


GH and sex hormones are critical regulators of body growth and composition, somatic development, intermediate metabolism, and sexual dimorphism. Deficiencies in GH- or sex hormone-dependent signaling and the influence of sex hormones on GH biology may have a dramatic impact on liver physiology during somatic development and in adulthood. Effects of sex hormones on the liver may be direct, through hepatic receptors, or indirect by modulating endocrine, metabolic, and gender-differentiated functions of GH. Sex hormones can modulate GH actions by acting centrally, regulating pituitary GH secretion, and peripherally, by modulating GH signaling pathways. The endocrine and/or metabolic consequences of long-term exposure to sex hormone-related compounds and their influence on the GH-liver axis are largely unknown. A better understanding of these interactions in physiological and pathological states will contribute to preserve health and to improve clinical management of patients with growth, developmental, and metabolic disorders.
La GH y las hormonas sexuales son importantes reguladores del crecimiento y la composición corporal, el desarrollo somático, el metabolismo intermediario y el dimorfismo sexual. Deficiencias en las actividades fisiológicas de estas hormonas, así como las interacciones de las hormonas sexuales con la GH, repercuten en la fisiología hepática tanto durante el desarrollo corporal como en la edad adulta. Las hormonas sexuales pueden actuar sobre el hígado por mecanismos directos, a través de sus receptores hepáticos, o indirectos, modulando las funciones de la GH a niveles endocrino y/o metabólico. Las hormonas sexuales pueden modular las acciones de la GH a nivel central, regulando su patrón de secreción hipofisaria, y periféricamente, modulando sus mecanismos de señalización intracelular. Las consecuencias endocrinas y/o metabólicas de la exposición prolongada a compuestos relacionados con hormonas sexuales, así como su influencia sobre el eje GH-hígado son, en gran medida, desconocidas. La comprensión de estas interacciones en diferentes estados fisiológicos y patológicos contribuirá a mantener la salud y mejorar el manejo clínico de los pacientes con transtornos del crecimiento, el desarrollo y el metabolismo.
GH is the main regulator of somatic growth, metabolism, and gender-differentiated functions in liver.1–6 GH is predominantly linked to linear growth during childhood, but continues to have important metabolic actions throughout life. Interestingly, deficiency of GH-GH receptor (GHR),6 estradiol (E2)-Estrogen Receptor α (ERα)7–11 or testosterone (T)/Androgen Receptor (AR)11–15 signaling in adults causes a similar metabolic-like syndrome (i.e., fatty liver, visceral adiposity, insulin resistance, decreased muscle mass), a phenotype that can be ameliorated by GH or sex hormones replacement. E2 and T can modulate GH actions in the liver by acting both centrally, regulating pituitary GH secretion,16,17 and, peripherally, modulating GH signaling.18 Most previous studies have been focused on the influence of E2 and T on gender-specific pituitary GH secretion, which has a great impact on hepatic transcriptional regulation. In addition, the liver is a direct target of sexual hormones because it expresses ERα and AR, and the signaling pathways linked to these receptors are connected with lipid and glucose homeostasis,11 liver growth and regeneration,19 body growth,20 drug-induced hepatotoxicity,21 hepatic carcinogenesis,22 or fertility.23 Notably, E2 has been shown to modulate GH actions in the liver through induction of Suppressor of Cytokine Signaling (SOCS)-2, which in turn negatively regulates GHR-Janus Kinase (JAK)-2-Signal Transducer and Activator of Transcription (STAT)-5 signaling pathway. Interactions between T and GHR-JAK2-STAT5b-SOCS2 signaling pathways might also play a relevant role to regulate hepatic metabolism.24,25 This phenomenon is clinically relevant because its importance in the regulation of endocrine, metabolic, and gender-differentiated actions of GH in the liver. Notably, disruption of the GHR-JAK2-STAT5-SOCS2 signaling pathway is associated with disorders in somatic growth5,6 and gender dimorphism,17,26 and with liver diseases such as non-alcoholic fatty liver, insulin resistance, fibrosis, or hepatocellular carcinoma.27–30 However, the specific roles of the E2/ER and T/AR signaling for the regulation of liver physiology and, particularly, GHR-JAK2-STAT5-SOCS2 signaling pathway are still largely unknown. Furthermore, the novel discovery of JAK2 as a negative regulator of ERα suggests a more complex level of crosstalk between E2/ERα and GH-mediated signaling pathways.31 In the general population, the endocrine and metabolic consequences of long-term exposition to sex hormones-related compounds and their influence on the pituitary (GH)-liver axis are also largely unknown. In this review, we will summarize the current status of the influence of sex hormones on GH actions in the liver. A better understanding of this complex interaction in physiological and pathological states will contribute to prevent health damage and improve clinical management of patients with growth, developmental and metabolic disorders.
