiperleptinemia reduce la ingesta, aumenta la actividad simpática y favorece la oxidación de ácidos grasos en los músculos13.
Los adipocitos del TAV tienen mayor capacidad de captación de glucosa que los del TAS, en probable relación con una mayor expresión de GLUT-414, lo que es un sustrato para alcanzar un almacenamiento de triglicéridos superior al del TAS.
El mayor grado de lipólisis del TAV favorece el flujo aumentado de ácidos grasos libres al hígado por vía portal, donde contribuyen a generar IR, esteatosis hepática y sus complicaciones metabólicas.
La densidad de receptores de andrógenos y glucocorticoides en el TAV es superior a la del TAS8, lo que es la base de su regulación endocrina. El tono estrogénico favorece la acumulación gluteofemoral de TAS, mientras que su desaparición en la menopausia promueve la de TAV15. La menopausia incrementa el tamaño de los adipocitos del TAV y su actividad lipolítica como consecuencia de la IR, mientras que en el TAS no se producen cambios respecto a la situación premenopáusica16. Así pues, la proporción entre las magnitudes de TAV y TAS aumenta tras la menopausia y la aproxima a la situación del varón8. El efecto inhibidor de los estrógenos sobre la producción de IL-617 también se reduce tras la menopausia, a pesar de que el adipocito se convierta en una fuente estrogénica a través de la aromatización de andrógenos. Consecuentemente los estrógenos, por este y otros mecanismos, pueden contribuir al efecto protector contra la enfermedad cardiovascular que presentan las mujeres premenopáusicas cuando se las compara con los varones de su misma edad18, lo que explicaría que el tratamiento hormonal sustitutivo en la menopausia ejerza un efecto protector contra la acumulación de TAV.
La actividad de la 11-betahidroxiesteroide deshidrogenasa tipo 1, que convierte la inactiva cortisona en cortisol, es más elevada en el TAV19, y da soporte a la hipótesis según la cual el hipercortisolismo tisular favorece el desarrollo de TAV y de síndrome metabólico, que muestra algunas características comparables con las del síndrome de Cushing.
Hay indicios de que la magnitud del TAV influye en la sensibilidad a la lipólisis de los adipocitos del TAS, que se ve elevada en pacientes con obesidad visceral20, lo que contribuiría a aumentar el contingente circulante de ácidos grasos libres (AGL). Por otra parte, algunos factores de transcripción presentes en el TAV se encuentran también en adipocitos del TAS. Los preadipocitos del TAS poseen mayor capacidad que los del TAV para diferenciarse en adipocitos pequeños, que muestran alta insulinosensibilidad y acumulan triglicéridos y AGL, evitando su depósito en otros tejidos como músculo, hígado, páncreas o miocardio, por lo que ejercen un papel protector contra la esteatosis orgánica y la lipotoxicidad, que son esenciales en el desarrollo de las complicaciones de la obesidad. Posiblemente, cuando los adipocitos del TAS ven superada su capacidad de almacenamiento, se vuelven insulinorresistentes, aumenta su capacidad lipolítica, liberan AGL y permiten el aumento del TAV y sus complicaciones.
En concordancia con estos aspectos, algunas observaciones clínicas otorgan al TAS un papel protector contra la enfermedad cardiovascular, estableciendo una relación negativa entre el TAS gluteofemoral y la arteriosclerosis21. Sin embargo, otros autores consideran que el TAS contribuye al desarrollo de inflamación y estrés oxidativo22. Es posible que, dependiendo de las características de sus adipocitos, en un momento determinado el TAS traduzca uno u otro efecto.
La sensibilidad del TAV es superior a la del TAS en la respuesta a la restricción calórica, que induce aumentos en la expresión de genes que codifican mecanismos promotores de la lipólisis como la lipasa sensible a hormonas, receptores beta-3 y proteína desacoplante 2 (UCP-2) en TAV, pero no en TAS23. Estos hallazgos justifican la gran mejoría de factores de riesgo cardiovascular que se produce en la obesidad con pérdidas ponderales relativamente escasas.
Las citocinas proinflamatorias y generadoras de IR como TNFα e IL-6 muestran interacciones con los AGL y la adiponectina. La adiponectina inhibe el factor de transcripción NF-κB y contrarresta los efectos proiinflamatorios de TNF e IL-624.
Los estudios comparativos de expresión génica en TAS y TAV llevados a cabo en pacientes con obesidad contribuyen a esclarecer la función de ambos compartimentos de tejido adiposo. Tanto el TAV como el TAS muestran sobreexpresión de genes relacionados con la inflamación en pacientes obesos25. Otros estudios revelan que algunas quimiocinas de la familia CC relacionadas con la atracción de células mononucleares a los tejidos inflamados, como son CCL5 y CCL11, se expresan más en TAV que en TAS, mientras CCL8 se expresa más en TAS26. La expresión del marcador de macrófagos CD68, así como la de TNFα, es similar en el TAS y el TAV de pacientes obesos26. Otros genes, como los que codifican los receptores de adiponectina, se expresan por igual en TAS y TAV27.
En general, el TAS y el TAV muestran una alteración en la expresión de genes implicados en la señalización insulínica, la lipogénesis y la inflamación, lo que indica que ambos compartimentos participan en las complicaciones metabólicas de la obesidad28. La ausencia de expresión de resistina en adipocitos aislados, en contraste con los resultados obtenidos en el análisis de TAS y TAV total, es indicio de su origen extraadipocitario25.
