





239ALTERACIÓN DE LA RESPUESTA DE CORTISOL MATUTINO A LA ADMINISTRACIÓN DE ALPRAZOLAM EN PACIENTES CON DEPRESIÓN
J.C. Galofré Ferrater*, J. Schlatter**, E. Santos Mazo*, S. Santos Palacios*, M.J. Gil Calvo*** y J. Salvador Rodríguez*
*Endocrinología y Nutrición, **Psiquiatría, ***Bioquímica Clínica Universitaria de Navarra. Pamplona.
La administración aguda de benzodiacepinas en sujetos normales reduce la actividad del eje hipotálamo-hipofiso-adrenal (HHA) mediante la inhibición de la secreción de CRH. Los pacientes con depresión generalmente muestran una respuesta alterada en los mecanismos de retroalimentación que se manifiesta mediante una hiperactividad del eje HHA. Con objeto de estudiar el efecto de la hipersecreción del CRH endógeno, hemos administrado una dosis de alprazolam (APZ) en un grupo de pacientes con depresión (n = 24; edad media: 42,2, rango 18-66) que fueron divididos según la duración de la enfermedad (larga: > 12 meses, n = 12; corta: < 12 meses, n = 12) y en sujetos normales (n = 19; edad media: 28,6, rango: 21-56). Se obtuvieron muestras seriadas de cortisol plasmático (mcg/dL) a las 08:00 h y 24:00 h en situación basal (día 1), tras la administración de placebo (día 2) y a las 80:00 h del tercer día (día 3) tras la administración de APZ (150 mg p.o.) a las 24:00 h del día 2. Se tomaron muestras de cortisol libre urinario (CLU) durante el período diurno (8:00 h - 24:00 h) y nocturno (24:00 h - 8:00 h) a lo largo del período de estudio. Los controles y pacientes mostraron un inequívoco ritmo circadiano en los valores plasmáticos de cortisol basal y tras la administración de placebo. Los valores de los controles fueron: Día 1: 8:00 h: 23,1 ± 7,8; 24:00 h: 6,4 ± 3,2. Día 2: 8:00 h 21,9 ± 6,7; 24 h: 6,6 ± 4,0. Según lo esperado, APZ indujo una reducción significativa en los valores plasmáticos de cortisol matutino (día 3: 18,9 ± 7,7) con respecto a los valores tras la administración de placebo en los sujetos normales (p < 0,01). Los valores en los pacientes con depresión fueron: Día 1: 8:00 h: 17,1 ± 5,1; 24:00 h: 4,7 ± 3,0. Día 2: 8:00 h: 16,4 ± 5,4; 24 h: 4,8 ± 3,1. En el conjunto de estos pacientes no se observaron cambios en los valores de cortisol tras la administración de APZ: día 3: 16,5 ± 5,1 (p = 0,9). El patrón de cortisol plasmático en los deprimidos de corta evolución fue idéntico a los de larga data. No se observaron cambios en la media de los valores de CLU (mcg/24h) nocturno tras la administración de APZ ni en los pacientes (18,2 vs. 19,3) ni en los controles (17,6 vs. 15,5). Se concluye que los pacientes con depresión muestran una disminución del efecto inhibitorio de APZ sobre los niveles plasmáticos del cortisol matutino, lo que sugiere una hiperactividad de CRH endógeno. Esta respuesta neuroendocrina específica, que podría utilizarse como marcador evolutivo de la enfermedad, no parece estar influida por la duración de la depresión.
240DIFERENCIAS EN LA COMPOSICIÓN DE LA DIETA EN MUJERES OBESAS CON O SIN SÍNDROME DE OVARIO POLIQUÍSTICO (SOP)
F. Álvarez Blasco, J.I. Botella Carretero, M. Luque Ramírez, G. García Romero de Tejada, J. Sancho Rof y H.F. Escobar Morreale
Endocrinología Hospital Ramón y Cajal. Madrid.
Introducción: La obesidad de distribución abdominal es un hallazgo frecuente en las pacientes con SOP. Las dietas ricas en grasas saturadas y pobres en fibra han sido relacionadas con la obesidad abdominal.
Objetivo: Valorar la existencia de diferencias en la ingesta alimentaria en pacientes obesas con o sin SOP.
Material y métodos: 49 mujeres obesas en edad fértil referidas a nuestro Hospital de manera consecutiva para pérdida de peso fueron evaluadas. El diagnóstico de SOP fue establecido por la demostración de oligoovulación e hiperandrogenismo clínico y/o bioquímico. El estudio incluía medidas antropométricas y determinación de niveles circulantes de andrógenos, SHBG y perfil lipídico. El hirsutismo fue valorado mediante la escala de Ferriman-Gallwey modificada, la resistencia insulínica por HOMA, y la grasa total (kg) y relativa (%) con un impedanciómetro manual. Todas las participantes completaron el cuestionario de frecuencia alimentaria CFA.1 Los datos se expresan como media ± DE y fueron analizados con la prueba t para muestras independientes o U de Mann Whitney.
Resultados: 16 pacientes de las evaluadas fueron diagnosticadas de SOP. No se encontraron diferencias entre las mujeres con o sin SOP en IMC (35,7 ± 8,4 vs. 35,1 ± 6,1 kg/m2), ICC (0,84 ± 0,1 vs. 0,83 ± 0,1), grasa corporal total (36 ± 9 vs. 35 ± 10 Kg) o relativa (38 ± 4 vs. 39 ± 5%). Las pacientes con SOP presentaron cifras elevadas de testosterona total y libre (59,3 ± 19,6 vs. 38,4 ± 14,5 ng/dL, p < 0,0001 y 39,8 ± 12,9 vs. 23 ± 8,4 ng/dL, p < 0,0001 respectivamente), DHEA-S (2055 ± 692 vs. 1494 ± 628 ng/mL, p < 0,008), androstendiona (4,1 ± 1,9 vs. 2,5 ± 1,3 mg/mL, p < 0,002) e hirsutismo (5,2 ± 4 vs. 1,7 ± 1,9, p < 0,011). No se encontraron diferencias en HOMA, SHBG y perfil lipídico. El consumo energético fue similar en las mujeres con o sin SOP (2067 ± 1051 vs. 2283 ± 1003 Kcal/d, respectivamente). No encontramos diferencias en la ingesta de proteínas (96 ± 50 vs. 104 ± 42 g/d), carbohidratos (239 ± 133 vs. 263 ± 124 g/d), grasa total (83,9 ± 39,2 vs. 94,4 ± 47,1 g/d), ácidos grasos saturados (24,7 ± 11,6 vs. 29,6 ± 16,5 g/d), ácidos grasos monoinsaturados (38,9 ± 20,2 vs. 43,2 ± 23,9 g/d), ácidos grasos poliinsaturados (16,6 ± 6,2 vs. 14 ± 6,8 g/d), colesterol (310 ± 145 vs. 353 ± 165 mg/d), fibra (19,4 ± 11,5 vs. 21,3 ± 9,5 g/d) o alcohol (1,6 ± 2,6 vs. 1,5 ± 3,2 g/d).