Regulation of pituitary GH secretionGH is a polypeptide mainly secreted by the somatotroph cells, but also produced in extra-pituitary tissues; therefore, GH also has paracrine-autocrine effects, distinct from its classic endocrine somatotropic effects.32,33 The regulation of pituitary GH secretion involves a complex neuroendocrine control system that includes the participation of several neurotransmitters and the feedback of hormonal and metabolic factors. Pituitary GH secretion is regulated by two hypothalamic peptides: GHRH and the inhibitory hormone somatostatin (SS). The balance of these peptides is, in turn, indirectly affected by many physiological stimulators (i.e., exercise, sleep, nutrients, thyroid hormones, sex hormones) and inhibitors (i.e., glucocorticoids, IGF-I, GH). The final integration of these signals occurs in the hypothalamus. Pituitary GH secretion is mainly reduced by the negative feedback of two circulating signals: pituitary GH itself, and liver-derived IGF-I stimulated by GH. In addition to hypothalamic and endocrine factors, other peripheral factors influence pituitary GH release (i.e., free fatty acids (FFA), insulin, glucose, amino acids, leptin, neuropeptide Y, ghrelin). These factors are primarily related to or derived from the metabolic status of the organism, which is consistent with the role of GH in regulating substrate metabolism, adiposity, and growth, and appear to coordinate the metabolic status of the organism with GH secretion. This is exemplified by adiposity, which is a powerful negative regulator of GH secretion: GH can stimulate FFAs mobilization, which inhibit GH release and complete a feedback loop among pituitary and adipose tissue. Conversely, leptin, which is also produced in adipose tissue, and ghrelin, from endocrine cells of the stomach, are inducers of pituitary GH secretion. Sex steroids are also physiological regulators of pituitary GH secretion.16,17 Neonatal and post-pubertal sex steroids control the ability of the hypothalamus to drive the sexually dimorphism of pituitary GH secretion seen in adulthood, both in rats and humans, which could explain gender-dimorphism in liver physiology. Sexual dimorphism in rodents seems to be regulated by E2 secretion in adult females and by T secretion, both neonatally and during adulthood, in males. Neonatal exposure to T imprints the male program of neuroendocrine control of the pulsatile pituitary GH secretion that is first seen at puberty, when the adult pattern of GH secretion becomes evident, and continues throughout adulthood. In post-pubertal rats, the blood male pattern consists of high and regular amplitude pulses spaced near 3–4h apart with no measurable trough levels. In contrast, the female pattern shows lower and erratic amplitude pulses, and GH is always present in blood. If such an androgen re-programming does not occur, the secretion pattern will remain as the feminine pattern. Interestingly, depletion of liver-derived IGF-I (LID)33 or SOCS2 deletion29 in male mice, or exposition of adult male rats to E234 can cause liver feminization of some of the GH-regulated biomarkers of gender dimorphism.
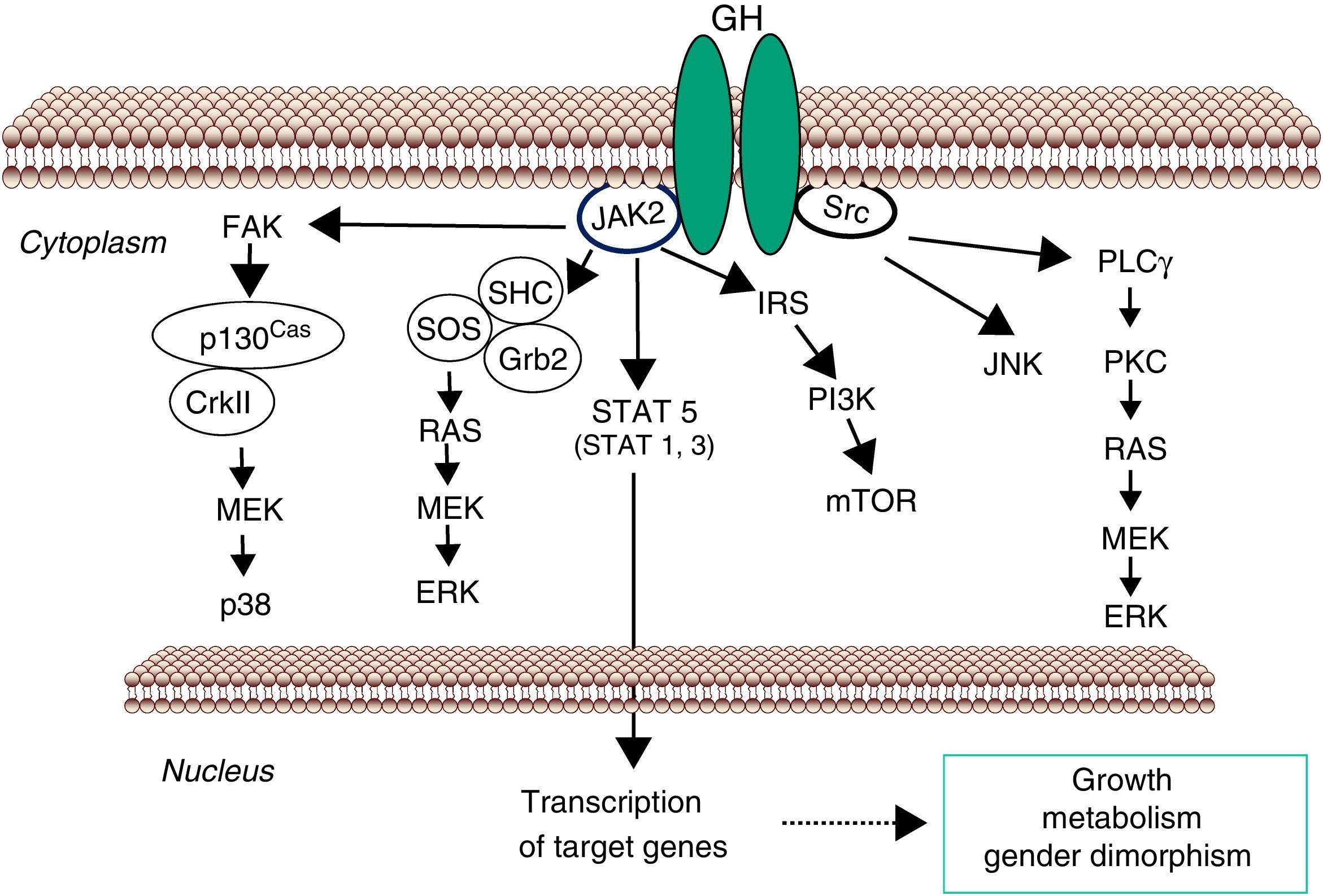
GH-STAT5 signaling in liver physiologyGH mediates its intracellular effects via the GHR which is ubiquitously expressed, especially in liver, fat, and muscle. GHR belongs to a family of receptors without intrinsic kinase activity. However, the GH molecule interacts with preformed dimmers of identical GHR pairs which results in a conformational change in the GHRs and associated tyrosine kinase JAK2 molecules3 (Fig. 1). This event unmasks the catalytic domain of JAK2, which is masked by its pseudokinase domain in the inactive state, and results in activation of adjacent JAK2 molecules by transphosphorylation. Activated JAK2 phosphorylates the GHR cytoplasmic domain on tyrosine residues and subsequent JAK2-dependent and -independent intracellular signal transduction pathways evoke cell responses including changes in gene transcription, proliferation, cytoskeletal re-organization, and lipid and glucose metabolism. STAT5 phosphorylation by JAK2 results in their dissociation from the receptor, dimerization, and translocation to the nucleus, where they modulate the transcription of target genes such as IGF-I, SOCS2, CYP2C12 and HNF6.35–38 STAT5b is a key transcription factor in GH regulation of target genes associated with body growth, intermediate metabolism (e.g., lipid metabolism) and gender dimorphism, even though STAT1, 3, and 5a have also been shown to be recruited by the GHR. In addition, many transcripts are regulated independently from STAT5b as a result of GHR activation of Src, ERK, and PI3K-mTOR signaling pathways.

