


El síndrome de Turner es definido por un conjunto de rasgos fenotípicos característicos resultantes de la alteración completa o parcial del cromosomaX.
Pacientes y métodosEstudio descriptivo retrospectivo en el que se analiza el diagnóstico, la evolución y la situación actual de pacientes controladas en los últimos 40años de síndrome de Turner en un hospital terciario (Bizkaia) mediante revisión de historias clínicas y encuestas telefónicas.
ResultadosEstudiamos 45 mujeres, con edad media actual de 22,95años (rango 2-38) y edad media de diagnóstico de 4,71años. El 63% presentaron mosaicismo en su cariotipo. El motivo de consulta más frecuente fue talla baja (54%), objetivándose un incremento de diagnóstico prenatal en los casos más recientes. Han recibido tratamiento con hormona de crecimiento el 72%, y el 26% además recibieron oxandrolona. En el 69% de las pacientes la talla final alcanzada fue baja. Han presentado fallo gonadal el 66%, recibiendo la mayoría tratamiento hormonal sustitutivo. Tres pacientes han tenido descendencia con óvulo de donante. El seguimiento de las 31 pacientes adultas es llevado a cabo fundamentalmente por endocrinología (37,5%) y/o ginecología (34,4%). En el ámbito psicosocial han precisado ayuda durante la escolarización el 22%, alcanzando el 80% estudios de nivel medio-alto. Dos pacientes han fallecido, una por disección de aneurisma aórtico y la otra paciente, con múltiples patologías, por insuficiencia respiratoria.
ComentariosLa talla baja es el motivo más frecuente de consulta en pacientes con síndrome de Turner. En la mayoría de los casos el cariotipo es mosaico. Las manifestaciones clínicas más frecuentes son talla baja y fallo gonadal. El 80% consiguen realizar estudios de nivel medio-alto. En la edad adulta el seguimiento es irregular y en ocasiones escaso, siendo claramente mejorable.
Turner syndrome is characterized by a great variability of clinical manifestations caused by a total or partial loss of X-chromosome.
Patients and methodsA retrospective, descriptive study of the diagnosis, course, and current status of patients with Turner syndrome followed up at our section over the past 40 years, based on review of medical records supplemented with a telephone survey.
ResultsForty-five female patients with a current mean age of 22.95years (range 2-38) and a mean age at diagnosis of 4.71 were included. Sixty-three percent of them showed a mosaic karyotype. Short stature was the most common reason for consultation (54%), with increased prenatal diagnosis in most recent cases. Seventy-two percent have been treated with growth hormone, together with oxandrolone in 26%. Final stature was short in 69% of patients. Gonadal failure was found in 66%; most of whom received replacement therapy. Three patients achieved pregnancy by oocyte donation. The 31 adult patients are mainly monitored by the endocrinology (37.5%) and/or gynecology (34.4%) departments. As regards psychosocial aspects, 22% required support during school, and 80% completed middle to high level education. Two patients died, one due to dissecting aortic aneurysm and the other one, who had multiple pathological conditions, from respiratory failure.
ConclusionsShort stature is the main cause of diagnosis in patients with Turner syndrome; most cases show genetic mosaicism. The most common clinical manifestations include short stature and gonadal failure. Eighty percent of patients complete middle or high education. In adulthood, follow-up is irregular, sometimes scarce, and clearly improvable.
El síndrome de Turner (ST) es una enfermedad genética causada por la ausencia completa o parcial del segundo cromosomaX, con o sin mosaicismo, que afecta aproximadamente a 1/2.500 recién nacidas con fenotipo femenino. Las manifestaciones clínicas más frecuentes son talla baja, disfunción gonadal, malformaciones cardiacas y renales, problemas óticos y oftalmológicos y determinados rasgos fenotípicos. La bibliografía demuestra que el tratamiento con hormona de crecimiento (GH) es fundamental para mejorar la talla baja, y en las pacientes con insuficiencia ovárica es necesario el tratamiento con estrógenos1. Retrasar la terapia estrogénica hasta los 15años para optimizar la talla potencial, como se recomendaba previamente, parece desaconsejable, ya que infravalora la importancia psicosocial de una maduración puberal acorde con la normalidad. Van Pareren et al.2 y Rosenfield et al.3 sugieren que iniciar la estrogenización a los 12años permite una evolución puberal normal sin interferir con el efecto positivo de GH sobre la talla adulta final. Existen diversas formas de administrar los estrógenos, siendo la oral la más frecuente hasta ahora. Sin embargo, en la actualidad se considera que los estrógenos transdérmicos o los inyectables depot podrían ser más convenientes, por ser alternativas más fisiológicas4-6. Es fundamental educar a las pacientes en la importancia de mantener la terapia hormonal sustitutiva (THS) hasta la edad normal de la menopausia para mantener la feminización y prevenir la osteoporosis7.