Conclusión: En nuestro estudio no encontramos diferencias en la ingesta alimentaria en mujeres obesas con o sin SOP.
Ref: 1. Vioque J, González L. Eur Cancer Prev 1991;1 (supl 1):19-20
Financiación: FIS 02/0741 & RGDM 03/212, Beca FPI CAM 01/0281/03
241DIFERENCIAS ENTRE FEOCROMOCITOMAS ESPORÁDICOS Y FAMILIARES
E. Hervás Abad*, C. Páramo Fernandez*, P. Álvarez-Vázquez*, A. Casteràs Román*, M. Samprón**, R. Luna Cano* y R.V. García-Mayor*
*Endocrinología, **Nefrología C H Universitario Xeral-Cíes. Vigo.
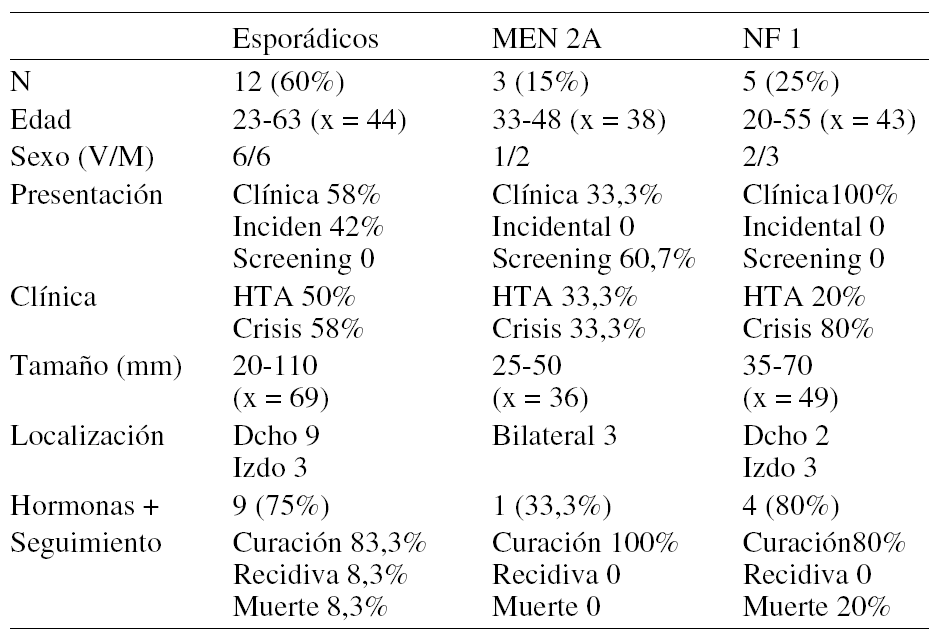
Introducción: Los feocromocitomas (FC) se clasifican en esporádicos (unilaterales, sin historia familiar, y sin endocrinopatía asociada) o familiares (bilaterales, heredados y con endocrinopatía asociada a MEN 2 A, 2B, VHL y NF 1).El conocimiento de los genes responsables de estos síndromes ha hecho posible el diagnóstico precoz cambiando su espectro clínico. Revisamos los 20 FC intervenidos entre EN-1980 y DIC-2004, 12 esporádicos y 8 en el contexto de síndromes familiares (3 MEN 2 A y 5 NF 1).
Resultados:

Conclusiones:1. Los FC esporádicos se diagnosticaron frecuentemente de forma incidental a pesar de presentar mayor agresividad que los FC asociados a MEN 2 A. 2. La detección precoz de MEN 2 A mediante screening genético diagnosticó formas incipientes y con menor significancia clínica. 3. Los FC asociados a NF 1 presentan un comportamiento tanto clínico como neoplásico agresivo. La incidencia en nuestra serie es mayor de la descrita en otras series.
242EFECTOS DEL TRATAMIENTO SUSTITUTIVO CON TESTOSTERONA SOBRE FACTORES DE RIESGO CARDIOVASCULAR NO CLÁSICOS EN PACIENTES ANDROPÁUSICOS CON DIABETES MELLITUS TIPO 2 (DM2)
R.M. Burgo López1, B. González Berrocal2, M. Almeida3, I. Alberca4, J.M. Miralles García1, J.M. González Buitrago2, A. Orfao3 y J.J. Corrales Hernández1
1Endocrinología, 2Bioquímica, 3Citometría de flujo, 4Hematología Hospital Universitario. Salamanca.
Los varones ancianos con DM2 constituyen un subgrupo de población con déficit parcial androgénico. En sujetos no diabéticos dicho déficit se asocia con un incremento en la prevalencia de factores de riesgo cardiovascular clásicos y en varones diabéticos se ha demostrado una correlación inversa entre concentraciones séricas de testosterona y la severidad de la enfermedad aterosclerótica carotídea. Sin embargo, se desconocen en la actualidad los efectos de la terapia sustitutiva con testosterona sobre los factores no clásicos de riesgo cardiovascular. Nosotros realizamos un estudio seriado prospectivo de los efectos de tratamiento androgénico sustitutivo y de su retirada sobre los siguientes factores no clásicos de riesgo cardiovascular: proteína C reactiva (PCR), lipoproteína a (Lp (a)), ferritina, homocisteína y fibrinógeno, en pacientes con DM2 de 2 a 22 años de evolución. El tratamiento consistía en 150 mg de enantato de testosterona (vía IM cada 15 días), aplicado a 13 pacientes que cumplían criterios clínicos y bioquímicos de déficit androgénico parcial y no tenían contraindicaciones para el tratamiento androgénico. Estos varones tenían una edad de 63,9 ± 5,5 años y un IMC de 27,3 ± 3,8 kg/m2. Ninguno de ellos había presentado enfermedad cardiovascular clínica. Los pacientes eran estudiados antes del tratamiento, durante el mismo (en los meses 1, 6 y 12) y 3 meses después de su retirada. Los resultados obtenidos fueron (media ± DE):

Durante este período de tratamiento los pacientes no experimentaron cambios ponderales ni se produjeron efectos adversos cardiovasculares ni prostáticos. No se detectaron cambios significativos en los valores de antígeno prostático específico. El tratamiento con testosterona indujo un incremento significativo en los niveles de testosterona total y libre y un descenso también significativo de las concentraciones de LH. Concluimos que el tratamiento sustitutivo con testosterona en pacientes andropáusicos con DM2, bajo las condiciones del estudio, es seguro. Ni dicho tratamiento ni su retirada inducen efectos adversos sobre los factores no clásicos de riesgo cardiovascular.