Signaling pathways used by GH to regulate growth and metabolism. GH binds to a preformed GHR dimmer which results in activation of JAK2 tyrosine kinase. Simultaneously, Src kinase is also activated. Canonical JAK2 signaling via STAT5 involves phosphorylation of key tyrosine residues in the cytoplasmic domain of GHR, which recruit STATs to the activated JAK2 and thus facilitating their tyrosine phosphorylation and subsequent dimerization and translocation to the nucleus to regulate gene transcription. ERK can be activated either by SRC and/or PLCγ and Ras, or by JAK2 via the adaptors proteins. The PtdIns 3-kinase and the serine-threonine-protein kinase mTOR pathway is activated by JAK2 via IRS phosphorylation. These signaling pathways influence transcription of genes involved in growth and metabolism. Abbreviations: ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; Grb, growth factor receptor-bound protein; IRS, insulin receptor substrate; JAK2, Janus Kinase 2; JNK, c-Jun N-terminal kinase; MEK, dual specificity mitogen-activated protein kinase kinase 2; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinae; PKC, protein kinase C; PLCγ, phospholipase Cγ; SHC, SH2-domain containing transforming protein; SOCS, suppressor of cytokine signaling; SOS, son of sevenless; SRC, proto-oncogene tyrosine-protein kinase Src; STAT, signal transducer and activator of transcription.
The analysis of the molecular mechanisms involved in inactivation of GHR-dependent signaling pathway is also imperative for understanding GH physiology. This is clearly illustrated in the case of hepatic GHR-JAK2-STAT5b activation where signal duration regulates gender differences in liver gene expression.38 Studies in primary hepatocytes and several cell lines have shown that GH-induced JAK2-STAT5b activation is transient, with maximal activation achieved within the first 30min of stimulation, followed by a period of inactivation. This period is characterized by an inability to achieve maximal JAK2-STAT5 activation by GH in the following 3–4h, unless GH is withdrawn from the media.39 As mentioned above, the male pattern of pituitary GH secretion in rats is episodic with peaks every 3–4h and no measurable trough levels. Consequently, intracellular activation of STAT5b is also episodic and periods with low GH circulating levels are required to achieve maximal activation of STAT5b. Female rats, which exhibit a more continuous GH secretion pattern with higher basal levels and smaller and irregular intermittent peaks show reduced STAT5b activation compared with males. As expected, protein phosphatases are involved in negative regulation of GH signaling. In addition, the level of cell surface GHRs can be influenced by transcriptional, translational and posttranslational factors (e.g., nutritional status, endocrine context, developmental stage, and sex steroids) which, thereby, regulate cell sensitivity to GH actions. GHR translocation is also directly inhibited by IGF-I, likely contributing to a local feedback loop to hamper GH sensitivity. An early step in the termination of GH-dependent signaling is removal of GHRs by mechanisms of endocytosis and ubiquitination. In this context, SOCS2, an ubiqutin ligase, has been shown to be a key component of negative regulation of GHR-JAK2-STAT5 signaling pathway.40 In general, SOCS2 protein level is constitutively low, but its expression is rapidly induced by GH which is followed by SOCS2 binding to GHR complex to promote its ubiquitination and subsequent proteasomal degradation. Clinically relevant, SOCS2 is a key negative regulator of GH-dependent control of body growth1 and lipid and glucose homeostasis.29 Furthermore, several cytokines, growth factors (e.g., insulin), xenobiotics (e.g., dioxin, statins), and sex hormones can induce SOCS2 levels which provides a mechanism for cross-talking where multiple factors (endo- and xenobiotics) can regulate GH activity.