Las mujeres con ST tienen mayor morbimortalidad, siendo la principal causa las enfermedades cardiovasculares. Tienen mayor riesgo de otras patologías como osteoporosis, hipotiroidismo, diabetes y dislipidemia1. El abordaje debe incluir a especialistas en pediatría, ginecología y obstetricia, endocrinología, cardiología, genética, patología otorrinolaringológica (ORL) y oftalmología, entre otros8. El desarrollo intelectual suele ser normal, salvo en las pacientes con un cromosomaX en anillo, que tienen mayor riesgo de presentar un déficit intelectual variable. Es habitual encontrar un perfil neurocognitivo específico con defectos en diferentes áreas del desarrollo relacionadas con la percepción visual y espacial, la comunicación no verbal, la coordinación motora, las habilidades perceptivas y la memoria visual9. En las guías internacionales recientes para el cuidado de las mujeres con ST se recomienda un control analítico anual que incluya glucemia, perfil hepático y lipídico y hormonas tiroideas, así como control de presión arterial (PA) frecuente, audiometría cada 2-3años e imagen cardiaca cada 5-10años7.
A continuación exponemos los resultados del estudio retrospectivo realizado para describir las características endocrinológicas y psicosociales de las pacientes con ST diagnosticadas en la sección de Endocrinología infantil del Hospital Universitario de Cruces en los últimos 40años y su situación en la edad adulta.
Pacientes y metodologíaSe ha realizado un estudio descriptivo retrospectivo en las pacientes diagnosticadas de ST en el Hospital Universitario de Cruces en los últimos 40años (tabla 1). La recogida de datos ha procedido de 2 fuentes: la revisión retrospectiva de las historias clínicas de la consulta de endocrinología pediátrica y la recogida actual de variables mediante una encuesta telefónica.
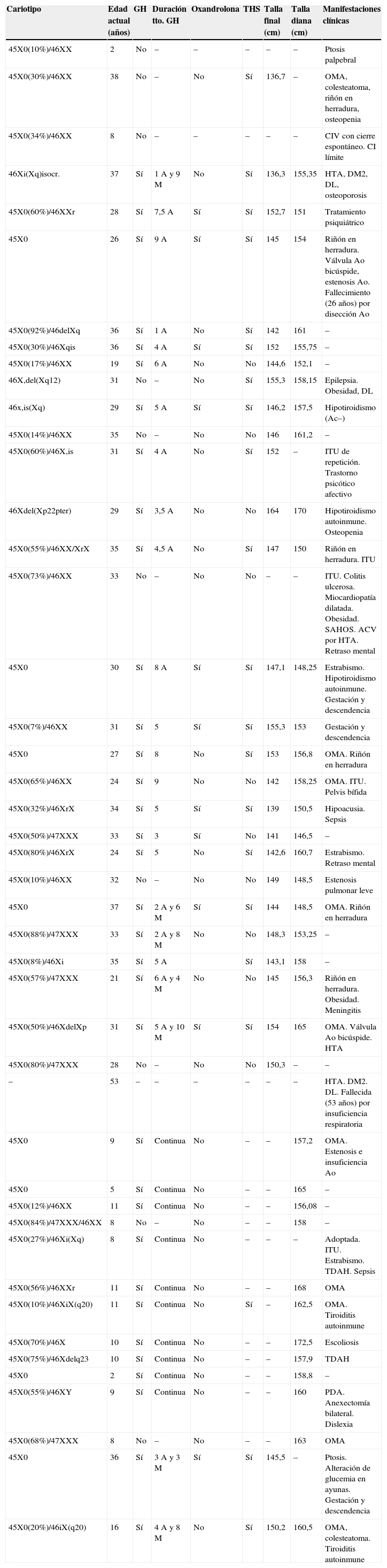
Resumen de datos individuales de las 45 pacientes con síndrome de Turner
| Cariotipo | Edad actual (años) | GH | Duración tto. GH | Oxandrolona | THS | Talla final (cm) | Talla diana (cm) | Manifestaciones clínicas |
|---|---|---|---|---|---|---|---|---|
| 45X0(10%)/46XX | 2 | No | – | – | – | – | – | Ptosis palpebral |
| 45X0(30%)/46XX | 38 | No | – | No | Sí | 136,7 | – | OMA, colesteatoma, riñón en herradura, osteopenia |
| 45X0(34%)/46XX | 8 | No | – | – | – | – | – | CIV con cierre espontáneo. CI límite |
| 46Xi(Xq)isocr. | 37 | Sí | 1 A y 9 M | No | Sí | 136,3 | 155,35 | HTA, DM2, DL, osteoporosis |
| 45X0(60%)/46XXr | 28 | Sí | 7,5 A | Sí | Sí | 152,7 | 151 | Tratamiento psiquiátrico |