Así pues, tanto el TAV como el TAS poseen características anatómicas y funcionales particulares (fig. 2), que determinan su papel clave en el desarrollo del síndrome metabólico.
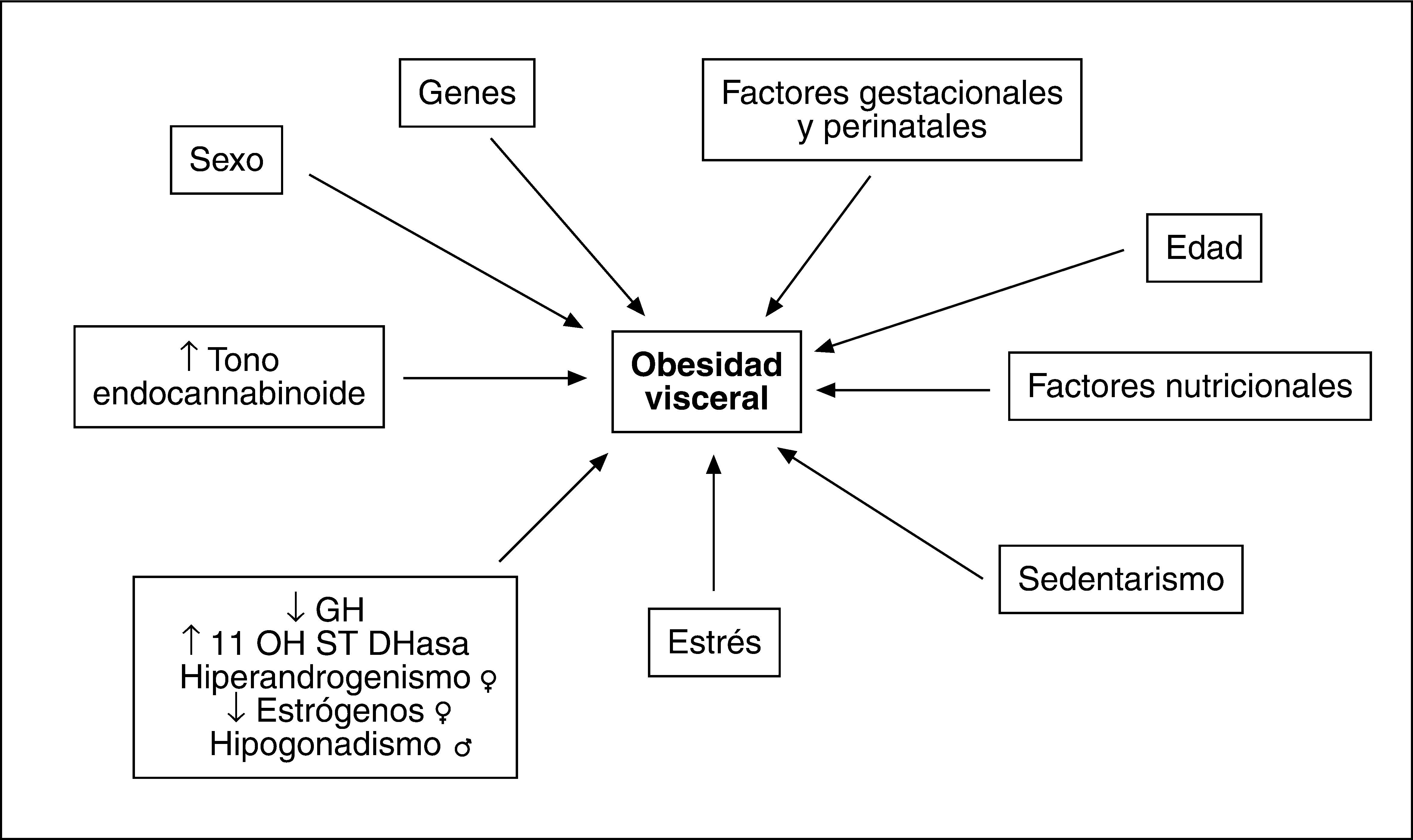
FACTORES ETIOLÓGICOS IMPLICADOS EN EL AUMENTO DE GRASA VISCERAL
La causa que subyace en el aumento del compartimento graso visceral reconoce un origen multifactorial (fig. 3). Aunque el desequilibrio en el balance energético a favor de la ingesta posee un protagonismo fundamental, se han descrito alteraciones genéticas que afectan al comportamiento alimentario. Hay indicios de que existen determinantes genéticos para la magnitud de la grasa visceral que afectan tanto a la secreción de adipocinas como a los receptores hormonales29,30. Obviamente hay influencias nutricionales y de hábitos de vida que promueven la acumulación de grasa y más concretamente de la visceral, como se deduce de los efectos del sedentarismo31.

Tanto la edad como el sexo y las hormonas sexuales son factores reguladores de la masa grasa visceral y de su efecto en la promoción de diferentes factores de riesgo cardiovascular32. El entorno hormonal es un elemento modulador de primer nivel que explica el aumento de grasa visceral en la mujer menopáusica respecto a la edad fértil y la mayor tendencia a la obesidad abdominal en el varón. En la mujer el patrón hiperandrogénico, que incluye la reducción de globulina transportadora de hormonas sexuales (SHBG), favorece la obesidad visceral33. Así, en las mujeres premenopáusicas se ha observado asociación entre ovario poliquístico, hiperandrogenismo y síndrome metabólico34, mientras que en las posmenopáusicas la concentración de testosterona, el índice androgénico libre y el estradiol se encuentran más elevados y la concentración de SHBG es más baja en las pacientes con síndrome metabólico33. El patrón androgénico se asocia con todos los factores individuales propios del síndrome según criterios diagnósticos ATP-III.