Body growthGH is predominantly linked with postnatal growth.32 Liver is the principal source of circulating IGF-I, and GH-dependent transcription of IGF-I is directly regulated by STAT5.1,5 Importantly, the mode of GH administration influences GH actions on liver. Intermittent (male pattern) GH administration to rodents is a more potent stimulus of body growth rate, IGF-I expression, and STAT5b activity in liver than is continuous (female pattern) administration. However, GH is more effective than IGF-I because GH exerts additional growth-promoting actions independent of IGF-I. Global disruption of STAT5b in mice causes loss of sexually dimorphic growth characteristics, so that the affected males reduced their size to female size, while female mice appeared unaffected. In addition, circulating IGF-I is reduced by 30–50% in affected male, but not in female mice. However, combined disruption of STAT5a/b significantly reduced body weight gain in females and suppressed body growth more than in STAT5b null male mice, approaching that observed in either GH or GHR deficient mice. These studies demonstrated that STAT5b is important for male-specific body growth, whereas STAT5a regulates body growth in both sexes. In addition to STAT5b, other transcription factors are related with body growth. This is exemplified by the glucocorticoid receptor, which is a critical co-activator of STAT5b in liver,41 or by interactions between E2/ER signaling and STAT5.42 Experiments in mice with SOCS2 disruption also support that STAT5b is critical for GH regulation of somatic growth.40,43 In addition to endocrine actions, paracrine involvement of STAT5a/b in the effects of GH on muscle is also evidenced by the loss of muscle IGF-I transcripts and mass seen in the muscle-specific deletion of STAT5a/b.44
Lipid metabolismThe main metabolic process affected by GH status is energy/fuel metabolism, particularly lipid/fat metabolism.1,5,45 A key physiological function of GH is the promotion of protein synthesis and inhibition of protein degradation in muscle, bone and other large organs, inhibiting glucose and aminoacids catabolism by promoting the utilization of lipids as energy source. These systemic effects of GH are achieved through inhibition of insulin actions and the promotion of fatty acid mobilization from adipose tissue and liver.1,4 In adipose tissue, GH is a lipolytic hormone and reduces fat mass. This is particularly evident in individuals who have accumulated excess fat during periods of GH deficiency.2,4 In the liver, GH promotes triglyceride synthesis and secretion; in addition to increasing lipogenesis (e.g., SREBP1), GH inhibits PPARα expression and decreases lipid oxidation.34,46 In the skeletal muscle, GH stimulates triglyceride uptake and lipid oxidation, which can be modified by factors such as nutrition, exercise, and sex steroid hormones. Conversely, GH deficiency (GHD) in adulthood causes a metabolic syndrome (i.e., increased visceral adiposity, fatty liver, decreased muscle mass, metabolic disturbances) that can be ameliorated by GH replacement. An ineffective GHR-JAK2-STAT5 signaling pathway results in fatty liver and adiposity in rodents and human which is due to enhanced lipogenesis and reduced triglyceride secretion as well as reduced lipolysis.6,27,28 This is supported by original findings that STAT5b-deleted male mice become obese in later life26 and that STAT5b deletion in a mature human was associated with obesity.47 In contrast, ablation of SOCS2 in mice, which increased STAT5 signaling, protected from high-fat diet-induced liver steatosis29. These findings highlight two physiological aspects of GHR-STAT5b signaling: (a) STAT5b plays a critical role in regulation of key enzymes or genes otherwise involved in lipid and energy balance. Clinically relevant, anti-obesity actions of GH are enhanced by the male pattern of pituitary GH secretion due to pulsatile STAT5 activation. (b) Despite normal plasma FFA and minimal adiposity, absent GHR signaling leads to fatty liver because of reduced STAT5 activation. Relevant to this review, agonists of Liver X Receptor (LXR), which cause hepatic steatosis,48 can inhibit GH-STAT5b signaling.49 This inhibition is mediated by SREBP1, a LXR target gene, through the downregulation of STAT5b gene transcription and stimulation of STAT5b protein degradation. These findings highlight the molecular interactions of LXR with GH-STAT5b signaling in liver.
Glucose metabolismGH stimulates the hepatic glucose production. GH increases glycogenolysis; however, it has either a stimulatory or no effect on gluconeogenesis. This may be a result of GH antagonism of insulin action leading to hepatic/systemic insulin resistance. In addition, IGF-I exerts profound effects on carbohydrate metabolism and may enhance insulin sensitivity by suppressing GH release.4 Therefore, activation of IGF-I signaling adds more complexity for understanding molecular mechanisms involved in GH-induced insulin resistance in vivo. GHD mice and the GHRKO mice have improved insulin sensitivity and up-regulated hepatic insulin signaling, suggesting that GH antagonizes insulin signaling locally in the liver.50 However, over-expressing the human GH gene in rats increases basal hepatic glucose uptake and glycogen content.51 GH-induced insulin resistance may be developed by the increased FFA mobilization from peripheral adipose tissue which can then affect liver insulin sensitivity, and lead to insulin resistance and up-regulation of gluconeogenic genes [e.g., glucose-6-phosphatase, phosphoenolpyruvate carboxykinase] in the liver with an important role in glucose homeostasis. Interestingly, the LID mice shows a 75% reduction in circulating IGF-I levels, 3–4 fold increase in circulating GH levels and insulin resistance, without significant increase in circulating FFA levels, arguing for the existence of a local cross-talk between GH and insulin signaling systems within the hepatocyte. Moreover, after crossing LID mice with GH transgenic mice, serum FFA levels were significantly increased and there was an improvement in insulin sensitivity due to higher hepatic, adipose tissue and skeletal muscle glucose uptake.52 It has been suggested that, in addition to FFA, the SOCS family of proteins, whose expression is induced by both GH and insulin in the liver, may contribute to insulin resistance.49,53 Recently, we have shown that SOCS2 deletion protected against fatty liver but, paradoxically, worsen insulin resistance in high-fat diet fed mice.29 In contrast, SOCS2 deletion protected against streptozotocin-induced type I diabetes in adult male mice (in press).
