




La mayor parte de las neoplasias que crecen en la silla turca son adenomas hipofisarios. El resto incluye tumores benignos (craneofaringiomas y meningiomas), tumores malignos (primarios: de células germinales, cordomas, linfomas, adenocarcinomas hipofisarios, o enfermedad metastásica: mama y pulmón principalmente) y otras entidades infrecuentes (quistes, abscesos, fístulas arteriovenosas, hipofisitis linfocitaria). Los schwannomas constituyen hasta el 8% de los tumores cerebrales primarios1, están habitualmente asociados a nervios craneales y presentan especial predilección por la porción vestibular del VIII par craneal a nivel del ángulo pontocerebeloso, pero la localización intraselar es excepcional.
Presentamos aquí la descripción del decimoséptimo caso de schwannoma intraselar indexado hasta la fecha desde el de Perone et al2, el cual simula un adenoma hipofisario no secretor tanto clínica como radiológicamente.
Una mujer de 56 años acudió a nuestra consulta para estudio de una masa hipofisaria. No refería antecedentes familiares de especial interés. Como antecedentes personales presentaba: migrañas, vértigo de Ménière en seguimiento por otorrinolaringología, que había solicitado una resonancia magnética nuclear (RMN) craneal con hallazgo de un tumor hipofisario, y colecistectomía por colelitiasis. Había tenido dos embarazos sin incidencias y la menopausia a los 50 años. La paciente no aquejaba alteraciones visuales, ni clínica sugestiva de hipo o hiperfunción hipofisaria. A la exploración física presentaba tensión arterial 110/70mmHg, una frecuencia cardiaca de 78 latidos por minuto, una talla 159cm, y peso de 62Kg, índice de masa corporal 24,5 Kg/m2. Estaba normohidratada y normocoloreada. No tenía rasgos acromegaloides ni sugestivos de síndrome de Cushing. No se palpaba bocio. La auscultación cardio-pulmonar fue normal, la exploración básica abdominal también lo era y no presentaba edemas en las extremidades inferiores ni signos de trombosis. En la campimetría por confrontación no había defectos notorios del campo visual. No existía galactorrea espontánea ni a la expresión del pezón. El informe de la RMN craneal (fig. 1) describía un macroadenoma hipofisario que se realzaba de forma homogénea tras la inyección de contraste y producía una ligera compresión sobre el quiasma óptico. No se identificaban lesiones ocupantes de espacio en ángulos pontocerebelosos ni alteraciones en los nervios craneales VII y VIII de ambos lados. Se solicitó una campimetría que puso de manifiesto una hemianopsia bitemporal.

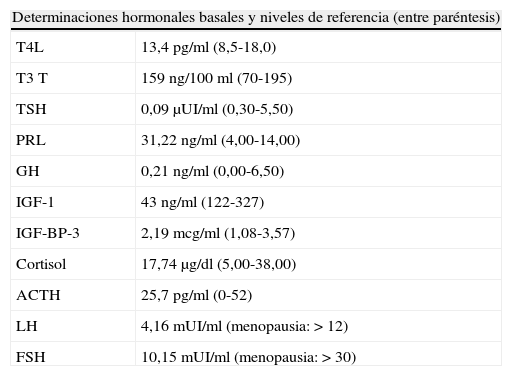
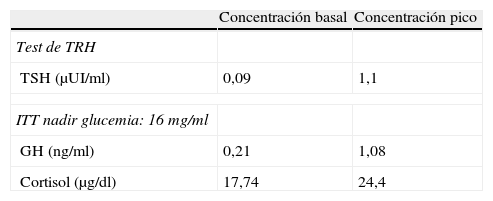
El estudio hormonal hipofisario basal (tabla 1) y tras estímulo con hormona liberadora de tirotropina (TRH) e insulina (ITT) (tabla 2) mostró un eje suprarrenal conservado, una leve hiperprolactinemia, concentraciones inapropiadamente bajas de gonadotropinas para una situación posmenopáusica, concentraciones bajas de factor de crecimiento insulínico tipo 1 (IGF-1) con ausencia de respuesta de hormona de crecimiento (GH) a la hipoglucemia inducida por insulina y concentraciones bajas de tirotropina (TSH), con respuesta escasa al estímulo con TRH pero con concentraciones circulantes de tetrayodotironina (T4) libre normales. La impresión diagnóstica fue de que se trataba de un adenoma hipofisario no funcionante con déficit secundario de tirotropina, hormona del crecimiento y gonadotropinas con hiperprolactinemia leve secundaria a compresión del tallo hipofisario.
