



Adrenocortical carcinoma (ACC) is a rare disease with a poor prognosis. The clinical experience acquired, even from a small number of cases, has improved understanding of this condition. The purpose of this study is to characterize patients with ACC followed up at a Portuguese reference center over the past 22 years.
MethodsRetrospective analysis of clinical records of patients with histopathological diagnosis of ACC followed up from 1992 to 2014.
ResultsThe study sample consisted of 22 patients, 20 of them female. Eleven patients were in stage II, four in stage III, and five in stage IV; 13 patients had functioning lesions. Adrenalectomy was performed in 20 patients, with complete tumor resection in 90% of the cases. During follow-up, eight patients experienced recurrence of local disease, and 12 distant metastases. Fourteen patients received mitotane, 35.7% (n=5) as adjuvant therapy and 64.3% (n=9) after recurrence; therapeutic plasma mitotane levels were achieved in 70% of patients. Stage III patients who received adjuvant therapy had longer survival time (13.5 vs. 2.5 months). Two patients were given chemotherapy associated to mitotane. Median survival was 11 months (0–257 months); it was slightly longer in younger patients or patients with non-functioning tumors. Six patients are still alive, four of them with no evidence of disease.
ConclusionDespite the overall poor prognosis, some patients with ACC may have a long survival time. Although complete tumor removal remains the only potentially curative treatment, diagnosis at a younger age, presence of non-functioning tumors, and mitotane treatment also seemed to be associated to longer survival in our patients.
El carcinoma de la corteza suprarrenal (ACC) es una enfermedad rara, con mal pronóstico. La experiencia clínica, incluso cuando se obtiene a partir de un número limitado de casos, ha mejorado el conocimiento sobre esta entidad. Este estudio tiene como objetivo caracterizar los pacientes con ACC seguidos en un centro de referencia portugués durante los últimos 22 años.
MétodosAnálisis retrospectivo de la historia clínica de los pacientes con diagnóstico histopatológico de ACC seguidos entre 1992 y 2014.
ResultadosSe incluyeron 22 pacientes, 20 de ellos mujeres. Once pacientes se encontraban en estadio ii, 4 en estadio iii y 5 en estadio iv, mientras que 13 pacientes mostraron lesiones funcionales. La adrenalectomía se realizó en 20 pacientes, con resección completa del tumor en el 90% de los casos. Durante el seguimiento 8 pacientes presentaron recurrencia de la enfermedad local y 12 metástasis a distancia. Catorce pacientes recibieron tratamiento con mitotano: adyuvante en el 35,7% (n=5) o después de la recidiva de 64,3% (n=9), alcanzándose niveles plasmáticos terapéuticos en el 70% de los casos tratados. Los pacientes en estadio iii que recibieron tratamiento adyuvante presentaron mayor supervivencia (13,5 frente a 2,5 meses). Dos pacientes fueron sometidos a quimioterapia asociada con mitotano. La mediana de supervivencia fue de 11 meses (0-257 meses), y resultó ligeramente mayor en los pacientes más jóvenes o con tumores no funcionantes. Seis pacientes continúan vivos, 4 de ellos sin evidencia de enfermedad.
ConclusiónA pesar del mal pronóstico global, algunos pacientes con ACC pueden presentar una larga supervivencia. Aunque la eliminación completa del tumor sigue siendo el único tratamiento potencialmente curativo, en nuestros pacientes el diagnóstico a una edad más joven, la presencia de tumores no funcionantes y el tratamiento con mitotano también parecen asociarse con una mayor supervivencia.
The adrenocortical carcinoma (ACC) is a rare disease, with an annual incidence of about 0.7–2 cases per million population.1,2 It mainly affects women aged 40–50 years; most of ACC are sporadic, although there are also some congenital and familial forms of this disease.3,4 ACC are larger than benign adrenal masses and measure about 10–12cm. ACC accounts for 2% of adrenal tumors that measure 4cm or less, 6% of tumors that measure from 4.1 to 6cm, and 25% of those tumors greater than 6cm.5 According to their functional type ACC may be classified as hormone secreting tumors or non-functioning tumors. Hormone-secreting ACC appear with manifestations of virilization, feminization or Cushing syndrome; on the other hand, non-functioning ACCs are usually diagnosed incidentally.6,7 Regarding its prognosis, unfortunately this tumor represents one of the most aggressive malignant endocrine neoplasia. In fact, the first reports of this disease date back to 1958 and referred that untreated patients have a lifespan of only 3 months.8
Although the interest in this disease has increased exponentially over the past few years, its rarity has limited a large-scale analysis of all possible factors with prognostic effects. Our current knowledge has been mainly obtained from clinical experience obtained from series of patients with ACC, even including a limited number of cases. The majority of published studies did not present data on long-term follow-up, which also limited insight about its evolution, including the rate of cure or late recurrence of the disease.
In this study the authors aim to present their experience in the diagnosis, therapeutic management and clinical outcome of patients with ACC followed in a Portuguese reference center over the last 22 years.