243EL GROSOR INTIMA-MEDIA CAROTÍDEO (CIMT) Y LA VASODILATACIÓN DEPENDIENTE DE ENDOTELIO (FMD) CORRELACIONAN DE FORMA INDEPENDIENTE CON LOS ANDRÓGENOS Y LA RESISTENCIA A LA INSULINA, EN MUJERES CON HIPERANDROGENISMO
M. Luque Ramírez*, C. Mendieta Azcona**, F. Álvarez Blasco*, J.I. Botella Carretero*, G. García Romero de Tejada* y H.F. Escobar Morreale*
*Endocrinología y Nutrición, **Cirugía Vascular Hospital Ramón y Cajal. Madrid.
Introducción: La insulinorresistencia es un hallazgo frecuente tanto en mujeres con síndrome del ovario poliquístico (SOP) como con hiperandrogenismo funcional. Estudios previos han demostrado aumento del CIMT y alteración de la FMD en estados de insulinorresistencia. En este trabajo piloto, hemos estudiado la contribución de la resistencia a la insulina y la hiperandrogenemia al CIMT y alteración de la FMD en mujeres hiperandrogénicas.
Pacientes y métodos: Se estudiaron 18 mujeres con hiperandrogenismo clínico y/o bioquímico; edad (media ± desviación estándar): 24 ± 7 años; IMC: 30 ± 6 kg/m2. Trece de ellas presentaban anovulación crónica, cumpliendo todos los criterios del NICHD para SOP. Se evaluaron una serie de variables clínicas y bioquímicas que incluyeron el índice cintura/cadera: 0,82 ± 0,10; colesterol total: 161 ± 30 mg/dL; LDL-colesterol: 45 ± 22 mg/dL; recuento leucocitario: 7133 ± 2345/mm3; testosterona total (TT): 61 ± 14 ng/dL; testosterona libre calculada (FT): 45 ± 12 pmol/L; leptina: 45 ± 22 ng/mL; presión arterial sistólica (SBP), diastólica (DBP) y media de 24 horas (MBP), mediante M.A.P.A.: 120 ± 10, 72 ± 7 y 87 ± 8 mmHg, respectivamente. Se estimó el índice de sensibilidad insulínica (ISI) a partir de una sobrecarga oral de glucosa estándar. El CIMT y FMD se determinaron mediante ecografía doppler. Se realizó un análisis de correlación de Pearson, después de confirmar la normalidad de las variables mediante la prueba de Kolmogorov-Smirnov.
Resultados: CIMT correlacionó con la TT (r = 0,43, P = 0,038), FT (r = 0,42, P = 0,042) y androstendiona (r = 0,59, P = 0,005), pero no con el ISI ni con la glucosa o la insulina en ayunas. FMD correlacionó inversamente con la glucosa basal (r = - 053, P = 0,013), SBP (r = - 0,46, P = 0,030), DBP (r = - 0,50, P = 0,021) y MBP (r = - 0,54, P = 0,012), y de forma directa con el ISI (r = 0,41, P = 0,045) y HDL-colesterol (r = 0,65, P = 0,002). No se observó correlación entre el FMD ni los andrógenos séricos.
Conclusiones: Tanto el hiperandrogenismo como la resistencia a la insulina contribuyen a la disfunción cardiovascular de las mujeres hiperandrogénicas, incluidas aquellas con criterios de SOP, a través de mecanismos independientes. Si nuestros datos se confirman en una serie más amplia de pacientes, indicarían la necesidad de un tratamiento combinado en estas mujeres encaminado tanto hacia la mejora de la sensibilidad hacia la insulina, como también hacia la disminución del hiperandrogenismo.
Financiación: FIS 02/0741 & RGDM 03/212
244EVOLUCIÓN RADIOLÓGICA DE ADRENALITIS TUBERCULOSA
P. Suárez Llanos*, P. Bacarizo*, A. Jara Albarrán* y M.R. Checa**
*Endocrinología, **Estudiante de 4º de Medicina, HGU Gregorio Marañón. Madrid.
Introducción: Actualmente la insuficiencia suprarrenal 1º es una enfermedad muy poco frecuente, de origen autoinmunne en el 80-90% de los casos. La adrenalitis tuberculosa es todavía la causa más frecuente en países en desarrollo, siendo al diagnóstico procesos de larga evolución, radiológicamente caracterizados por atrofia o disminución glandular y presencia de calcificaciones. Está descrita la presencia de aumento glandular, típicamente bilateral, en fases agudas o subagudas de fallo adrenal secundario a tuberculosis, generando problemas diagnósticos. Se presentan dos casos con dicha evolución.
Casos clínicos:Caso 1: Varón de 77 años diagnosticado de crisis adrenal con hiperpigmentación característica, hipotensión severa y síndrome constitucional de 2 años de evolución. Datos de laboratorio: pH 7.24, bicarbonato 5,9 mmol/l, Na+ 126 mmol/l, K+ 8,1 mmol/l, cortisol plasmático 2,55 µg/dl (N 5-25) y tras Synacthen 1,9 µg/dl. ACTH 468 pg/ml, DHEA-S < 15 µg/dl (120-420), 4-Androstendiona (4-A) 0,3 ng/ml (N 0,3-3,1). Test Mantoux positivo, BAAR en esputo negativo. Ac antiadrenales negativos. Caso 2: Varón de 65 años que debuta con cuadro similar sin hiperpigmentación. En la analítica destaca: Na+ 128 mmol/l, K+ 3,7 mmol/l, CLU 13,0 µg/24 horas (N 20-90), cortisol plasmático 4,4, tras estímulo 5,4 µg/dl. ACTH de 446 pg/ml, DHEA-S < 30 µg/dl, 4-A < 0,1 ng/ml. Mantoux de 30 mm, Ac antiadrenales negativos en 3 ocasiones. El estudio inicial con TC mostró aumento bilateral de ambas glándulas suprarrenales, con morfología nodular en el 1º caso y difusa en el 2º. Tras 2 años de seguimiento la evolución radiológica mostró una franca involución de las glándulas adrenales, con calcificaciones puntiformes en el caso 1. Con tratamiento sustitutivo, ambos pacientes se mantuvieron asintomáticos y con buen control analítico.