La deficiencia de somatotropina (GH) favorece la obesidad visceral, aspecto reversible con tratamiento sustitutivo35. La reducción en la secreción de GH que se produce con el paso de los años puede favorecer el aumento de la adiposidad visceral y el aumento en la tasa de síndrome metabólico que ocurre en edades avanzadas. Asimismo, la hiperactividad del eje hipotalamohipofisoadrenal (HPA) y la consiguiente hipercortisolemia que se ha demostrado relacionada con la obesidad visceral favorecen la acumulación grasa centrípeta y la obesidad abdominal36. Además, se ha descrito una sobreexpresión de 11-β-hidroesteroide deshidrogenasa tipo 1, que convierte en las células la inactiva cortisona en cortisol en adipocitos de grasa visceral37, lo que amplificaría aún más el efecto glucocorticoideo y favorecería la obesidad visceral.
El papel del estrés debe subrayarse especialmente38,39. Uno de los mecanismos postulados implica a la activación del eje HPA, que además de producir hipercortisolemia, inhibe los ejes somatotropo y gonadal, lo que estimula el desarrollo de la grasa visceral. La simultánea activación del sistema nervioso simpático favorece la lipólisis del tejido omental y la síntesis de IL-6. Los adipocitos de la grasa visceral son muy sensibles al efecto lipolítico de las catecolaminas, lo que favorece la generación de AGL. La exposición al estrés genera aumento de grasa visceral en adultos jóvenes sanos38, y ello confirma la relación entre el estrés y el desarrollo de obesidad visceral y síndrome metabólico.
El descubrimiento del sistema endocannabinoide, en el que se incluyen ligandos endógenos como anandamida y 2-araquidonilglicerol y receptores específicos para estas moléculas, como CB-1 y CB-2, con amplia distribución en diferentes tejidos del organismo, y su posible implicación en la regulación del comportamiento alimentario y en el desarrollo de OA y sus complicaciones ha aportado un nuevo elemento en la fisiopatología de la obesidad40. La activación del receptor CB-1 en el hipotálamo y el sistema límbico se asocia a aumento de la ingesta calórica41. Además, la sobreexpresión de este receptor en los adipocitos inhibe la síntesis de adiponectina42 mientras que en el hígado favorece la expresión de enzimas estimuladoras de la síntesis de ácidos grasos43 y en los músculos inhibe la captación de glucosa y promueve la resistencia insulínica44. Su activación en el tubo digestivo interfiere con las señales de saciedad y favorece la ingestión de alimentos. Así pues, el aumento del tono endocannabinoide a través de la síntesis de sus principales mediadores45 y de la expresión y la activación de receptores CB-1 se asocia al desarrollo de OA y efectos relacionados con sus complicaciones metabólicas, como la resistencia insulínica, la intolerancia a los hidratos de carbono y la dislipemia. La implicación de este sistema en la fisiopatología de la obesidad abre una ventana terapéutica muy importante que tiene repercusión potencial en diversos mecanismos que participan en el desarrollo de OA y sus complicaciones.