Gender dimorphismAs mentioned above, sex hormones imprint a sex-dependent pattern of pituitary GH hormone secretion which is a major player in establishing and maintaining gender dimorphism in liver physiology.17,54 From neonatal period of life, gonadal steroids play a critical role to maintain liver response to GH in adulthood. Neonatal exposure to T is crucial and the full response to androgens in adulthood is dependent on neonatal imprinting by T. The male characteristic metabolism in liver in adulthood is dependent on continuous androgen exposure. In female rats, gonadectomy has little impact on hepatic steroid metabolism; estrogen treatment (or exposition), however, feminizes hepatic metabolism in male rats. Most of the hepatic gender dimorphism can be explained by the female-specific pattern of pituitary GH secretion, through the induction of female-predominant transcripts and suppression of male-predominant ones; 20–30% of all hepatic genes have a sex-specific expression pattern in rodents. Genome-wide screens of gene expression have shown that GH- and sex-dependent regulation of hepatic gene expression affects several families of hepatic genes involved in endo- and xenobiotic metabolism and metabolic functions (e.g., lipid metabolism). In addition, a number of other hepatic transcripts encoding plasma proteins, enzymes, transcription factors and receptors involved in the metabolism of proteins, carbohydrates, lipids, or signaling regulation have been also found to be up- and/or down-regulated by the different patterns of GH or sex-steroid exposure.34,38 There is consensus on the response to sex-different pattern of pituitary GH secretion as the major cause of gender dimorphism in liver. However, it is likely that other factors are behind some hepatic sex differences. Potential mechanisms that could contribute to this “liver sexuality” are the pituitary-independent effects of estrogens through interaction with ERα and GH-JAK2-STAT5b signaling pathway in liver. STAT5b is a key player in this scenario and it is responsible for the masculinization of the male liver.26,38 Conversely, other transcription factors (e.g., HNF6 and HNF3b) are more efficiently activated in female liver or by the continuous GH administration.55,56 As mentioned above, SREBP1c induction, as well as hepatic triglyceride synthesis and VLDL secretion, and PPARα inhibition can be observed in the liver after continuous GH administration.34,46
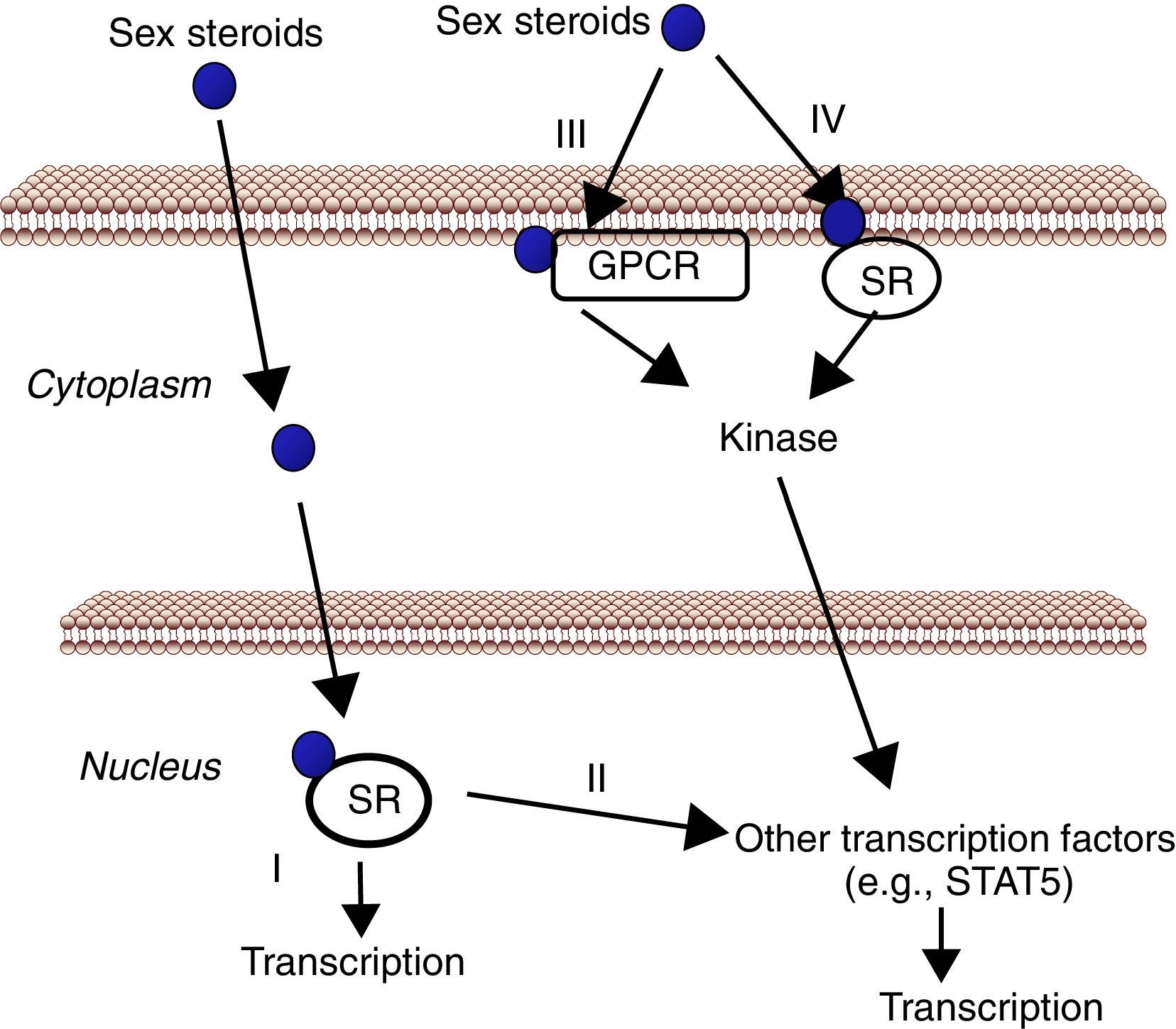
The liver, a physiological target for sex steroidsAs mentioned above, the liver represents a target tissue for sex hormones where physiologically and therapeutically relevant interactions between E2/T- and GH-dependent signaling can be developed. Estrogens and androgens can signal through multiple receptors (Fig. 2). Estrogen signaling can be mediated by multiple receptors.57 Most of the known estrogenic effects are mediated via direct interaction of estrogen with the DNA-binding transcription factors, ERα and ERβ. This classical estrogen signaling occurs through a direct binding of ER dimers to estrogen responsive elements in the regulatory regions of estrogen target genes, followed by activation of the transcriptional machinery at the transcription start site. In addition, E2 can modulate gene expression by a second mechanism in which ERs interact with other transcription factors (e.g., STAT5), through a process referred to as transcription factor cross-talk. E2 may also elicit effects through non-genomic mechanisms, which involve the activation of downstream kinase pathways via membrane-localized ERs. An orphan G protein-coupled receptor (GPR)-30 in the cell membrane has also been reported to mediate non-genomic and rapid estrogen signaling. Finally, E2 has a similar affinity for ERα and ERβ and they are activated by a wide range of ligands including Selective Estrogen Receptor Modulators (SERMs) (e.g., raloxifen) as well as many other xenobiotic compounds. Tissue expression of ERs is also a critical determinant of estrogenic actions. ERβ is expressed in the ovary, prostate, lung, gastrointestinal tract, bladder, and hematopoietic and the central nervous systems, while ERα is mainly expressed in reproductive tissues, kidney, bone, white adipose tissue, and liver. This indicates that specific actions of estrogens in liver can be mimicked by using selective ERα agonists such as propyl-pyrazole-triol (PPT).58 Therefore, the above mentioned data indicate that the mechanisms involved in ER signaling are influenced by tissue ER distribution, the target gene, and the activity or crosstalk with other signaling pathways.