| 45X0 | 26 | Sí | 9 A | Sí | Sí | 145 | 154 | Riñón en herradura. Válvula Ao bicúspide, estenosis Ao. Fallecimiento (26 años) por disección Ao |
| 45X0(92%)/46delXq | 36 | Sí | 1 A | No | Sí | 142 | 161 | – |
| 45X0(30%)/46Xqis | 36 | Sí | 4 A | Sí | Sí | 152 | 155,75 | – |
| 45X0(17%)/46XX | 19 | Sí | 6 A | No | No | 144,6 | 152,1 | – |
| 46X,del(Xq12) | 31 | No | – | No | Sí | 155,3 | 158,15 | Epilepsia. Obesidad, DL |
| 46x,is(Xq) | 29 | Sí | 5 A | Sí | Sí | 146,2 | 157,5 | Hipotiroidismo (Ac–) |
| 45X0(14%)/46XX | 35 | No | – | No | No | 146 | 161,2 | – |
| 45X0(60%)/46X,is | 31 | Sí | 4 A | No | Sí | 152 | – | ITU de repetición. Trastorno psicótico afectivo |
| 46Xdel(Xp22pter) | 29 | Sí | 3,5 A | No | No | 164 | 170 | Hipotiroidismo autoinmune. Osteopenia |
| 45X0(55%)/46XX/XrX | 35 | Sí | 4,5 A | No | Sí | 147 | 150 | Riñón en herradura. ITU |
| 45X0(73%)/46XX | 33 | No | – | No | No | – | – | ITU. Colitis ulcerosa. Miocardiopatía dilatada. Obesidad. SAHOS. ACV por HTA. Retraso mental |
| 45X0 | 30 | Sí | 8 A | Sí | Sí | 147,1 | 148,25 | Estrabismo. Hipotiroidismo autoinmune. Gestación y descendencia |
| 45X0(7%)/46XX | 31 | Sí | 5 | Sí | Sí | 155,3 | 153 | Gestación y descendencia |
| 45X0 | 27 | Sí | 8 | No | Sí | 153 | 156,8 | OMA. Riñón en herradura |
| 45X0(65%)/46XX | 24 | Sí | 9 | No | No | 142 | 158,25 | OMA. ITU. Pelvis bífida |
| 45X0(32%)/46XrX | 34 | Sí | 5 | Sí | Sí | 139 | 150,5 | Hipoacusia. Sepsis |
| 45X0(50%)/47XXX | 33 | Sí | 3 | Sí | No | 141 | 146,5 | – |
| 45X0(80%)/46XrX | 24 | Sí | 5 | No | Sí | 142,6 | 160,7 | Estrabismo. Retraso mental |
| 45X0(10%)/46XX | 32 | No | – | No | No | 149 | 148,5 | Estenosis pulmonar leve |
| 45X0 | 37 | Sí | 2 A y 6 M | Sí | Sí | 144 | 148,5 | OMA. Riñón en herradura |
| 45X0(88%)/47XXX | 33 | Sí | 2 A y 8 M | No | No | 148,3 | 153,25 | – |
| 45X0(8%)/46Xi | 35 | Sí | 5 A | Sí | 143,1 | 158 | – | |
| 45X0(57%)/47XXX | 21 | Sí | 6 A y 4 M | No | No | 145 | 156,3 | Riñón en herradura. Obesidad. Meningitis |
| 45X0(50%)/46XdelXp | 31 | Sí | 5 A y 10 M | Sí | Sí | 154 | 165 | OMA. Válvula Ao bicúspide. HTA |
| 45X0(80%)/47XXX | 28 | No | – | No | No | 150,3 | – | – |
| – | 53 | – | – | – | – | – | – | HTA. DM2. DL. Fallecida (53 años) por insuficiencia respiratoria |
| 45X0 | 9 | Sí | Continua | No | – | – | 157,2 | OMA. Estenosis e insuficiencia Ao |
| 45X0 | 5 | Sí | Continua | No | – | – | 165 | – |
| 45X0(12%)/46XX | 11 | Sí | Continua | No | – | – | 156,08 | – |
| 45X0(84%)/47XXX/46XX | 8 | No | – | No | – | – | 158 | – |
| 45X0(27%)/46Xi(Xq) | 8 | Sí | Continua | No | – | – | – | Adoptada. ITU. Estrabismo. TDAH. Sepsis |