Conclusión: El diagnóstico a posteriori de la etiología tuberculosa se ha basado en el estudio específico, signos radiológicos y negatividad de Ac antiadrenales, así como en la evolución hacia la disminución o atrofia glándular. Las calcificaciones, sin ser específicas de tuberculosis, sí son altamente sugerentes y descartan el origen autoinmune.
245FEOCROMOCITOMAS FAMILIARES ASOCIADOS A MUTACIONES EN LA SUCCINATO DESHIDROGENASA
C. Fuentes Gómez, M.J. Goñi Iriarte, J.P. Martínez de Esteban, E. Menéndez Torre, J. Basterra Gortari, E. Anda Apiñániz, R.M. Rodríguez Erdozáin y M. García Mouriz
Endocrinología Hospital de Navarra. Pamplona.
Introducción: La prevalencia de los feocromocitomas familiares es del 13-50% de todos los feocromocitomas. Recientemente se han encontrado mutaciones en el gen de la enzima succinato deshidrogenasa (complejo II de la cadena respiratoria mitocondrial) asociados también a feocromocitomas y paragangliomas esporádicos o familiares. Se han identificado mutaciones en las subunidades B, C y D de la SDH que dan lugar a los síndromes asociados a paragangliomas tipo 4 (PGL-4), tipo 3 (PGL-3) y tipo 1 (PGL-1) respectivamente
Material y métodos: Presentamos tres familias con feocromocitomas familiares asociados a mutaciones en SDH. Familia 1: Varón diagnosticado de paraganglioma paraaórtico con metástasis costales. Su hija es diagnosticada de paraganglioma abdominal paraaórtico. Familia 2: Mujer intervenida de paraganglioma de glomus carotídeo. Su sobrino es diagnosticado e intervenido posteriormente de un paraganglioma del X par craneal. Familia 3: Mujer con paraganglioma de glomus carotídeo. Su padre fue diagnosticado anteriormente de metástasis ósea de paraganglioma.
Resultados: Se realiza estudio genético de las familias encontrándose, en la familia 1, mutación en SDH B, c. 558-3 C/G en exon 5 en el caso índice y en sus dos hijas. Familia 2, mutación para la SDH C, exon 1, en la paciente índice, uno de sus dos hijos, dos de sus hermanos y en su sobrino. La familia 3 está pendiente de estudio genético
Conclusiones: El porcentaje de feocromocitomas familiares ha aumentado encontrándose mutaciones en la succinato deshidrogenasa asociadas a la aparición de estos tumores, lo que plantea el estudio genético de los feocromocitomas aparentemente esporádicos. Las mutaciones en SDH D (PGL-1) están relacionadas más frecuentemente con paragangliomas del cuello. La SDH B (PGL-4) aparece principalmente en tumores abdominales y se comportan de forma más agresiva. La SDH C (PGL-3) está asociada a paragangliomas no funcionantes de cabeza y cuello.
246HIPERPLASIA SUPRARRENAL CONGÉNITA, FORMA NO CLÁSICA, EN LA EDAD PEDIÁTRICA: PRESENTACIÓN CLÍNICA Y VALORACIÓN DEL CRECIMIENTO
C. Gutiérrez, M.D. Ollero, M. López-Capapé, N. Dedieu, R. Barrio y M. Alonso
Unidad de Endocrinología y Diabetes Pediátrica Hospital Universitario Ramón y Cajal. Madrid.
Introducción: El déficit de 21-hidroxilasa (21-OH) es la causa más frecuente de hiperplasia suprarrenal congénita (HSC). En las formas tardías, la expresión clínica del cuadro es variable y con frecuencia se pone de manifiesto en la edad pediátrica. La talla adulta en estos pacientes se sitúa alrededor de 1 desviación estándar (DE) y puede no alcanzar la talla genética.
Pacientes y métodos: Análisis retrospectivo de 18 pacientes pediátricos diagnosticados de HSC, forma no clásica, por déficit de 21OH. El diagnóstico se estableció mediante determinaciones de 17OH progesterona basal (17OHPb) y tras estímulo con ACTH, y se confirmó posteriormente con la realización de estudio genético, excepto en 4 pacientes. En 9 pacientes se analizó la talla final alcanzada respecto a la población general y su talla genética (tablas de Hernández). Tres de estos pacientes no habían recibido tratamiento, mientras que los 6 restantes sí se trataron.
Resultados: Las características de los pacientes al diagnóstico se resumen en la tabla 1. En la tabla 2 se analizan los datos de los 9 pacientes que completaron crecimiento según recibieron o no tratamiento.


Conclusiones: La mutación Val281Leu es la más frecuente. La pubarquia es el motivo de consulta más habitual. Al diagnóstico todos los pacientes presentaban avance madurativo óseo. Los pacientes que fueron tratados alcanzaron una talla final mejor que los pacientes que no recibieron tratamiento.
247IMPORTANCIA DEL SEGUIMIENTO MULTIDICIPLINAR DEL SÍNDROME DE TURNER EN LA EDAD ADULTA
M. García Gómez* y C. Luzuriaga Tomás**
*Endocrinología y Nutrición Hospital Universitario Marqués de Valdecilla. Santander, **Endocrinología Pediátrica Hospital Universitario Marqués de Valdecilla. Univesidad de Cantabria. Santander.
Introducción: El síndrome de Turner (ST) se caracteriza por una anomalía numérica o estructural del cromosoma X, afectando a 1/2500 niñas nacidas vivas.Cursa con talla baja, disgenesia gonadal y aumento de morbimortalidad, principalmente por enfermedad endocrina y cardiovascular.
Objetivo: Evaluar el tipo de atención médica en un grupo de pacientes adultas diagnosticadas en edad pediátrica, sus características clínicas y patología.