Estudio de la función basal hipofisaria y tras estímulo
| Determinaciones hormonales basales y niveles de referencia (entre paréntesis) | |
| T4L | 13,4pg/ml (8,5-18,0) |
| T3T | 159ng/100ml (70-195) |
| TSH | 0,09μUI/ml (0,30-5,50) |
| PRL | 31,22ng/ml (4,00-14,00) |
| GH | 0,21ng/ml (0,00-6,50) |
| IGF-1 | 43ng/ml (122-327) |
| IGF-BP-3 | 2,19mcg/ml (1,08-3,57) |
| Cortisol | 17,74μg/dl (5,00-38,00) |
| ACTH | 25,7pg/ml (0-52) |
| LH | 4,16mUI/ml (menopausia: >12) |
| FSH | 10,15mUI/ml (menopausia: >30) |
ACTH: corticotropina; FSH: folitropina; GH: somatotropina; IGF-BP-3: proteína 3 de unión al receptor del factor de crecimiento insulínico; IGF-1: factor de crecimiento insulínico tipo 1; LH:lutropina; PRL: prolactina; T4L: tiroxina libre; TSH: tirotropina; T3T: triyodotironina total.
Resultados del estímulo de tirotropina (TSH) con hormona liberadorada de tirotropina (TRH) y del test de estímulo de somatotropina (GH) y cortisol con insulina (ITT)
| Concentración basal | Concentración pico | |
| Test de TRH | ||
| TSH (μUI/ml) | 0,09 | 1,1 |
| ITT nadir glucemia: 16mg/ml | ||
| GH (ng/ml) | 0,21 | 1,08 |
| Cortisol (μg/dl) | 17,74 | 24,4 |
La paciente fue remitida a neurocirugía para exéresis tumoral que se realizó por abordaje transesfenoidal. El estudio intraoperatorio sugirió la posibilidad de un adenoma hipofisario, pero el estudio definitivo en parafina mostró un tejido hipofisario de patrón trabecular y acinar sin atipias, con expresión variable de GH, prolactina (PRL) y en menor grado TSH, hormona foliculoestimulante (FSH) y hormona adrenocorticotropa (ACTH). La conclusión diagnóstica fue de tejido hipofisario no neoplásico, concordante con hiperplasia difusa de hipófisis, predominantemente de células secretoras GH y PRL.
Se realizó una segunda RMN cerebral indicando la persistencia de la tumoración con crecimiento intra y supraselar, que causaba compresión del quiasma óptico sin invadir senos cavernosos. La masa, de tamaño máximo 1,5cm, era homogénea tanto en su estructura como en su captación; loa hallazgos eran compatibles con macroadenoma.
Se procedió a una segunda intervención quirúrgica con resección radical del tumor mediante craneotomía frontal derecha. El estudio patológico del tumor mostró el siguiente inmunofenotipo: positivo para vimentina y proteína S-100, y negativo para actina, desmina, citoqueratinas y EMA, diagnóstico de neurinoma/schwannoma.
La paciente desarrolló panhipopituitarismo anterior y posterior postquirúrgico, por lo que se le prescribió tratamiento sustitutivo con hidroaltesona, levotiroxina, hormona de crecimiento y desmopresina. El campo visual inicialmente afectado se recuperó en su mayor parte, persistiendo algunos pequeños defectos periféricos (más en el ojo izquierdo que en el derecho).
El curso evolutivo ha sido excelente hasta la fecha, habiendo transcurrido ya diez años de seguimiento.
El caso, interesante por su excepcionalidad en cuanto a incidencia se refiere, comparte lo común de las características clínicas y radiológicas de los demás publicados. Tal como Perez et al revisan3, la mayoría de los pacientes presentan defectos en el campo visual producidos por la extensión supraselar del tumor con modesta hiperprolactinemia secundaria, y existen datos de hipopituitarismo en la mitad. Radiológicamente, los schwannomas en la RMN son hipointensos-gris mate en las imágenes ponderadas en T1 y, generalmente, hiperintensos en T2, con realce homogéneo tras la administración de contraste. La mayoría presenta extensión supraselar. En general, son tumores de crecimiento lento y curso indolente. No obstante, puede ocurrir una transformación maligna4. El tratamiento es quirúrgico, en principio por vía transesfenoidal, pero la excisión completa es infrecuente debido a su consistencia más dura y su mayor vascularización por lo que se han propuesto otros abordajes5,6.
Macroscópicamente, el schwannoma, también llamado neurinoma, neurilemoma, neuroma o neurolemoma está bien circunscrito, es pseudoencapsulado y presenta, como se ha comentado, una vascularización importante, lo que le hace más vulnerable al sangrado. Microscópicamente, es característico el patrón alternante entre áreas con patrón de Antoni de tipo A y B. Las áreas con patrón de Antoni de tipo A están compuestas por compactas células fusiformes, de límites citoplasmáticos imprecisos, cuyos núcleos se disponen en empalizada dejando un centro más claro, formando los «cuerpos de Verocay» (fig. 2.A.). Las áreas con patrón de Antoni de tipo B son menos celulares, se pueden ver cambios microquísticos, células inflamatorias y fibras de colágena, así como grandes espacios vasculares. Respecto a las técnicas inmunohistoquímicas, las células son positivas para la proteína S-100 (marcador de tumores de vaina nerviosa y melanoma) (fig. 2.B.). El caso descrito fue positivo para vimentina, marcador entre otros tumores de meningiomas, y para la proteína S-100. Los meningiomas son negativos para S-100 y positivos para EMA. Tampoco se trataba de un melanocitoma por datos histomorfológicos –ausencia de pigmento - aunque son positivos para S-100, por lo que quedaban excluidos las dos alternativas diagnósticas principales a considerar: meningioma fibroso y, en segundo lugar, melanocitoma.