| 45X0(56%)/46XXr | 11 | Sí | Continua | No | – | – | 168 | OMA |
| 45X0(10%)/46XiX(q20) | 11 | Sí | Continua | No | Sí | – | 162,5 | OMA. Tiroiditis autoinmune |
| 45X0(70%)/46X | 10 | Sí | Continua | No | – | – | 172,5 | Escoliosis |
| 45X0(75%)/46Xdelq23 | 10 | Sí | Continua | No | – | – | 157,9 | TDAH |
| 45X0 | 2 | Sí | Continua | No | – | – | 158,8 | – |
| 45X0(55%)/46XY | 9 | Sí | Continua | No | – | – | 160 | PDA. Anexectomía bilateral. Dislexia |
| 45X0(68%)/47XXX | 8 | No | – | No | – | – | 163 | OMA |
| 45X0 | 36 | Sí | 3 A y 3 M | Sí | Sí | 145,5 | – | Ptosis. Alteración de glucemia en ayunas. Gestación y descendencia |
| 45X0(20%)/46iX(q20) | 16 | Sí | 4 A y 8 M | No | Sí | 150,2 | 160,5 | OMA, colesteatoma. Tiroiditis autoinmune |
Ac: anticuerpos; ACV: accidente cerebrovascular; Ao: aórtico; CI: coeficiente intelectual; CIV: comunicación interventricular; DL: dislipidemia; DM: diabetes mellitus; HTA: hipertensión arterial; ITU: infección del tracto urinario; PDA: persistencia del ductus arterioso; TDAH: trastorno de déficit de atención e hiperactividad.
Hemos recogido los siguientes datos referentes a la etapa pediátrica: edad de diagnóstico, motivo de consulta, resultado del cariotipo, datos antropométricos (basales y evolutivos), escolaridad normal o con apoyo, tratamiento con GH asociada o no a oxandrolona, efectos adversos de esta terapia, existencia o no de fallo gonadal y tratamiento o no con THS, existencia de patología inmune, oftalmológica, ORL, renal y cardiaca.
Se ha completado la recogida de datos con una encuesta telefónica realizada en las pacientes adultas (mayores o igual a 16años) para obtener la siguiente información: talla adulta; existencia o no de una planificación de transición desde pediatría a los especialistas de adultos; seguimiento médico actual o ausencia del mismo; tipo de especialista que les controla; conocimiento de que se les hayan efectuado desde el alta de endocrinología pediátrica algunas exploraciones complementarias (densitometría, perfil lipídico y glucídico, registro de PA); tratamiento o no con THS; existencia o no de descendencia; estudios académicos y posición social alcanzados; relaciones sociales, de pareja y posible fallecimiento.
Hemos clasificado a la muestra global de pacientes en subgrupos en relación con su edad actual (mayores de 30años; de 16 a 30años; menor de 16años) para comparar algunas de las variables estudiadas, para destacar las diferentes tendencias terapéuticas a lo largo de los años y para ver su posible influencia en la talla final.