Material y métodos: Revisión retrospectiva de las pacientes adultas diagnosticadas de ST en nuestro hospital a partir de 1982. Evaluación auxológica, clínica, analítica y del metabolismo hidrocarbonado, con análisis del área bajo la curva tras sobrecarga oral de glucosa, 3h, para glucosa (ABC-G) e insulina (ABC-I), antes y durante el tratamiento con hormona de crecimiento (GH).
Resultados: 19 pacientes de 24,5 ± 4,2 años, el cariotipo más frecuente (45%) fue el 45 XO. Diagnosticadas a los 7,57 ± 3,57 años, tratadas con GH; SDS talla al inicio 2,91 ± 0,68, talla final 150,1 ± 4,6 cm, SDS 1,94 ± 0,9 (p < 0,05). Pubertad espontánea (31,5%), con menarquia a los 13,5 ± 2,06 años. Pubertad inducida a los 14,2 ± 1,2 años, dosis etinil-estradiol inicial 68 ± 44 ng/kg. Hipotiroidismo subclinico en el 21,5% de los pacientes, anticuerpos antitiroideos en 15,7%. Osteoporosis en 10,5%; osteopenia en 5,2%. No diabetes mellitus, 5,2% presentaron intolerancia a la glucosa. El ABC-G pre GH vs post GH, p > 0,05 (20340 ± 3477,2 vs 20268,75 ± 3730,8), el ABC-I pre GH vs post GH, p < 0,005 (6787,6 ± 3735,7 vs 11622 ± 7385). El 21% presentaba hipercolesterolemia. Retraso psicomotor en 10,5%. Sordera neurosensorial 10,5%. Deformidades óseas (cubitus valgus, clinodactilia, campodactilia, pies planos) en 26,3%.Riñón en herradura 10,5%. Un 42,1% mostraba fenotipo severo. Sólo tres pacientes, (15,7%), eran seguidas por endocrinología de adultos y 6 pacientes, (31,5%), por ginecología.
Conclusiones: En nuesta serie es poco frecuente la alteración del metabolismo de hidratos de carbono, demostrándose un incremento de secreción de insulina durante el tratamiento con GH, que corrige la hiperglucemia. Aparte de la talla baja (motivo principal de consulta) y la disgenesia gonadal, existe una elevada morbilidad endocrinológica, asociándose otras patologías que precisan atención especializada, y no son seguidas por un equipo multidisciplinar. Creemos que debe ser el endocrinólogo, el médico de referencia de este equipo y el responsable de estas pacientes en la edad adulta, para mejorar así su calidad y expectativa de vida.
248PARAGANGLIOMAS: TAMBIÉN TUMORES NEUROENDOCRINOS
E. Hervás Abad*, C. Páramo Fernandez*, A. Casteràs Román*, P. Álvarez-Vázquez*, A. Argibay**, R. Luna Cano* y R.V. García-Mayor*
*Endocrinología, **Medicina Interna C H Universitario Xeral-Cíes. Vigo.
Introducción: Los paragangliomas (PG) son tumores neuroendocrinos derivados del tejido cromafín extra-adrenal. El síndrome PG tipo 1 (PGL-1) incluye los PG localizados en cabeza y cuello, frecuentemente multifocales y afuncionantes, y el tipo 4 (PGL-4) los infracervicales, con producción hormonal, mayor posibilidad de malignidad y de asociar otras neoplasias como carcinoma papilar de tiroides o renal. La mutación responsable del PGL-4 se encuentra en el gen de la SDH subunidad B y tiene baja penetrancia mientras la mutación de la SDH subunidad D, que causa el PGL-1, tiene alta penetrancia.
Objetivos: Analizamos las características de los PG diagnosticados en nuestro centro en cuanto a presentación, manifestaciones clínicas, estudio bioquímico, anatomopatológico y evolución.
Pacientes: 12 PG fueron intervenidos entre 1980 y 2003 en nuestro centro, 9 mujeres y 3 hombres con edad media de 44 años (17 y 74 años).
Resultados:1) Presentación: efecto masa en 7 pacientes (58%), crisis HTA en 3 pacientes (25%) e incidental en 1 paciente. 2) Manifestaciones clínicas: el 75% presentaban clínica adrenérgica (HTA mantenida o crisis adrenérgicas) y el 58% sintomatología compresiva local. 3) Tamaño: entre 7 y 110 mm (media 52 mm). 4) Localización: 25% en cabeza y cuello (glomus carotídeos y glomus timpánico) y 75% por debajo del cuello (paravertebral dorsal y lumbar, yuxtaadrenales y aortico-simpático). 5) Estudio hormonal: + en los 4 pacientes a los que se les realizó. 6) Inmunohistoquímica: cromogranina A + en la totalidad de los pacientes a los que se les realizó. 7) Evolución: en 8 pacientes fue favorable, 2 pacientes murieron a causa de la extensión del tumor y 1 persiste parcialmente debido a se localización intracraneal.
Conclusiones:1) Los PG se asociaron frecuentemente a clínica adrenérgica y producción catecolamínica a pesar de lo cual en un alto porcentaje ni fueron remitidos ni evaluados por un servicio de Endocrinología ya que no fueron considerados "tumores neuroendocrinos" (sobre todo aquellos que se diagnosticaron por efecto compresivo). 2) En nuestra serie los PG localizados por debajo del cuello son tumores más productores, malignos y asociados a otras neoplasias que los localizados en cabeza y cuello. 3) Creemos que el estudio genético de la mutación de la SDH subunidad B y D de todos los PG, y de sus familias en caso de ser portadores, es indispensables en este tipo de tumores neurosecretores.
249SIGNIFICACIÓN CLÍNICA DE ALTERACIONES EN EL GEN CYP21B EN LA POBLACIÓN PEDIÁTRICA GALLEGA
P. Cabanas Rodríguez*, L. Loidi F. de Troconiz**, J. Barreiro Conde*, L. Castro Feijoó*, T. Arévalo Saade*, M. López Franco*, F. Vasconcelos Espada*, C. Quinteiro García**, F. Domínguez Puente** y M. Pombo Arias*
*Unidad de Endocrinología, Crecimiento y Adolescencia. Dto. Pediatría. Hospital Clínico Universitario de Santiago de Compostela. Universidad de Santiago de Compostela. Santiago de Compostela, **Unidad de Medicina Molecular. Fundación Pública Galega de Medicina Xenómica. Hospital Clínico Universitario de Santiago de Compostela. Santiago de Compostela.