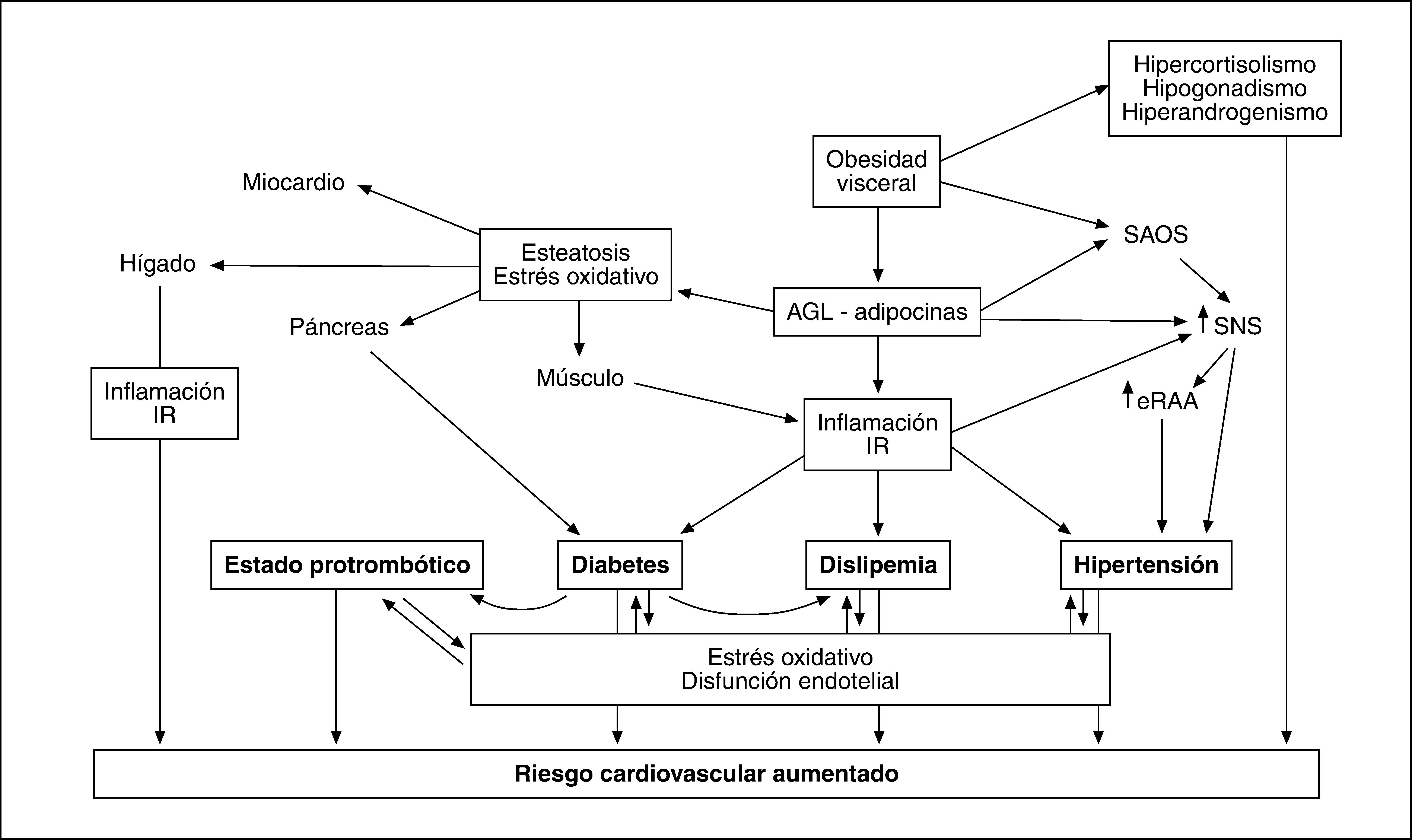
CAPACIDAD PATÓGENA DE LA OBESIDAD ABDOMINALEl RCM derivado de la OA y visceral se encuentra íntimamente ligado al síndrome metabólico, en el que la adiposidad central tiene un papel fundamental. Existen diversos mecanismos que explican la capacidad patógena de la obesidad visceral. La generación de un estado proinflamatorio y su relación bidireccional con la IR, frecuente en el síndrome metabólico, contribuye a generar hipertensión, dislipemia, disglucosis y una situación protrombótica que contribuye a aumentar la morbimortalidad cardiovascular. El aumento de producción de elementos implicados en el tono presor, como el angiotensinógeno, contribuye a activar el sistema renina-angiotensina, lo que —junto con otros factores46 como el aumento del tono simpático en el que participa la hiperinsulinemia, que a su vez promociona la reabsorción de sodio en el túbulo contorneado proximal de la nefrona47— constituye el sustrato de la hipertensión arterial.
Los efectos de la reducción de la grasa abdominal, y más específicamente visceral, en las citocinas proinflamatorias y el consiguiente aumento de insulinosensibilidad confirman que el exceso de TAV y sus mediadores son elementos clave en la inflamación y la consiguiente IR48.
Aunque ocupe otra localización anatómica, el tejido adiposo epicárdico (TAE) también es una fuente de ácidos grasos libres, adipocinas y citocinas proinflamatorias49,50. Su relación funcional con el TAV y su asociación con alteraciones funcionales cardíacas, posiblemente en parte debido a su proximidad anatómica, y otros factores de riesgo cardiovascular han hecho que este parámetro medible radiológicamente esté emergiendo como un elemento de interés en la estratificación del riesgo cardiometabólico49,50. Recientemente se han propuesto puntos de corte del grosor de TAE de 9,5 y 7,5mm en varones y mujeres, respectivamente, como valores predictores de síndrome metabólico51. Por la facilidad de su medición mediante ecografía y su significado funcional, la valoración del TAE puede ser un método alternativo a la estimación de grasa visceral. Así pues, los resultados publicados avalan un papel significativo del TAE en la fisiopatología de las complicaciones de la obesidad que posee connotaciones tanto diagnósticas como de seguimiento terapéutico.
Adipocitocinas, inflamación e insulinorresistenciaHoy está plenamente demostrado que el tejido adiposo, que incluye adipocitos, macrófagos, fibroblastos y otros tipos celulares, es una fuente de numerosas moléculas de diverso perfil de acción52. Muchas de ellas tienen carácter proinflamatorio; otras, efecto proagregante o favorecedor de hipertensión, son proangiogénicas, inmunomoduladoras y potenciadoras de resistencia insulínica, y algunas tienen acción insulinosensibilizante53.
Algunas adipocinas como leptina y adiponectina son producidas preferentemente por adipocitos, mientras otras de carácter inflamatorio, como TNFα e IL-6 que participan en los fenómenos de inflamación e IR, son segregadas predominantemente por los macrófagos.