Metodología estadísticaLa comparación de 2 variables categóricas se ha realizado empleando la prueba estadística Chi-cuadrado o su correspondiente corrección de Fisher. Para la comparación de medias se ha utilizado la t de Student o el test de ANOVA. Para la realización de estos cálculos se ha usado el programa estadístico SPSS vs 21.0
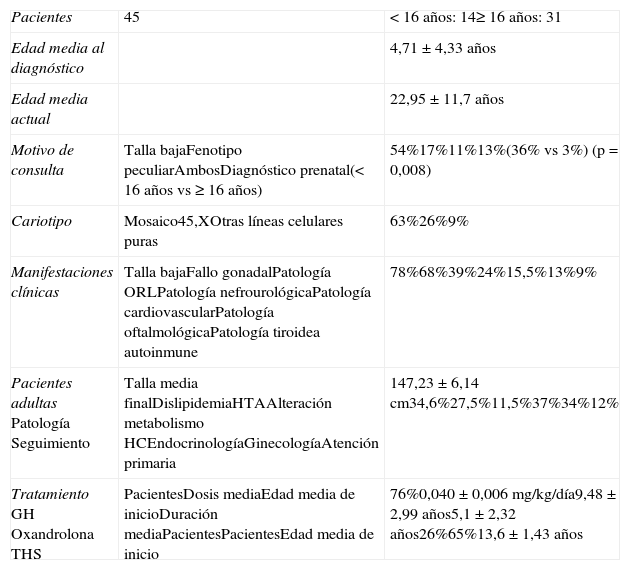
ResultadosEstudiamos a 45 pacientes con ST (tabla 2). Su edad media actual es de 22,95±11,7años (rango 2-38años) y edad media al diagnóstico de 4,71±4,33años. El 61% son mayores de 16años (n=31). El motivo de consulta más frecuente ha sido talla baja (54%), seguido por fenotipo peculiar (17%), ambas manifestaciones asociadas (11%) y diagnóstico prenatal (13%). Cabe destacar que comparando los 2 grupos de pacientes (pediátrico versus adultas) el diagnóstico prenatal por amniocentesis fue mayor en las pacientes jóvenes (36% vs 3%; p=0,008).
Características iniciales y de seguimiento de las pacientes con síndrome de Turner
| Pacientes | 45 | <16 años: 14≥16 años: 31 |
| Edad media al diagnóstico | 4,71±4,33 años | |
| Edad media actual | 22,95±11,7 años | |
| Motivo de consulta | Talla bajaFenotipo peculiarAmbosDiagnóstico prenatal(< 16 años vs ≥ 16 años) | 54%17%11%13%(36% vs 3%) (p=0,008) |
| Cariotipo | Mosaico45,XOtras líneas celulares puras | 63%26%9% |
| Manifestaciones clínicas | Talla bajaFallo gonadalPatología ORLPatología nefrourológicaPatología cardiovascularPatología oftalmológicaPatología tiroidea autoinmune | 78%68%39%24%15,5%13%9% |
| Pacientes adultasPatologíaSeguimiento | Talla media finalDislipidemiaHTAAlteración metabolismo HCEndocrinologíaGinecologíaAtención primaria | 147,23±6,14cm34,6%27,5%11,5%37%34%12% |
| TratamientoGHOxandrolonaTHS | PacientesDosis mediaEdad media de inicioDuración mediaPacientesPacientesEdad media de inicio | 76%0,040±0,006mg/kg/día9,48±2,99 años5,1±2,32 años26%65%13,6±1,43 años |
El cariotipo se ha determinado en linfocitos de sangre periférica, tras cultivo durante 72h y diversos tratamientos, con el estudio al microscopio de un mínimo de 20 metafases, ampliado a 50 en el caso de mosaicismos. El 63% de las pacientes presentan mosaicismo (un caso 45,X/46,XY). Cabe destacar que el cariotipo de 5 de las 6 pacientes con diagnóstico prenatal es mosaico. De los casos restantes, el 26% son 45,X y un 9% otras líneas celulares puras: 46,Xi(Xq), 46,Xdel(Xq) o 46Xdel(Xp).