Introducción: La hiperplasia suprarrenal congénita (HCS) es un trastorno de la estereidogénesis suprarrenal, de herencia AR y penetrancia variable. El déficit enzimático de la 21-hidroxilasa es el más frecuente. En los últimos años el estudio del gen implicado (CYP21B) y la relación genotipo-fenotipo han despertado gran interés.
Objetivos: Estudiar las alteraciones en el gen CYP21B en los pacientes diagnosticados de hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa, valorar sus características en nuestra población y su significación clínica.
Metodología: Se analizaron los datos clínicos, hormonales y el genotipo específico en cada caso. El estudio genético se realizó mediante secuenciación del gen CYP21B tras amplificación por PCR y Southern Blot.
Resultados: Se estudiaron 55 pacientes, 44 (80%) mujeres. Presentaron la forma clásica pierde sal 5 (9,09%). Los motivos de consulta más frecuentes fueron: pubarquia (49,1%), virilización (18,2%) e hirsutismo (10,9%). La edad ósea al diagnóstico estaba adelantada 1,4 años respecto a la cronológica. El 45% presentaron una pubertad adelantada. Se detectaron 112 mutaciones en los alelos estudiados: 67 V281L; 14 i2G; 9 Q318X; 4 del 8 pb; 3 P453S; 3 I172N; 3 F306+T; 3 R356W; 2 R444X; 1 P30L;1 P45+c; 1 P464L y 1 D322G.
Conclusiones: Se observa una menor incidencia en varones, en probable relación con una menor preocupación por los signos de virilización; sin embargo, es importante la detección precoz. La mutación más prevalente en nuestra población ha sido V281L, en concordancia con otros estudios. La primera vez que se han descrito las mutaciones R444X, D322G, P464L y P45+c ha sido en nuestra población; de ellas la R444X, es una mutación recurrente en la población gallega. En conclusión, la predicción del fenotipo por genotipo puede apoyar la actitud terapéutica, aunque siempre considerando la clínica y la genética en conjunto.
250SÍNDROME DE CUSHING POR HIPERPLASIA ADRENAL MACRONODULAR ACTH INDEPENDIENTE, CON INCOMPLETA SUPRESIÓN, Y DE PRESENTACIÓN UNILATERAL
S. Maraver Selfa*, A. Gentil Baldrich*, C. Morales Portillo*, C. Campos Martin*, A. Fernández-Arguelles García*, M. Diaz**, M. Diaz Galvez*, J. Villar** y E. Herrera Justiniano*
*Endocrinología y Nutrición, **Anatomía Patológica, HU Virgen Macarena. Sevilla.
Introducción: La Hiperplasia adrenal macronodular ACTH independiente es una forma muy infrecuente de síndrome de Cushing. Afecta a ambas glándulas adrenales y su tratamiento es la cirugía bilateral. Recientemente se han descrito expresión anómalas de receptores en la corteza adrenal, como causa de la enfermedad.
Material y método: Paciente de 48 años que consultó por afectación del estado general, ganancia de peso y abotargamiento facial. La exploración demostró BMI de 26,4 Kg/m2, rubeosis facial y tensión arterial de 140/100. Se valoró cortisol plasmático basal y tras dexametasona, 1 y 8 mg, cortisol libre en orina de 24 h, ACTH, y perfil secretorio de cortisol en 24h, con determinaciones cada 20 minutos con bomba de extracción continua, así como estudio de imagen (RNM y gammagrafía suprarrenal).
Resultados: Cortisol plasmático basal (640 nmol/l) fue normal o en valores ligeramente altos, pero con ausencia de ritmo (675 nmol/l) y con incrementos postprandiales (945, 942, 900, 1032 nmol/l), independientemente de los niveles muy bajos de ACTH (5, 6,2, 9,8 y 16 pg/ml) y no frenando con 1 mg (308 nmol/l), pero sí con 8 mg (50 nmol/l) de dexametasona. La RNM mostró lesión focal de 3 cm en adrenal izquierda con hipercaptación en la gammagrafía, siendo normal y con bloqueo isotópico la glándula contralateral. Se realizó adrenalectomía unilateral objetivándose glándula de 26 gr con múltiples nódulos, el mayor de 4,3 cm y otro de 1,4 cm o menores (0,4-0,7 cm); los nódulos más pequeños tienen límites poco precisos y se continúan con una corteza hiperplásica. El estudio complementario a los ocho meses demostró remisión clínica y funcional.
Conclusiones: La hiperplasia adrenal macronodular ACTH independiente puede presentarse de forma asíncrona y con exploración funcional con ausencia de ritmo y supresión incompleta del eje HPA, por síndrome de Cushing intermitente secundario a la ingesta.
251SÍNDROME DE OVARIO POLIQUÍSTICO Y FACTORES DE RIESGO CARDIOVASCULAR
J. Escalada San Martín, M.A. Vicente Vicente, L. Irigoyen Cucalón y R. Ezquerra Larreina
Endocrinología y Nutrición Hospital Santiago Apóstol. Vitoria.
El síndrome del ovario poliquístico (SOP) es la endocrinopatía más frecuente de la mujer en edad reproductiva. En los últimos años diversos estudios han demostrado que dichas pacientes constituyen un grupo de alto riesgo de síndrome metabólico.
Objetivo: Analizar los factores de riesgo cardiovascular asociados al SOP.
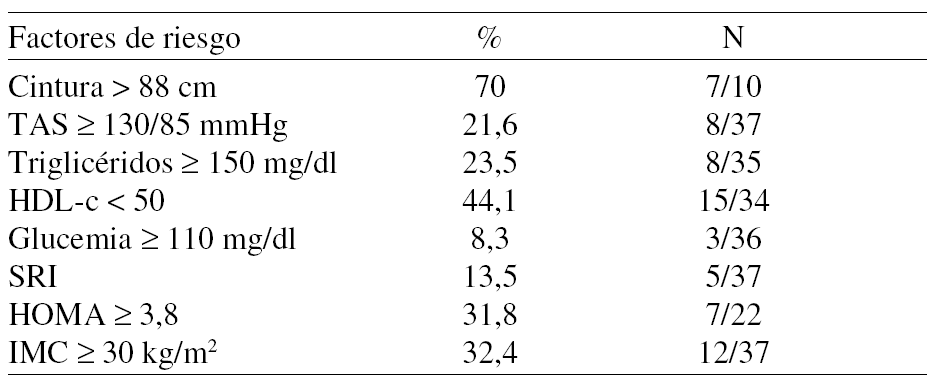
Material y métodos: Estudio retrospectivo de recogida de datos clínicos (índice de masa muscular, tensión arterial, diámetro de cintura) y analíticos (glucemia basal, insulinemia basal, perfil lipídico) de pacientes diagnosticadas de SOP (criterios NIH 1990) en nuestras consultas de Endocrinología entre los años 1998 y 2004. Se ha analizado la prevalencia de factores de riesgo cardiovascular, de síndrome de resistencia a la insulina (SRI si ≥ 3 criterios del ATP III) y HOMA patológico (≥3,8).