 (I). In addition, estrogens and androgens can modulate gene expression by a second mechanism in which SR interact with other transcription factors (e.g., STAT5) (II). There are also evidences that sex steroids may also elicit effects through non-genomic mechanisms, which involve the activation of downstream kinase pathways via a G protein-coupled receptor (GPCR) (III) or SR (IV) localized in the cell membrane.")
Cell signaling by sex steroids. Estrogens and androgens can signal through multiple receptors. Most of their known effects are mediated via direct interaction of sex steroids with the DNA-binding transcription factors, nuclear steroid receptor, SR (ER, AR) (I). In addition, estrogens and androgens can modulate gene expression by a second mechanism in which SR interact with other transcription factors (e.g., STAT5) (II). There are also evidences that sex steroids may also elicit effects through non-genomic mechanisms, which involve the activation of downstream kinase pathways via a G protein-coupled receptor (GPCR) (III) or SR (IV) localized in the cell membrane.
Androgen signaling can also be mediated by genomic and non-genomic mechanisms.14,59 The major circulating androgen, T, is synthesized in testicular Leydig cells and released into blood, where it binds to steroid hormone-binding globulin (SHBG) to facilitate the transport. Within target cells, T is converted to DHT, a more active androgen, by 5α-reductase. Similarly to classical E2 signaling, most of the known androgenic effects are mediated via direct interaction of T/DHT with the DNA-binding transcription factor AR which plays an important role in regulating androgen action in men and women. Androgen-activated AR regulates the transcription of a variety of target genes through the interaction with different coregulators which contribute to tissue specificity of androgen actions. There are also evidences that T/AR may also elicit effects through non-genomic mechanisms: (1) the AR is palmitoylated, which regulates its localization to the membrane; (2) the AR can be localized in lipid rafts within the membrane; (3) membrane-localized AR mediates rapid G protein signaling by androgens, and (4) androgen activation of membrane-localized AR leads to rapid transactivation of the EGF receptor which is followed by activation of Akt and MAPK pathways and subsequent nuclear AR-mediated signaling. However, whether altered androgen/AR signaling dysfunctions may play a role in the pathophysiology of different metabolic phenotypes or influence somatotropic-liver axis remains, in comparison with E2/ER signaling, largely unknown.
Body growth and compositionThe impact of sex steroids on body growth and composition is complex.16 The increase in pubertal growth velocity associated with increased GH secretion has been traditionally attributed to T in boys, and to E2 or adrenal androgen secretion in girls. However, during last years, research has indicated that E2 may be the principal hormone stimulating the pubertal growth spurt in both sexes.60,61 Interestingly, ERα, but not ERβ, mediates important effects of estrogens in the skeleton of male mice during growth and maturation.38 A phenotype similar to ERαKO mice can be also found in aromatase-deficient (ArKO) male rats, which cannot produce estrogens. Notably, E2 can rescue skeletal growth rates in the absence of GHR (i.e., in GHRKO mice) associated with an increase in hepatic and serum IGF-I,62 a novel mechanism of GHR-independent stimulation of hepatic IGF-I production. This is also supported by E2 induction of IGF-I gene expression in hypothyroid male rat liver, a model with low or undetectable levels of circulating GH.34 In addition, gender-related differences in body composition emerging at the time of pubertal growth are in part mediated by sex steroids modulating the GH-IGF-I axis. In adulthood, oral E2 administration to postmenopausal women can provoke a reduction of circulating IGF-I levels concomitantly with an increase of GH, whereas transdermally delivered E2 has been shown to elevate both GH secretion and IGF-I concentrations.63 Similarly, oral administration of pharmacological doses of estrogen to hypopituitary patients inhibits GH-regulated endocrine and metabolic effects (i.e., circulating IGF-I levels, lipid oxidation, and protein synthesis are suppressed, with a reciprocal elevation of carbohydrate oxidation). E2 modulation of GH signaling is also exemplified by the greater increase in lean mass and decrease in fat mass, or the greater increase in bone turnover and bone mass, induced by GH treatment in GH-deficient male compared to female patients.64,65 These effects on metabolism and body composition are attenuated by transdermal administration, suggesting that these route-dependent effects are consequence of first-pass effect of oral estrogen leading to direct inhibition of GH-JAK2-STAT5-IGF-I signaling pathway.