La manifestación clínica más frecuente fue la talla baja, que aparece en el 78%. Todas estas pacientes menos una recibieron tratamiento con GH. La dosis administrada se encuentra entre 0,030-0,054mg/kg/día, con una media de 0,040±0,006mg/kg/día. La edad media de inicio de tratamiento es de 9,5±2,99años. La duración del tratamiento fue muy variable (rango: 1-9años), con una media de 5,1±2,32años. No se han registrado efectos secundarios a esta terapia.
En el 26% de los casos se asoció oxandrolona. La proporción de pacientes que recibió oxandrolona fue superior en el subgrupo de mayor edad (<16años 0%, 16-30años 27,3%, ≥30años 47,4%; p=0,005). La oxandrolona se suspendió en 3 pacientes por efectos adversos graves: hirsutismo leve en una y adelanto de la edad ósea en otras 2.
La media de la talla final alcanzada fue 147,23±6,14cm (rango: 136,3-164cm), estando el 69% de los casos por debajo de percentil 3. La desviación media respecto a la talla diana es de −8,22±6,45cm. No existen diferencias en la talla final alcanzada a lo largo del tiempo (mujeres con edad menor o mayor de 30años: 148,7±6,4 vs 146,3±6,0cm). Tampoco hemos encontrado diferencias al comparar las que han usado oxandrolona y las que no (147,1±5,3 vs 147,3±6,8cm). No existe diferencia estadísticamente significativa entre las pacientes con cariotipo 45X y el resto de cariotipos (147,65±3,68 vs 147,16±6,50cm).
El fallo gonadal está presente en el 68% de las pacientes. Todas las pacientes que lo padecieron, excepto un caso por negativa familiar, han recibido THS con una pauta inicial de 50ng/kg/día de etinilestradiol por vía oral, aumentando las dosis cada 4-6meses, hasta alcanzar en 18-24meses 20μg/día. La edad media de inicio del tratamiento ha sido de 13,6±1,43años (rango: 11 y 16años). Al comparar la edad de inicio de tratamiento entre los subgrupos de pacientes hemos objetivado que en los últimos años la terapia se indica a edades cada vez más tempranas (subgrupo <16años: 11,65±0,91años; subgrupo: 16-30años: 12,55±0,42años; subgrupo ≥30años: 14,33±1,43 años; p=0,004). Tres pacientes han tenido descendencia con óvulo de donante.
Las siguientes manifestaciones asociadas son la patología ORL, prevalente en el 39% de las pacientes (otitis media de repetición, pérdida de audición leve-moderada y colesteatoma), la nefrourológica en el 24% (riñón en herradura o infecciones del tracto urinario) y la oftalmológica en el 13%. Respecto a la patología autoinmune, cabe destacar que 2 pacientes han desarrollado tiroiditis autoinmune y otras 2 presentan hipotiroidismo autoinmune.
En relación con la patología cardiovascular asociada, 6 pacientes han presentado problemas cardiacos congénitos: 3 alteraciones aórticas (válvula aórtica bicúspide y estenosis e insuficiencia aórtica leve), una comunicación interventricular, una estenosis pulmonar leve y un paciente con persistencia de ductus arterioso. Una séptima paciente ha sido diagnosticada en la etapa adulta de miocardiopatía dilatada. De las 45 pacientes, 2 han fallecido: una mujer de 26años por un aneurisma y disección aórtica, y otra de 53años por un cuadro de insuficiencia respiratoria aguda en el contexto de derrame pleural masivo.
Centrándonos en el subgrupo de las 31 pacientes adultas, la transición de endocrinología infantil a adultos se ha realizado tras alcanzar la talla final y con el desarrollo puberal completo, aunque no se ha realizado ninguna planificación de la transición. Actualmente son controladas de forma heterogénea: el 37% por especialistas en endocrinología, el 34% por ginecólogos y el 12% en atención primaria.
Disponemos de datos médicos completos en 26 pacientes. De ellas, el 34,6% presentan alteración del perfil lipídico, el 27,5% hipertensión arterial (HTA), y respecto al metabolismo de los hidratos de carbono, está alterado en 3 mujeres. A 6 pacientes se les ha realizado control ecocardiográfico en los últimos 5años, aunque únicamente a una paciente se le ha realizado resonancia magnética. El 62% continúan con THS. De las 6 pacientes en las que se ha realizado densitometría, se ha detectado un caso de osteopenia y otro de osteoporosis.