Resultados: Se han analizado datos de 37 pacientes con una edad de 23 ± 5,4 años, con los siguientes hallazgos.

Los 3 pacientes con glucemia ≥ 110 mg/dl fueron sometidos a sobrecarga oral de glucosa (75 gr) encontrando una alteración de la glucemia basal, una intolerancia a hidratos de carbono y una dibates mellitus 2. Además, 5 de los 37 pacientes (13,5%) tenían antecedentes de diabetes tipo 2 en familiares de primer grado.
Conclusiones: En nuestra serie, las pacientes con SOP presentan una elevada prevalencia de factores de riesgo cardiovascular y de resistencia insulínica, especialmente teniendo en cuenta la edad de las pacientes y el bajo porcentaje de obesidad de nuestra serie. Este hallazgo es un dato más para incidir en la modificación de dichos factores de riesgo mediante medidas higiénico-dietéticas y/o farmacológicas.
252
TRATAMIENTO HORMONAL PARA REASIGNACIÓN DE SEXO EN 402 TRANSEXUALES DURANTE 24 MESES: MODIFICACIONES ANTROPOMÉTRICAS Y METABÓLICAS
E. Dominguez*, J.F. Martín Lázaro** y M.J. Lucio Pérez***
*Servicio Central de Diabetes y Embarazo Instituto Nacional de Endocrinología y Hospital Ginecobstétrico Ramón González Coro. La Habana, **Unidad de Lípidos. Servicio de Bioquímica. Hospital Ramón y Cajal. Madrid, ***Servicio de Endocrinología. Hospital Ramón y Cajal. Madrid. Univ Alcalá.
La transexualidad es un fenómeno muy antiguo, presente en muchas culturas, pero en las últimas décadas, la demanda para la reasignación de sexo en transexuales se ha incrementado considerablemente, como lo demuestran diferentes estudios de prevalencia e incidencia. El tratamiento hormonal representa un importante papel en este proceso, que debe eliminar, idealmente, los caracteres sexuales secundarios del sexo original e inducir los del sexo opuesto tan rápido y completa como sea posible. Para analizar las modificaciones antropométricas y analíticas producidas por el tratamiento hormonal durante 24 meses estudiamos a 402 transexuales, 223 de hombre a mujer (H-a-M) y 179 de mujer a hombre (M-a-H), entre 19 y 56 años. El diagnóstico de transexualidad fue realizado por un psicólogo o psiquiatra. Después de evaluación clínica, antropométrica, bioquímica y hormonal eran seleccionados para tratamiento hormonal esteroideo y revisados cada 3-6 meses. Después de 24 meses, y antes de la cirugía de reasignación de sexo, la administración de estrógenos combinados con acetato de ciproterona, un antiandrógeno con actividad progestágena, en H-a-M produjo frente al inicio un significativo incremento en el índice de masa corporal (IMC) (26,2 ± 1,8 frente a 23,1 ± 2,4 kg/m2; p = 0,001), en el porcentaje de masa grasa (%MG) (bioimpedanciometría) (20,1 ± 8,9 frente a 15,1 ± 6,3%; p = 0,002), en la circunferencia de la cintura (CCi) (82,3 ± 5,1 frente a 73,9 ± 3,7 cm; p = 0,021) y en la circunferencia de la cadera (CCa) (98,5 ± 3,4 frente a 91,5 ± 6,0 cm; p = 0,004); lo mismo ocurrió en los valores plasmáticos de colesterol total (CT) (163,6 ± 35,3 frente a 160,5 ± 15,9 mg/dl; p = 0,033), de triglicéridos (TG) (67,8 ± 19,1 frente a 63,5 ± 27,8 mg/dl; p = 0,031) y de HDL-colesterol (HDL-c) (54,7 ± 12,1 frente a 51,2 ± 8,9 mg/dl; p = 0,033), sin cambios en los valores de glucosa ni creatinina. Por su parte, el tratamiento con andrógenos durante 24 meses en M-a-H produjo un incremento del IMC (25,8 ± 2,4 frente a 24,3 ± 2,1 kg/m2; p = 0,030), sin embargo una reducción significativa del %MG (24,6 ± 8,9 frente a 29,6 ± 14,5; p = 0,012), de la CCi (78,4 ± 5,6 frente a 81,2 ± 2,2 cm; p = 0,043) y de la CCa (93,7 ± 6,9 frente 98,9 ± 8,3 cm; p = 0,013). En cuanto a los valores plasmáticos, el tratamiento androgénico produjo un incremento significativo de CT (191,5 ± 51,9 frente a 155,4 ± 27,9 mg/dl; p = 0,002) y de TG (136,5 ± 61,7 frente a 72,2 ± 29,6 mg/dl; p = 0,001), y un descenso de HDL-c (39,5 ± 8,9 frente a 57,8 ± 11,2 mg/dl; p = 0,012); además incrementó los valores de glucosa (99,5 ± 22,1 frente a 88,3 ± 11,5 mg/dl; p = 0,002) y de creatinina (1,31 ± 0,12 frente a 1,10 ± 0,19 mg/dl; p = 0,015). Concluimos que el tratamiento hormonal para reasignación de sexo en transexuales, además de los efectos deseados, produce importantes cambios antropométricos y metabólicos que modifican el riesgo cardiovascular.
253UTILIDAD DE LA MONITORIZACIÓN AMBULATORIA DE PRESIÓN ARTERIAL EN EL MANEJO PREOPERATORIO DE UN PACIENTE CON FEOCROMOCITOMA
J.J. Chillarón*, J. Puig*, J.J. Sancho**, J. Flores*, M.J. Carrera*, A. Sitges** y J.F. Cano*
*Endocrinología y Nutrición, **Cirugía Endocrinológica Hospital del Mar. Barcelona.