En la obesidad, la secreción de moléculas como la proteína quimiotáctica de monocitos (MCP-1 o CCL-2) por el adipocito favorece la infiltración por células inflamatorias en un ejemplo de interacción bilateral (cross-talk) entre adipocitos, macrófagos, células endoteliales y monocitos, de importancia clave en la fisiopatología de la obesidad abdominal54-56. La elevación de marcadores biológicos como proteína C reactiva (PCR), alfa-1-glucoproteína, ácido siálico y amiloide A en pacientes con obesidad abdominal caracteriza al patrón de inflamación de bajo grado propio de este trastorno. Tanto el TNFα como las IL, especialmente la IL-6 producida por los macrófagos, fibroblastos, células endoteliales y adipocitos y estimulada por el sistema nervioso simpático, son de los principales mediadores del aumento de proteínas inflamatorias. Su concentración elevada se asocia con resistencia insulínica y disfunción endotelial y predice diabetes mellitus tipo 2 e infarto de miocardio57,58. Aunque la administración de IL-6 inhibe la respuesta de insulina a la estimulación por glucosa, el efecto diabetógeno proviene sobre todo de su efecto inhibidor de la señalización intracelular del receptor de insulina en hepatocitos59. También promueve el aumento de AGL y la reducción de adiponectina, circunstancias ambas que favorecen la IR60,61.
La observación de que la neutralización del TNFα induce un aumento de la insulinosensibilidad62 en la rata obesa sentó la base de su efecto promotor de IR, aspecto confirmado en estudios clínicos63. Tanto la disminución de adiponectina como el aumento de AGL y la interferencia con la señalización intracelular de insulina, a través de reducir la actividad de la tirosincinasa, son mecanismos potenciales del efecto en la IR del TNFα64. El TNFα da lugar a cambios proinflamatorios en células endoteliales y de músculo liso vascular65 y estimula la producción de moléculas de acción vasoconstrictora como endotelina-1 y angiotensinógeno, lo que contribuye al desarrollo de hipertensión arterial. De hecho, la concentración de TNFα se relaciona con la presión arterial y la IR en humanos66, por lo que constituye un elemento humoral clave en la fisiopatología del síndrome metabólico. Además, estimula la producción de IL-6 y PCR, con lo que se cierra un circuito reverberante generador de inflamación e IR67.
Uno de los mecanismos por los que las adipocinas promueven la inflamación es estimulando la producción hepática de PCR. Además de ser un marcador de inflamación, la PCR contribuye directamente a generar daño vascular, activación endotelial y trombosis68. Se ha demostrado su capacidad de inhibir la sintetasa del óxido nítrico y aumentar las moléculas de adhesión y el NF-κB el estrés oxidativo y la apoptosis endotelial, por lo que es un factor proarteriosclerótico69.
Otra adipocitocina de gran importancia es la adiponectina, también identificada como Acrp30, AdipoQ, apM1 o GBP28. La producen exclusivamente los adipocitos, tanto del tejido adiposo blanco como del pardo, y su secreción, en contraste con el resto de las adipocinas, se encuentra reducida en la obesidad. Tiene efecto insulinosensibilizante, antiinflamatorio y antiaterogénico70. Su papel en las complicaciones derivadas de la OA es muy probable, dado que la hipoadiponectinemia se asocia con la existencia de síndrome metabólico con mayor fuerza que cualquier marcador inflamatorio71. Tanto el sexo (más reducida en varones) como la dieta rica en hidratos de carbono y el estrés oxidativo reducen su concentración70. Su efecto insulinosensibilizador se relaciona con la activación de la cinasa AMPK, que además favorece el catabolismo de AGL y el gasto energético, en parte a través de la activación de receptores PPARα. La concentración circulante de adiponectina en humanos es muy elevada y se correlaciona negativamente con el IMC y la grasa visceral72. El mecanismo implicado en el efecto reductor de adiponectina que sucede en la obesidad no se conoce bien, aunque se ha propuesto que la hiperproducción de TNFα por el tejido adiposo visceral tiene efecto inhibidor de la síntesis de adiponectina por los adipocitos del tejido subcutáneo73, lo que justifica los hallazgos en plasma. Tanto la IL-6 como los glucorticoides también inhiben la secreción de adiponectina. La hipoadiponectinemia se asocia a resistencia insulínica. De hecho, el ratón knock-out para adiponectina muestra resistencia insulínica corregible mediante tratamiento sustitutivo74. Su concentración elevada parece ejercer un efecto protector contra el desarrollo de diabetes mellitus tipo 275, mientras que los pacientes con enfermedad coronaria muestran valores en plasma disminuidos76. Diversos estudios indican un papel protector de la adiponectina contra el desarrollo de enfermedad cardiovascular72,76, que es un factor clave en las comorbilidades del síndrome metabólico. Asimismo, la hipoadiponectinemia acompaña con frecuencia a la disfunción endotelial y a la hipertensión arterial77,78. En cuanto a su papel antiaterogénico, se acumula en el espacio subendotelial del vaso dañado, donde inhibe la adhesión monocitaria, la expresión de moléculas de adhesión como la molécula de adhesión intercelular 1 (ICAM-1) y selectina-E79 y la proliferación de células musculares lisas inhibiendo la AMPK80.
Otras adipocinas como la visfatina también se encuentran implicadas en la patogenia del síndrome metabólico. Aun cuando se la considera un origen preferencial por parte del tejido adiposo visceral, la expresión de ARNm de visfatina en tejido visceral y subcutáneo es similar81. A pesar de que tiene efecto insulinosensibilizador actuando sobre el propio receptor de insulina, estimula la producción de IL-1, IL-6 y TNFα, por lo que se debe considerarla una citocina proinflamatoria82. El papel que la visfatina desempeña en la fisiopatología de la OA y el síndrome metabólico está aún por esclarecerse.