 Fotomicrografía que muestra áreas de Antoni A compuestas por compactas células fusiformes (H&E, 100x). (B) Inmunohistoquímica positiva para la proteína S-100, con patrón citoplasmático dominante, de color pardo por el cromógeno utilizado (diaminobencidina) (proteína S100, 100x).")
(A) Fotomicrografía que muestra áreas de Antoni A compuestas por compactas células fusiformes (H&E, 100x).
(B) Inmunohistoquímica positiva para la proteína S-100, con patrón citoplasmático dominante, de color pardo por el cromógeno utilizado (diaminobencidina) (proteína S100, 100x).
En lo que se refiere a aspectos endocrinológicos del caso, es cuestionable la presencia de hipotiroidismo secundario con concentraciones de T4L de 13pg/ml pese a la escasa respuesta de TSH a la prueba de estímulo con TRH. En el hipertiroidismo la respuesta también está disminuida, pero la paciente no tenía patología tiroidea ni sintomatología compatible.
La anatomía patológica reveló una hiperplasia difusa de hipófisis a expensas de células somato y lactotropas, lo cual concuerda con el ligero aumento de PRL en sangre (si dejamos al margen una posible compresión del tallo hipofisario), pero es incongruente con las concentraciones bajas de IGF-1 y la respuesta de GH durante la ITT. Podría pensarse en haber realizado un segundo test de estímulo de GH pero la paciente no consintió en ello. Además, hay que considerar que el procedimiento quirúrgico «per se» pudiera modificar el comportamiento inicial, por lo que funcionalmente asumimos el déficit de GH que inmunohistoquímicamente luego no se confirmó. Es frecuente hallar en la práctica clínica este tipo de discordancias entre comportamiento bioquímico y resultado de la inmunohistoquímica.
El primer diagnóstico anatomo-patológico vino condicionado por un probable sesgo de biopsia, pudiendo corresponder a tejido hipofisario hiperplásico perilesional con impresión diagnóstica intraoperatoria de probable adenoma no confirmada posteriormente. La segunda cirugía, ante la persistencia de masa en la RMN, sí fue diagnóstica de schwannoma. Alternativamente, podría corresponder a un caso de incipiente evolución de una collision sellar lesion. Koutourousiou et al7 describieron 8 casos de adenomas hipofisarios que se presentan simultáneamente con una segunda lesión selar (de origen adenomatoso o no), las denominadas «collision sellar lesions», en su serie de 548 pacientes intervenidos de adenoma hipofisario por vía transesfenoidal. La cohorte incluía a 5 mujeres y 3 varones con una edad media de 48 años (rango de edad de 38 a 76 años). En base a los datos clínicos y radiológicos, todos los pacientes fueron sometidos a cirugía transesfenoidal con el diagnóstico preoperatorio de adenoma hipofisario sin sospecha de una lesión dual. Entre ellos hay un caso único de schwannoma intraselar coexistiendo con un adenoma secretor de GH que fue publicado con posterioridad8. Se trataba de un varón de 38 años con cefalea y disminución de la líbido que fue diagnosticado de acromegalia. En la RMN presentaba un tumor hipofisario de 19mm con extensión supraselar. La histología demostró un adenoma inmuhistoquímicamente positivo para GH y en la periferia un schwannoma hipofisario. El paciente fue seguido durante 2 años sin sufrir recurrencia tumoral. Los autores concluyen en su revisión que el diagnóstico preoperatorio de una lesión dual es muy difícil, puesto que la presentación clínica y radiológica en la mayoría de los casos es de adenoma hipofisario; el diagnóstico definitivo es merced al estudio histológico. A día de hoy, se desconoce el mecanismo patogénico de estas lesiones duales.
Por último, cabe hacer mención a la cuestión sobre el origen de los schwannomas intraselares, puesto que las células de Schwann no se localizan en el sistema nervioso central. Maartens et al9 separan los casos publicados en verdaderos schwannomas intraselares y en schwannomas paraselares - estos últimos constituyen la mayoría-, basándose en la hipótesis de Russel y Rubinstein10 de que los neurinomas crecen a partir de células de Schwann localizadas en plexos nerviosos próximos a la silla turca. Otras explicaciones propuestas son: se trata de células de Schwann perivasculares11, son células de Schwann ectópicas10, derivan de células pluripotenciales12 o son migraciones de células de la cresta neural que forman focos de células de Schwann (schwannosis) en el parénquima del sistema nervioso central11. El origen del presente caso es aún más incierto, si cabe, al tratar de explicar la coexistencia (¿casual?) de la hiperplasia de células de GH y PRL.
En resumen, la posibilidad remota de un schwannoma intraselar existe en el diagnóstico diferencial de las lesiones hipofisarias, siendo la inmunohistoquímica trascendental en el diagnóstico definitivo de estas raras masas selares. Los casos publicados son aún escasos para establecer con seguridad el perfil del schwannoma de la silla turca.