Introducción: La hipertensión arterial en el feocromocitoma se presenta de forma mantenida en el 50% de casos, en paroxismos en el 33% y alrededor de un 10% es silente. La monitorización ambulatoria de presión arterial (MAPA), puede aportar información valiosa en cuanto al número de crisis diario y a la duración e intensidad de las mismas. Presentamos un caso clínico de un varón con un feocromocitoma supuestamente silente.
Caso clínico: Varón de 69 años, sin antecedentes de interés. Su hijo de 45 años tras ser intervenido de feocromocitoma y carcinoma medular de tiroides fue diagnosticado de MEN 2A. El estudio genético detectó en el padre la misma mutación. Clínicamente asintomático. No paroxismos de sudoración, cefaleas o palpitaciones. TA 130/80 mm Hg; Fc 80 ppm en varias determinaciones. Los datos analíticos mostraban: Glu 150 mg/dL; Ca 9,3 mg/dL; Colest total 248 mg/dL; Adrenalina orina 24 h 257 mcg (N: 0,01-18); noradrenalina orina 24 h 85 mcg (N: 0,01-76); Metanefrina orina 24 h 1178 mcg (N: 52-341), normetanefrina orina 24 h 8645 mcg (N: 88-444); AVM orina 24 h 12,8 mcg (N: 2-9); En la TC abdominal se evidenció una masa suprarrenal izquierda de 4,8 cmX 4,8 cm con márgenes bien definidos. Se realizó monitorización ambulatoria de presión arterial, con TA media de 111/75 mm de Hg, y Fc media de 80 lpm, demostrando 2 picos hipertensivos de 178/126 mm Hg, Fc 127 lpm y 178/109, Fc 111 lpm, tras lo que se inició tratamiento con nifedipino 10 mg/8 horas. A los 7 días de iniciado el tratamiento, en una nueva MAPA, la TA media era de 111/67 mm de Hg, Fc media de 86 lpm, con TA máxima de 151/97 mm de Hg, Fc 114 lpm, por lo que se aumentó el tratamiento con nifedipino a 20 mg/8 horas. Previo a la cirugía la MAPA sólo mostró un pico hipertensivo (> 160/90 mmHg), y 5 determinaciones de frecuencia cardíaca superior a 100 lpm puntuales. Se aumentó el tratamiento con nifedipino a 30 mg/ 8 horas. No presentó en ningún momento hipotensión ortostática. Durante la cirugía laparoscópica presentó un sólo pico hipertensivo (175/90 mmhg) en el momento de manipulación del tumor, y precisó labetalol en una ocasión para controlar la frecuencia cardíaca, requiriendo un aporte de líquidos de 4300 cc en las primeras 24 horas para mantener cifras tensionales correctas. A las 48 horas fue dado de alta sin complicaciones y con TA estable sin tratamiento.
Conclusión: En el feocromocitoma "asintomático" la MAPA puede ser un método útil para detectar paroxismos hipertensivos y para el control del tratamiento prequirúrgico. En este paciente, el tratamiento con nifedipino fue una alternativa eficaz en el manejo preoperatorio
254VALIDACIÓN DE UN MÉTODO PARA CUANTIFICAR CORTISOL EN SALIVA. APLICACIÓN CLÍNICA
J.M. Navarrete Carranza*, M.E. Torregrosa Quesada*, R. Alfayate Guerra*, M. Mauri Dot*, S. Aznar Rodriguez**, J.R. Dominguez Escribano***, S. Lorenzo Garcia* y A. Picó Alfonso**
*Laboratorio de Hormonas Hospital General Universitario de Alicante. Alicante, **Sección de Endocrinología Hospital General Universitario de Alicante. Alicante, ***Sección de Endocrinología Hospital Universitario de San Juan. Alicante
Introducción: La pérdida del ritmo circadiano de cortisol es una característica del síndrome de Cushing. Concentraciones de cortisol plasmático a las 23 horas superiores a 1,8 µg/ dl tienen una sensibilidad diagnóstica muy próxima al 100%, pero su valoración exige ingreso hospitalario. El cortisol salivar es un reflejo de la concentración plasmática de cortisol libre, la forma biológicamente activa del cortisol circulante, permitiendo por tanto el estudio del ritmo de cortisol en casa. Sin embargo, los métodos utilizados para la determinación de cortisol plasmático no tienen suficiente sensibilidad para detectar las concentraciones de cortisol en saliva.
Objetivos: Validar analítica y clínicamente un método para determinar la concentración de cortisol en saliva.
Métodos: Las muestras de saliva se recogieron en dispositivos Salivette(Sarstedt) siguiendo las instrucciones del fabricante. Los tubos se centrifugaron a 1000g durante 2 minutos, congelándose la saliva eluida a 20º C hasta su procesamiento. Para la determinación del cortisol salivar se utilizó un RIA (Coat-A-Count Cortisol, DPC) diseñado para suero. Se han realizado modificaciones en la curva estándar así como en el tiempo de incubación con la finalidad de aumentar la sensibilidad analítica de la técnica.
Sujetos: Se determinó cortisol salivar matinal (8:00) y nocturno (23:00) en dos grupos de sujetos: 40 individuos sanos y 3 pacientes diagnosticados de síndrome de Cushing, dos de ellos de enfermedad de Cushing y el tercero de adenoma suprarrenal, confirmados histológicamente post cirugía.
Resultados: Valores de referencia en población sana:
Cortisol salivar matinal (08 h): 0,57 ± 0,54 µg/ dl (0,11-2,6)
Cortisol salivar nocturno (23 h): 0,04 ± 0,03 µg/ dl (< 0,03-0,13)
Imprecisión analítica: Coeficiente de variación intraserial 5,6%, coeficiente de variación interserial 6,4%. Sensibilidad analítica: 0,03 µg/ dl.
Los resultados en los 3 pacientes fueron para cortisol salivar matinal 4,01, 0,81 y 0,45 µg/ dl, y para cortisol salivar nocturno 1,76, 0,46, 0,48 µg/ dl respectivamente. Las concentraciones de cortisol salivar nocturno en los pacientes estudiados fueron superiores a las obtenidas en las personas sanas.
Conclusiones: El RIA modificado ha demostrado una precisión y sensibilidad analíticas adecuadas para la detección de cortisol salivar. El cortisol salivar a las 23 h puede utilizarse como prueba de despistaje de síndrome de Cushing, si bien sería necesario completar el estudio con una casuística mayor. La recogida de saliva es un procedimiento simple, no estresante y puede realizarse fácilmente de forma ambulatoria, sin necesidad de ingreso hospitalario.