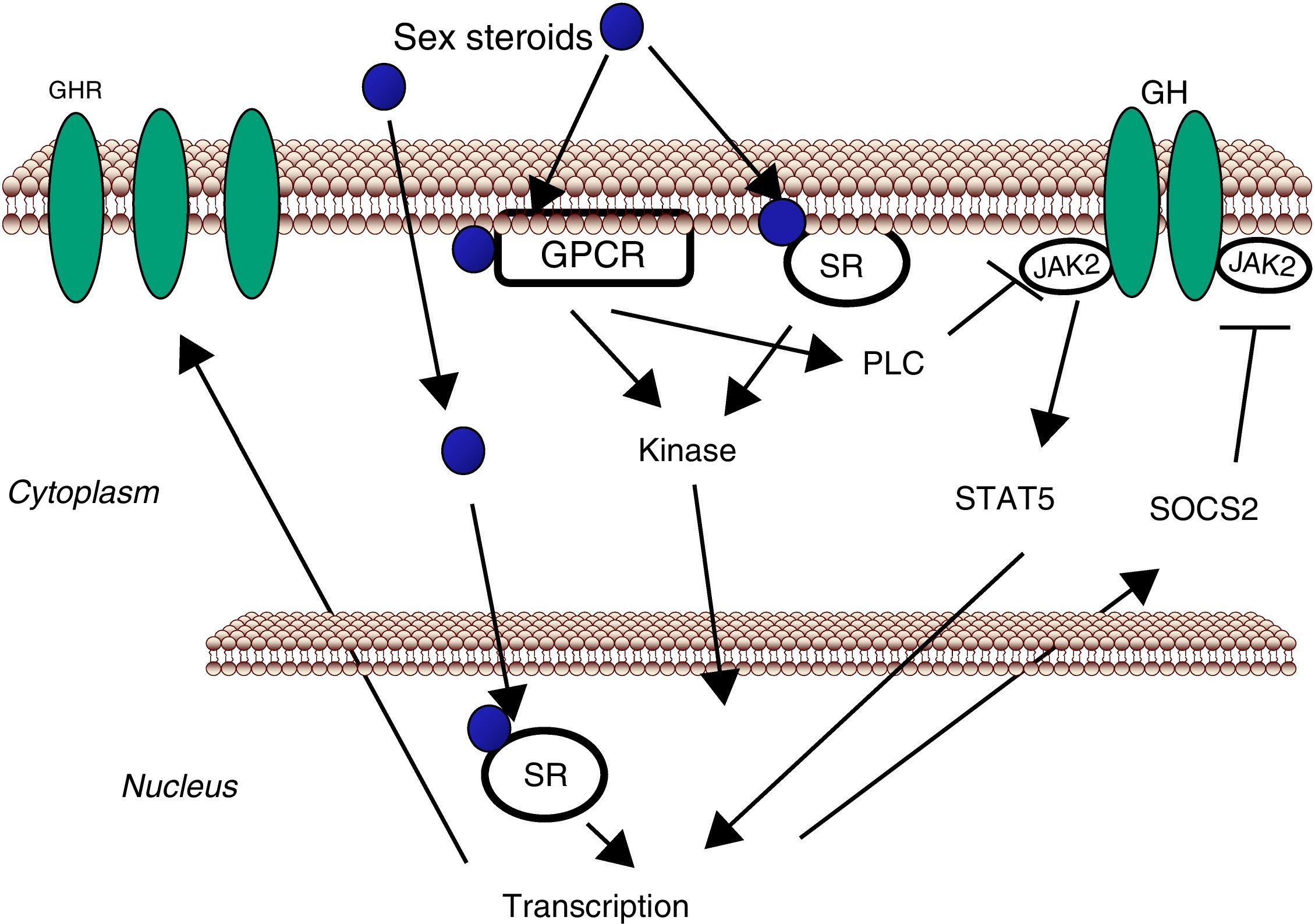
Sex steroids can modulate cell sensitivity to GH through multiple mechanisms (Fig. 3). Estrogens can modulate GH actions in liver by acting peripherally, modulating GH responsiveness, which include changes in hepatic GHR expression and crosstalk with GH-activated JAK2-STAT5 signaling pathway. Particularly, estrogens can induce SOCS-2 and SOCS-3 expression which in turn negatively regulate GHR-JAK2-STAT5 signaling pathway leading to reduction in transcriptional activity in liver.18 Recently, we have shown that subcutaneous administration of nearly physiological doses of E2 to hypothyroid male rats dramatically influenced the hepatic transcriptional response to pulsatile (male pattern) GH administration including expression of genes related to GH-regulated endocrine, metabolic, and gender-differentiated functions.54 Together with induction of a female-pattern of gene expression, E2 inhibited GH-regulated STAT5b targeted genes in liver which was associated with increased mRNA expression of several members of SOCS family. Hypothetically, other members of the negative regulators of STAT family may also contribute to E2 interaction with GH signaling in liver. This is explained by ERα stimulation of PIAS3 expression which inhibits STAT3 DNA-binding. Interestingly, as mentioned above, a direct interaction between ER and STAT5 may also regulate STAT5-dependent transcriptional activity42 and influence GH physiology. Therefore, besides E2 regulation of pubertal growth and the female-pattern of pituitary GH secretion, induction of negative regulators of JAK2-STAT5 signaling pathway in vivo is a very relevant mechanism that, in part, could explain how E2 modulates hepatic transcriptional response to GH. T can also modulate the GH-IGF-I axis. Androgen-mediated signaling has shown to be a critical determinant of body composition in adult men promoting growth of lean mass and suppressing fat deposition.66–70 Multiple evidences have shown that the growth promoting and metabolic effects of T need positive interplay with GH-IGF-I axis. Notably, linear growth in GH-deficient children receiving GH replacement is further stimulated by androgen treatment, and GH is required for full androgen growth-promoting effect. T enhances the growth of boys with hypogonadism and of those with hypopituitarism during GH treatment; however, the effects of T on somatic growth are poor in boys with hypopituitarism without concomitant GH replacement. The critical role of T-GH interactions on body composition is also exemplified in adult GH-deficient men, in whom lean body mass remains subnormal even after adequate androgen replacement. In hypopituitary adults who are not receiving GH replacement, T exerts no effect on circulating IGF-I and both hormones, however, are necessary to exert an optimal effect on circulating IGF-I. Finally, the observation that the effects of GH replacement are more marked in men than in women provides further evidence that T might enhance the anabolic effects of GH in vivo.