La leptina, además de su efecto inhibidor del apetito y estimulador del gasto calórico, tiene efectos promotores de la migración de monocitos y macrófagos y estimuladores de otras citocinas, genera efectos proliferativos, proinflamatorios, protrombóticos y favorecedores del estrés oxidativo, por lo que se la considera uno de los mediadores entre tejido adiposo e inflamación83,84.
La resistina no es una verdadera adipocina pues, aunque se produce en el tejido adiposo85, su origen se relaciona con la fracción estromal y no con los adipocitos25. Aunque en animales de experimentación se ha visto implicada en la fisiopatología de la IR86, no hay tal evidencia en humanos, y se la observa relacionada con la inflamación y sus marcadores biológicos87,88. El PAI-1 también se produce en exceso por los adipocitos en la obesidad visceral y contribuye al establecimiento de un estado protrombótico que caracteriza la obesidad visceral89.
Otras citocinas también participan en la fisiopatología de la inflamación. La IL-10, secretada por células inmunitarias, parece tener efecto protector contra la disfunción endotelial y la arteriosclerosis90,91. La disminución de IL-10 facilita la inestabilidad de la placa arteriosclerótica y el desarrollo de síndrome metabólico92.
De otras adipocinas de identificación más reciente, como omentina93 y lipocalina 294, aún no se ha concretado su papel en la fisiopatología de la obesidad y la IR, al igual que otras moléculas como la proteína transportadora de retinol 4 y la caveolina-195,96.
El papel de los ácidos grasos libresDiversas alteraciones metabólicas relacionadas con la obesidad abdominal pueden explicarse por una producción excesiva y el consiguiente aporte al hígado, a través de la vena porta, de AGL procedentes del TAV97. En principio, la magnitud de la grasa visceral y el flujo de AGL que alcanza el hígado son proporcionales. La concentración plasmática de AGL es un 20% mayor en obesos que en no obesos, y la contribución de la lipólisis esplácnica a la llegada de AGL al hígado es función del volumen de grasa visceral, aspecto más significativo en mujeres98. Esta es una de las razones por las que el tejido adiposo visceral es un predictor de las complicaciones metabólicas de las comorbilidades de la obesidad más importante que el subcutáneo. Los adipocitos hipertróficos del compartimento visceral son resistentes al efecto antilipolítico de la insulina, lo que favorece el aumento de la salida de AGL vía portal y origina en el hepatocito un aumento de secreción de lipoproteínas ricas en triglicéridos y reducción de la degradación de apoproteína B y de la extracción hepática de insulina. La transferencia de triglicéridos a partículas de lipoproteínas de baja densidad (LDL) y lipoproteínas de alta densidad (HDL) y su ulterior lipólisis por la lipasa hepática da lugar a partículas HDL y LDL pequeñas y a disminución de la concentración de HDL, configurando la dislipemia aterogénica que acompaña al síndrome metabólico5. El flujo aumentado de AGL participa en la fisiopatología de la esteatosis no sólo hepática y muscular, sino también cardíaca y pancreática, lo que produce las consiguientes alteraciones funcionales.
Disfunción endotelial y estrés oxidativoLa OA se asocia a disfunción endotelial99, manifiesta por alteración de marcadores bioquímicos como moléculas de adhesión, trombomodulina y endotelina1100. La disfunción endotelial, un marcador inicial del desarrollo de arteriosclerosis, se manifiesta ya en niños y adolescentes obesos99 y es reversible con la reducción ponderal obtenida mediante dieta hipocalórica y ejercicio físico90.
La obesidad se ha demostrado asociada con marcadores de estrés oxidativo. Concentraciones elevadas de AGL aumentan el estrés oxidativo, que a su vez produce alteraciones en la regulación de adipocinas proinflamatorias y protrombóticas, como IL-6, TNFα, PAI-1 y MCP-1, así como antiinflamatorias y de efecto insulinosensibilizador, como adiponectina101. Las relaciones funcionales del estrés oxidativo con la activación del factor de transcripción NF-κB configuran uno de los mecanismos más importantes en la fisiopatología del riesgo cardiovascular propio del síndrome metabólico102.
TRADUCCIÓN CLÍNICA DE LA OBESIDAD ABDOMINALLa OA, evaluada por el PC, se asocia a una mayor probabilidad de adquirir factores de riesgo cardiovascular y de muerte de cualquier causa cuando se ajusta por el valor de IMC103,104, con un riesgo 1,44 y 2,26 veces superior respectivamente105. Un estudio prospectivo reciente106 permite establecer que la obesidad visceral estimada mediante tomografía computarizada (TC) es un predictor independiente de mortalidad.
Los pacientes con síndrome metabólico, para cuyo diagnóstico la OA es un factor consustancial, tienen hasta 3 veces más riesgo de enfermedad coronaria y accidente cerebrovascular107. Una publicación reciente agrupa 37 estudios, que totalizan 43 cohortes y muestran un riesgo de enfermedad cardiovascular y muerte 1,78 veces superior al de la población normal; observan que la asociación es más intensa en mujeres108.
Aunque la obesidad en sí misma es causa de hipertensión arterial, los cambios evolutivos en el PC son predictivos de la cifra futura de presión arterial y de la incidencia de hipertensión, independientemente del IMC109. Es bien conocida la capacidad predictiva del desarrollo de diabetes mellitus, dislipemia aterogénica y otras complicaciones que la OA tiene110 que los resultados del estudio IDEA confirman111. Estos datos indican que la OA se asocia a factores de riesgo cardiovascular, y permiten otorgar a la evaluación del PC un valor pronóstico significativo y, por ende, es posible una intervención preventiva.