MethodsTwenty-two patients with ACC were diagnosed and monitored at the Endocrinology, Diabetes and Metabolism Department of Coimbra Hospital and University Centre from 1992 to 2014. The authors performed a retrospective analysis, through the review of individual medical records, of clinical features, diagnostic approach, therapeutic regimens and evolution of these patients.
DefinitionsThe diagnosis and therapeutic approach of ACC patients were based on the guidelines of our own institution, and more recently the implementation of the guidelines of ENSAT (European Network for the Study of the Adrenal Tumours) Group.9
ACC was considered as functioning when the patients presented clinical manifestations caused by hormonal hypersecretion and/or evidence of elevated serum hormone levels.
The staging system used was based on the TNM system for ACC, of AJCC/UICC Association (American Join Committee on Cancer/International Union Against Cancer), available since 2009.3
The extent of surgery was defined as adrenalectomy alone vs. multi-organ resection (resection of the adrenal gland and adjacent organs). Based on the surgical records, the removal of the lesion was reported as complete (R0), incomplete (R1) or indeterminate (Rx).
Survival time was calculated from the date of diagnosis to the date of death or the date of the last evaluation of the patient.
Recurrence of disease was based on clinical, laboratory and radiological evidence of disease, and did not require a histological confirmation.
Statistical analysisStatistical analysis was performed using a commercially available software package (SPSS 21.0 for Windows: SPSS Inc., Chicago, IL, USA). For the overall sample characterization, quantitative variables were expressed by median, followed by the range of variation; and qualitative variables by their absolute and relative frequencies. The survival time curves were calculated according to the Kaplan–Meier method and were compared using the log-rank test. A p value <0.05 was considered statistically significant.
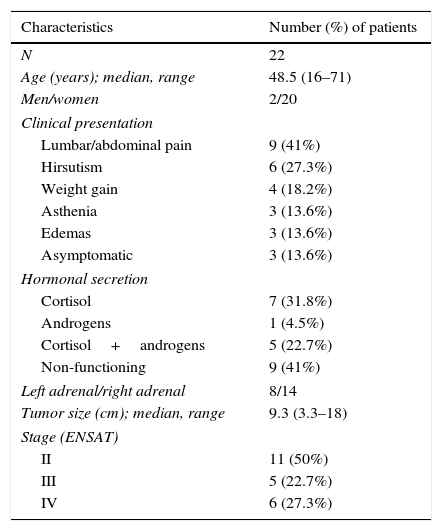
ResultsInitial clinical characteristicsTable 1 outlines the baseline clinical characteristics of the patients in our sample. Twenty of the 22 patients were women. The average age at onset of the disease was 48.5 years. The main complaints that lead to diagnosis were abdominal pain and hirsutism; the remaining patients presented nonspecific complaints as edema, weight gain and asthenia, among others. In three patients the lesion was identified incidentally.
Initial clinical characteristics.
| Characteristics | Number (%) of patients |
|---|---|
| N | 22 |
| Age (years); median, range | 48.5 (16–71) |
| Men/women | 2/20 |
| Clinical presentation | |
| Lumbar/abdominal pain | 9 (41%) |
| Hirsutism | 6 (27.3%) |
| Weight gain | 4 (18.2%) |
| Asthenia | 3 (13.6%) |
| Edemas | 3 (13.6%) |
| Asymptomatic | 3 (13.6%) |
| Hormonal secretion | |
| Cortisol | 7 (31.8%) |
| Androgens | 1 (4.5%) |
| Cortisol+androgens | 5 (22.7%) |
| Non-functioning | 9 (41%) |
| Left adrenal/right adrenal | 8/14 |
| Tumor size (cm); median, range | 9.3 (3.3–18) |
| Stage (ENSAT) | |
| II | 11 (50%) |
| III | 5 (22.7%) |
| IV | 6 (27.3%) |
Thirteen (68%) of the 22 patients studied had a functioning adrenal carcinoma: 7 (31.8%) with isolated Cushing's syndrome, 5 (22.7%) with Cushing syndrome associated with hyperandrogenism and 1 (4.5%) with isolated hyperandrogenism. Only two of the patients with Cushing's syndrome received medical treatment prior to surgery; both had been treated with metyrapone with no reported complications. All patients with hyperandrogenism either isolated or in association with Cushing's syndrome presented with elevated levels of dehydroepiandrosterone-sulphate, androstenedione and total testosterone. Two patients with isolated hyperandrogenism also had elevated levels of 17-hydroxyprogesterone. There were no reports of symptoms related to other hormonal overproduction such as hypokalemia, weight loss or catabolic syndrome, among others.
To assess prognosis staging is mandatory. Stage I is defined by the presence of an isolated tumor measuring less than 5cm; stage II is defined by the presence of an isolated tumor measuring more than 5cm; stage III is defined by the invasion of the proven tumor into the surrounding tissue or the finding of venous tumor thrombus in the vena cava or renal vein as well as positive lymph nodes; and finally stage IV is defined by the presence of distant metastases regardless of the tumor size or lymph nodes metastases.3 Most of our patients were not at an advanced stage of disease; 11 (50%) patients were in stage II, 5 (22.7%) in stage III and only 6 (27.3%) in stage IV. The tumor was detected almost exclusively through abdominal computed tomography (CT) (with or without contrast); only four patients were identified through abdominal magnetic resonance imaging (MRI). The tumor size ranged from 3.3 to 18cm, with an average diameter of 9.3cm. Six patients presented invasive disease [kidney (2); diaphragm (2); inferior vena cava+kidney (1); and inferior vena cava+diaphragm (1)] at the time of diagnosis. Four patients had distant metastasis to the liver and lymph nodes; one of them also presented pulmonary lesions.