Lipid and glucose metabolism
Sexual dimorphism exists in lipid and glucose metabolism.11,54 E2 has a physiological role in the control of lipid and glucose metabolism in both human and rodents; deficiency of E2/ERα signaling can lead to a metabolic syndrome-like phenotype (i.e., fatty liver, adiposity, insulin resistance).7–9 This is exemplified in postmenopausal women who have higher tendency to develop metabolic syndrome than premenopausal. ERα deficiency or decreased levels of aromatase activity leads to the development of visceral adiposity, insulin resistance, and hyperinsulinemia both in male humans and mice. This metabolic syndrome-like phenotype is reversible by E2 treatment in ERαKO and ArKO mice. The beneficial influence of E2 in relation to normalizing lipid and glucose homeostasis is also evidenced in ob/ob and high-fat diet fed mice, models of obesity and type 2 diabetes. Importantly, treatment of ob/ob mice with PPT can improve glucose tolerance and insulin sensitivity, which supports the critical role of ERα signaling in the control of glucose homeostasis. Estrogenic signaling via GPR-30 has also been implicated in insulin production and glucose homeostasis. ERα mainly mediates anti-lipogenesis, reduction of adiposity and improvement of insulin sensitivity; ERβ-dependent activity, however, may be detrimental for the maintenance of normal glucose and lipid homeostasis. In ERαKO mice, insulin resistance is mainly localized to the liver, including increased lipid content and hepatic glucose production. Surprisingly, when ERα was selectively ablated in liver (LERKO), LERKO mice did not recapitulate the observed ERαKO mice phenotype (i.e., adiposity, glucose intolerance, insulin resistance), even when challenged with a high fat diet. This suggests the presence of unidentified compensatory mechanism/s or that hepatic insulin resistance occurs as a secondary effect upon ablation of E2 signaling in other cell types. Interestingly, selective ablations of ERα in the hypothalamic brain region or in hematopoietic/myeloid cells give rise to an increase in body weight and reduced glucose tolerance. The antilipogenic effects of E2 in liver are in part due to activation of PPARα- and inhibition of LXRα-dependent signaling pathways which is followed by increased fatty acid oxidation and inhibition of lipogenic genes (e.g., SREPB1c, Apo E).34 Activation of LXRα-dependent signaling increases triglyceride accumulation in the liver; E2/ERα signaling, in contrast, reduces lipogenic pathway expression and fatty liver induced by LXR activation in mouse.58 Similarly to E2/ERα deficiency, a decrease of androgen/AR signaling is also associated with a metabolic syndrome-like phenotype (i.e., truncal adiposity, fatty liver, increased triglycerides/cholesterol, reduced HDL, insulin resistance-type 2 diabetes) and this is improved after T replacement therapy.13,70,71 However, the specific role of the androgen/AR signaling for the regulation of liver metabolism is, in comparison with E2/ERα, still largely unknown. Results derived from various AR knockout mouse models reveal tissue-specific AR signaling that is involved in regulation of lipid metabolism (i.e., inhibits lipogenesis, prevents liver steatosis) and promotes anabolic growth in peripheral tissues.70 In male mice, the deletion of AR (ARKO) causes late-onset obesity whereas the liver-specific ARKO (LARKO) male mice have increased insulin resistance and steatosis, with decreased β-oxidation, upon high-fat diet. Clinically relevant, increased insulin resistance and impaired glucose tolerance have also been observed in men with T deficiency.13,70,71 In addition, some AR polymorphisms, with reduced AR activity, are associated with an excess of body fat and fat distribution pattern in both sexes.14 Notably, T treatment can reduce visceral-fat in mice and men, and improves non-alcoholic fatty liver disease.11–14,70–72 However, most E2/ERα actions that control body weight and lipid/glucose metabolism are relevant in both female and male, suggesting that T aromatization in E2, acting on ERα, might also contribute to energy homeostasis in males.
In summary, a decrease in E2/ERα- or T/AR signaling is associated with metabolic disorders (i.e., metabolic syndrome-like phenotype with adiposity and hepatic steatosis) which resemble deficiency of GHR-JAK2-STAT5 signaling. Interestingly, these metabolic disorders can be partially prevented, or ameliorated, by E2/T and/or GH replacements which suggest that these hormones regulate overlapping cellular networks related with physiological control of lipid and glucose homeostasis.
ConclusionsEstrogen/ERα- and androgen/AR-dependent signaling play a critical role in liver physiology and pathology in both females and males. Physiologically and therapeutically relevant are sex hormones interactions with GH-regulated endocrine (e.g., IGF-I), metabolic (e.g., lipid and glucose metabolism), and sex-differentiated (e.g., endo- and xenobiotic metabolism) functions in the liver. The influence of sex hormones is executed at the level of pituitary GH secretion and the regulation of GHR-JAK2-STAT5-SOCS2 signaling pathway. Therefore, the pituitary (GH)-gonadal (E2 and T)-liver axis is relevant due to the physiological roles that these hormones have in mammals, and the widespread use of sex hormones-related compounds in humans. In the general population, the endocrine and metabolic consequences of long-term exposition to sex hormones-related compounds and their influence on the pituitary-liver axis are largely unknown. Thus, understanding this complex interaction in physiological and pathological states could contribute to prevent health damage and improve clinical management of patients with growth, developmental and metabolic disorders.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the authors that have made a contribution to the understanding of the crosstalk between sex hormones and GH signaling in liver. We apologize to those whose work deserves to be cited but unfortunately are not quoted because of space limitations. The research program in the author's lab was supported by grants-in-aid from the Spanish Ministry of Economy and Competitivity (MINECO) with the funding of European Regional Development Fund-European Social Fund (SAF2012-37344), Canary Islands Government (ACIISI PI2010), and Alfredo Martin-Reyes Foundation (Arehucas)-FICIC.