Además de promover la inflamación de bajo grado y la IR, la excesiva producción de AGL y de adipocinas por el TAV participa en otros mecanismos fisiopatológicos que contribuyen a incrementar el riesgo cardiovascular (fig. 4).

La OA se asocia a hepatopatía grasa no etílica (HGNE), que se produce por infiltración adiposa de los hepatocitos, en la que el flujo incrementado de AGL procedente de la grasa visceral tiene un papel significativo112. Independientemente de su asociación con el síndrome metabólico, la HGNE puede contribuir por sí sola a favorecer el riesgo cardiovascular y metabólico generando IR, dislipemia, inflamación y estrés oxidativo, que a su vez se ven implicados en el deterioro de la función hepática, con lo que se cierra un círculo vicioso113. La reducción de adiponectina se relaciona con la gravedad histológica independientemente de la OA o de los componentes del síndrome metabólico114.
Análogamente al depósito de grasa en el hígado, el páncreas y el músculo esquelético, la frecuente disfunción cardíaca que acompaña a la obesidad puede estar, al menos parcialmente, relacionada con el depósito intramiocárdico de triglicéridos, que es 5-6 veces superior que en sujetos no obesos115, y favorecer la aparición de insuficiencia cardíaca. En consonancia con estos datos, el tratamiento de la obesidad mediante bypass gástrico reduce la masa ventricular y mejora su función116.
El depósito adiposo centrípeto se asocia a síndrome de apneas obstructivas del sueño, que es otro elemento potenciador del RCM porque favorece la activación del sistema nervioso simpático, la hipertensión arterial y el aumento de citocinas proinflamatorias y protrombóticas.
La OA se ha mostrado ligada a otros procesos patológicos como diferentes tipos cáncer117,118 y la infertilidad119.
La condición metabólica de la obesidad es función del tejido adiposo visceralAsumiendo que el TAV es fundamental en el desarrollo del síndrome metabólico y sus complicaciones, es factible que haya disociación entre la magnitud de la obesidad y la dimensión del TAV, de modo que existan pacientes con aumento de TAV e IMC normal. A este tipo de pacientes se los denomina normoponderales metabólicamente obesos (NPMO)120 y en ellos se observa aumento de TAV > 100cm2 medido con TC y rasgos analíticos de IR. Su prevalencia puede alcanzar un 13-18%121. Tanto el bajo peso al nacer como la raza (frecuente en japoneses y estadounidenses asiáticos) son factores que favorecen el desarrollo de este trastorno.
El cuadro opuesto, es decir, los obesos metabólicamente normales (OMN), son individuos con IMC > 30 que no muestran signos de IR y tienen valores bajos de TAV. Suelen iniciar la obesidad en la infancia y su perfil metabólico es favorable, con HDL elevadas, triglicéridos normales y ausencia de signos de IR. Pueden alcanzar al 20% de la población obesa121. En este contexto, se ha observado una relación entre el estado de salud cardiorrespiratoria y la magnitud del TAV independientemente del IMC o del PC122.
Los resultados obtenidos tras extirpación quirúrgica del TAV tanto en modelos experimentales como en humanos sustentan su contribución a la comorbilidad inflamatoria y metabólica123.
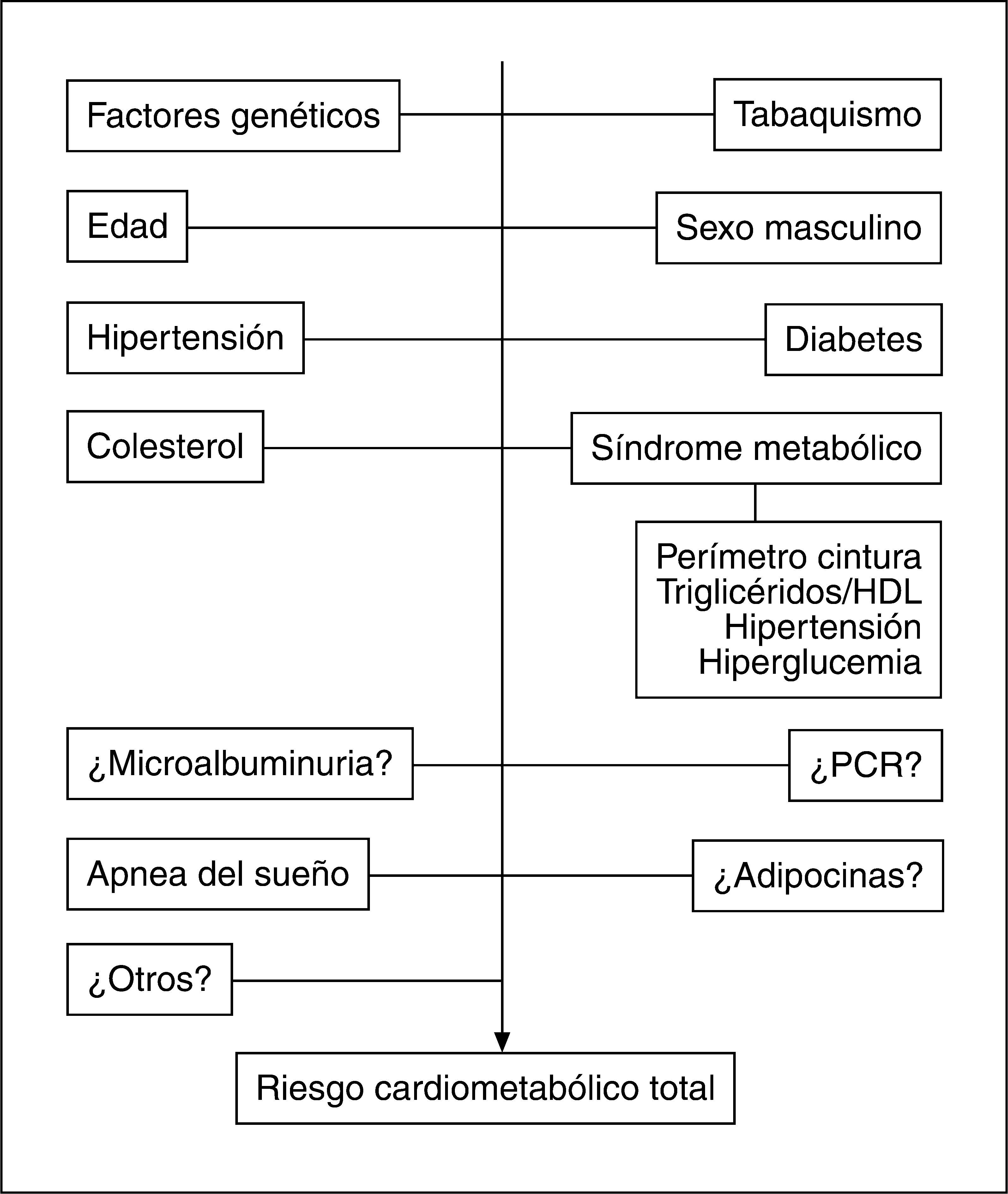