A nivel cognitivo precisaron ayuda durante la escolarización el 22% de las pacientes, y 2 de ellas padecen un retraso mental importante, siendo una de ellas portadora de mosaicismo 45,x/46,xrx. Hemos valorado el nivel socioemocional en 24 mujeres. La mayoría de ellas han alcanzado estudios de nivel medio-alto (80%). Respecto a la capacidad de relación, el 6% refieren tener escasas relaciones sociales y un 41% de las pacientes tienen o han tenido pareja en algún momento.
DiscusiónEn el ST, tanto la correlación entre el genotipo y el fenotipo como la edad del diagnóstico están sujetas a gran variabilidad. Cuando el diagnóstico se realiza por amniocentesis9 es difícil predecir el fenotipo del feto. Se ha demostrado que las mujeres portadores de la línea celular pura 45X tienen mayor riesgo de muerte fetal o mayor expresividad clínica del ST10. En este estudio se observa un aumento de casos diagnosticados mediante amniocentesis comparando las niñas con las adultas, hecho que refleja la mayor proporción de amniocentesis que se realiza actualmente en nuestro entorno. El cariotipo más frecuente en este estudio es un mosaicismo, a diferencia de lo publicado previamente por otros autores, que encuentran una mayor prevalencia de cariotipo 45X. Una posible explicación para esta diferencia puede ser que los embarazos con alteración 45X se interrumpan voluntariamente en nuestro medio, hecho que no podemos demostrar.
En la serie publicada por Savendahl y Davenport11 el 15% de los diagnósticos posnatales se realizó en la etapa neonatal y el 38% en la edad adulta, por lo que la edad media de 4,7años de diagnóstico en nuestro estudio es aceptable. Dado que el motivo de consulta más frecuente es la talla baja, remarcamos la importancia de incluir el cariotipo en el estudio de niñas con talla baja, de acuerdo con lo recomendado en la guía de práctica clínica del grupo de estudio del ST publicada por Bondy7 en 2007.
Está establecido que el tratamiento con GH es efectivo para aumentar la talla final12 de estas pacientes, pero existe controversia respecto a la dosis y la edad óptima de su inicio8,13. La talla media de esta serie es similar a la publicada en el estudio canadiense, que es el único estudio aleatorizado y controlado que mostró un beneficio medio de 6,5cm con el tratamiento con GH en estas pacientes12. En nuestro trabajo la dosis media se encuentra en el intervalo de la dosis recomendada7,9.
La talla media de nuestra serie es inferior a la de otros trabajos, como el estudio multicéntrico norteamericano realizado por Rosenfeld et al.14 en el que las pacientes tratadas solo con GH alcanzan una talla final media de 150,4±5,5cm, mientras que las que reciben GH y oxandrolona alcanzan 152,1±5,9 cm, con inicio precoz y duración prolongada del tratamiento.
En la guía clínica de Bondy7 se recomienda valorar dosis mayores de GH y asociar un esteroide anabólico no-aromatizable como la oxandrolona en pacientes con más de 9años y/o con talla baja extrema. En nuestro estudio, el subgrupo de pacientes en el que se asoció oxandrolona no alcanzó mayor talla final, hecho por el que se dejó de asociar esta terapia hace 15años.
En un estudio reciente, Menke et al.15 muestran que el tratamiento de GH combinado con oxandrolona a dosis bajas (0,03mg/kg/día) aumenta moderadamente la talla final alcanzada, con un perfil de seguridad aceptable, excluyendo una pequeña deceleración en el desarrollo mamario.
Solo el 20-30% de las mujeres con ST presentan pubertad espontánea9. Acorde con este dato, el 69% de las pacientes de nuestra serie tuvieron un hipogonadismo primario y recibieron terapia estrogénica. La disminución de la secreción de estradiol tanto en niñas como en adultas jóvenes puede ocasionar una menor densidad mineral ósea (DMO). La THS es un tratamiento crucial para evitar un rápido descenso de la DMO, para inducir el máximo pico de masa ósea en las jóvenes con ST sin desarrollo puberal espontáneo7 y para el mantenimiento posterior de una adecuada masa ósea8. En esta serie solo se ha realizado densitometría a 6 de las pacientes, detectándose un caso de osteopenia y otro de osteoporosis. Tres de las pacientes han tenido descendencia con óvulo de donante. El embarazo en las mujeres con ST es una situación de riesgo por aumento de la morbilidad cardiovascular, hecho del que deben de ser informadas adecuadamente16,17.