Lugar de la obesidad abdominal en el concepto y la valoración del riesgo cardiometabólico totalEl papel de la OA como contribuyente directo al riesgo metabólico y cardiovascular aún no está sistemáticamente incorporado a la valoración clásica del RCM. Por una parte, los puntos de corte de la PC para las diferentes razas son controvertidos4. Por otro lado, el debate surgido en relación con el papel del síndrome metabólico en la predicción del riesgo de diabetes mellitus y enfermedad cardiovascular aún persiste6. Esencialmente, se cuestiona la contribución al riesgo cardiovascular del síndrome metabólico y sus componentes cuando se tiene en cuenta los factores de riesgo clásicos incluidos en sistemas tradicionales como el de Framingham. Sin embargo, recientemente ha emergido el concepto de RCM propuesto por la Asociación Americana de Diabetes y la Asociación Americana del Corazón124 para determinar el riesgo de diabetes mellitus y enfermedad cardiovascular aunando los factores de riesgo tradicionales y los incluidos en el síndrome metabólico, cuyo valor predictivo de las alteraciones citadas está demostrado. Despres et al125 han propuesto la valoración del RCM total añadiendo a los factores tradicionales el estado de síndrome metabólico. Por lo tanto, tratar de sustituir los sistemas tradicionales de predicción del riesgo cardiovascular por el concepto de síndrome metabólico es inadecuado, pero considerar los factores de riesgo incluidos en los criterios del síndrome metabólico parece correcto (fig. 5), dado que estos trastornos no se incluyen en las escalas clásicas de valoración y su capacidad predictiva de enfermedad cardiovascular es, cuando menos, similar a la de la hipertensión o el tabaquismo126.

Dado que la OA sin alteraciones metabólicas acompañantes no puede considerarse necesariamente como un factor de riesgo cardiovascular o de diabetes, es necesario fenotipificar a los individuos con OA para establecer cuál es su riesgo real. De este modo, la hipertrigliceridemia, que junto con el aumento del PC predice la resistencia insulínica, se constituye en un elemento de interés a la hora de evaluar la trascendencia de la OA125. Un consenso reciente127, elaborado para establecer la importancia clínica del PC, considera que este parámetro puede ser especialmente útil en pacientes con IMC normales o indicativos de sobrepeso, dado que pueden mostrar un RCM aumentado que no se sospecharía midiendo sólo el IMC.
No obstante, es imprescindible contar cuanto antes con estudios prospectivos que informen sobre el carácter predictivo del desarrollo de diabetes y enfermedad cardiovascular que tienen los puntos de corte de PC asignados actualmente a las distintas poblaciones étnicas y así conocer si la OA en sí misma constituye un elemento predictor y un heraldo del desarrollo de síndrome metabólico y consecuentemente de diabetes y enfermedad cardiovascular. En tal caso, la intervención terapéutica en fases iniciales previas a la aparición de complicaciones metabólicas podría tener una relación de coste-beneficio favorable. Asimismo, la incorporación de otros marcadores como el tamaño de las partículas LDL, índices de resistencia insulínica, adiponectina o PCR puede mejorar la actual capacidad predictiva de RCM (fig. 5).
En resumen, la obesidad abdominal desempeña un papel clave en el desarrollo de síndrome metabólico y las complicaciones cardiovasculares y metabólicas. El compartimento visceral, a través de la secreción portal de AGL y citocinas, parece ser el más relacionado con el desarrollo de diabetes mellitus, dislipemia, esteatosis hepática, inflamación y estado protrombótico. Aun cuando no disponemos de elementos precisos de medida, la estimación del PC es un procedimiento sencillo y universalmente aplicable para establecer los riesgos de la obesidad y vigilar su evolución.