Una de las consecuencias más importantes de la haploinsuficiencia del cromosomaX son las anomalías cardiovasculares. La incidencia de lesiones cardiovasculares varía del 23 al 45% según las series18,19 y depende del tipo de exploración complementaria utilizada, siendo la angiorresonancia la prueba que más lesiones diagnostica20. Las anomalías más frecuentes son la válvula aórtica bicúspide (13-34%) y la coartación de aorta (4-14%). La prevalencia que hemos encontrado en este estudio ha sido inferior (13%). Matura y col. demostraron que las mujeres jóvenes con ST tienen un riesgo elevado de disección aórtica, y es preciso hacer un seguimiento estricto, con mediciones del tamaño aórtico indexado17,21. La prevalencia de HTA en nuestro trabajo ha sido del 27,5%, dato acorde con lo publicado en la bibliografía22,23.
La prevalencia de alteraciones ORL, renales y oftalmológicas en este grupo de pacientes es concordante con lo publicado en otras series7-9. Llama la atención la escasa prevalencia que hemos encontrado de patología autoinmune a pesar del cribado anual realizado en todas las niñas, menor que la descrita por otros24.
El control médico de las pacientes con ST en la edad adulta es heterogéneo y en muchas ocasiones insuficiente, dada la elevada morbilidad que presentan. Se recomienda que sean referidas a unidades especializadas en ST, y se ha de prestar particular atención a las mujeres con bajo nivel de educación y socioeconómico1. Devernay et al.1 objetivaron un seguimiento inadecuado en sus pacientes durante la fase de transición. En nuestro estudio la derivación de las pacientes a especialistas de adultos no se ha realizado de forma homogénea, hecho debido en parte al largo periodo de tiempo que abarca el estudio.
Las guías de práctica clínica más recientes del ST recomiendan un control y seguimiento multidisciplinar de estas pacientes realizando periódicamente: examen clínico (general, cardiaco y ORL), control de PA, estudio ecocardiográfico y/o angiorresonancia magnética cardiaca, audiometría, densitometría, control de THS y valoración psicosocial7. El especialista de endocrinología de adultos se ha propuesto como el más adecuado para integrar este seguimiento1, siendo así en el 37% de las pacientes de esta serie. Dado que a la mayoría de las mujeres adultas no se les han realizado las exploraciones recomendadas por los expertos25, es preciso mejorar la etapa de transición de estas pacientes y su seguimiento en la etapa adulta.
Estudios epidemiológicos revelan una tasa de mortalidad 3 veces superior en las pacientes con ST respecto a la población general en base a la prevalencia de HTA y de enfermedades cardiovasculares21,26,27. Coincidiendo con lo publicado, en esta serie han fallecido 2 mujeres en edad temprana.
Según algunos autores9,28,29, las mujeres con ST tienen dificultades en los ámbitos psicosocial y cognitivo: mayor riesgo de aislamiento social, inmadurez, trastornos relacionados con la ansiedad y dificultad para las relaciones en pareja, con retraso en el abandono del hogar paterno y con inicio sexual tardío. Sin embargo, la mayoría de las mujeres con ST adquieren un buen nivel de educación y encuentran empleos retribuidos, como en nuestra serie.
Con este estudio queremos destacar la necesidad de mejorar tanto la fase de transición como el seguimiento en pacientes adultas, ya que muchas de ellas siguen un control insuficiente, sobre todo en el aspecto cardiovascular, precisando la realización de resonancia magnética en todas las pacientes. Además, creemos que la alta tasa de pacientes que alcanzan estudios de nivel medio-alto puede ser un dato optimista a la hora de informar a las pacientes y a sus familias.
Las limitaciones del estudio son debidas fundamentalmente a su naturaleza retrospectiva, así como a la dispersión temporoespacial de las pacientes, con seguimiento en distintos centros sanitarios y a lo largo de 40años, lo que dificulta la recogida de datos y la homogeneidad de la muestra.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