Therapeutic regimens and clinical outcomeTwenty (90.9%) patients were submitted to adrenalectomy through laparotomy (Table 2): 15 patients were subjected to isolated adrenalectomy and 5 had a simultaneous excision of lymph nodes (2), spleen (1), liver (1), and diaphragm (1). Total resection was reported in 18 (90%), excluding two patients presenting tumor invasion of surgical margins. The mortality rate resulting from surgical intervention was nil. Two patients did not undergo adrenalectomy for not presenting surgical conditions.
Treatment and clinical evolution.
| Characteristics | Number (%) of patients |
|---|---|
| N | 22 |
| Adrenalectomy | 20 (%) |
| Isolated | 15 (68.2%) |
| Resection multi-organ | 5 (22.7%) |
| Not performed | 2 (9.1%) |
| Recurrence/metastasis | |
| Local recurrence | 8 (40%) |
| Lymph nodes | 10 (50%) |
| Lung | 5 (25%) |
| Liver | 5 (25%) |
| Bone | 2 (10%) |
| Absent | 5 (25%) |
| Mitotane treatment | |
| Adjuvant | 5 (22.7%) |
| After recurrence | 9 (40.9%) |
| Not performed | 8 (36.4%) |
| Clinical evolution | |
| Death related to disease | 12 (54.6%) |
| Death from another cause | 3 (13.6%) |
| Persistent disease | 3 (13.6%) |
| No evidence of disease | 4 (18.2%) |
| Survival (months); median, range | 11 (0–257) |
Postoperative histopathological evaluation revealed adrenal masses with median weight 740.7g (62–2200g) and diameter 11.8cm (5–28cm). Invasive disease was studied in all the organs excised, as well as the presence of metastatic disease in lymph nodes. The Weiss criteria were only assessed and recorded in 11 (50%) patients, all of them submitted to surgery over the last 8 years. Each patient recorded a median of four Weiss criteria (3–7). Seven patients had also information regarding Ki67 proliferative index presenting a median value of 7%, ranging from 4% to 12%.
Patients were followed through periodic imaging reevaluations (chest CT and abdominal CT/MRI) at 3–4 months’ intervals, in association with serial hormonal monitoring. The reevaluation period has become progressively longer in the absence of recurrence signs. All patients maintained follow-up in our center. The recurrence of the disease was studied using additional studies such as ultrasound imaging methods, PET-18FDG and/or bone scintigraphy, among others.
During follow-up, 8 (40%) of the 20 operated patients presented local recurrences and 12 (60%) had distant metastases. The average time to local recurrence was 7.5 months (2–23 months) and the average time to metastases was 3.5 months (1–28 months). The most common sites of metastases are lung (10), liver (5), lymph nodes (5) and bone (2).
Adjuvant treatment and treatment of metastatic or recurrent diseaseAfter the surgery, 5 (25%) patients received treatment with mitotane: 2 patients in stage II, 2 in stage III and 1 in stage IV of the disease. These patients were all women, with average age of 31.5 years and almost all of them had non-functioning tumors. Even though the differences in individual clinical characteristics preclude the thorough assessment of the effectiveness of this treatment, stage III patients treated with mitotane presented a longer survival time (13.5 vs. 2.5 months) when compared to patients of the same stage who did not receive treatment with this drug.
In 9 (45%) patients, treatment with mitotane was only introduced after local recurrence or distant metastasis. There were no significant differences in survival time of these patients compared to those with the same disease stage who did not receive medical treatment. Both patients diagnosed in stages II and IV, who were prescribed chemotherapy (etoposide, cisplatin and doxorubicin – EDP), kept local and distant metastatic disease progression with a survival time of 48 and 12 months, respectively.
There were no significant differences in mitotane dose when prescribed as an adjuvant or in the context of disease recurrence. However, the prescribed dose differed considerably from individual to individual. The median initial dose was of 2.75g (1–7g) and the maximum was 5.6g (3–12.5g). The median cumulative dose was 531.5g (95–5890g) for an average period of 12.3 months (1–68 months) of treatment. Mitotane plasma levels were evaluated in 10 of the 14 patients treated: 7 of them (70%) reached therapeutic serum levels in 4.5 months (3–6 months).
Only one (7.1%) patient who received mitotane did not report any drug side effect. Most of the patients reported nauseas (50%, n=7), skin lesions (21.4%, n=3), paresthesia (21.4%, n=3), diarrhea (14.3%, n=2) and nonspecific cognitive impairment (14.3%, n=2). Two patients even had to discontinue treatment due to intolerance. There were some cases of therapeutic failure detected at follow-up that may be attributed to intolerance, or cases where the remaining drug was returned to the family after patient's death.
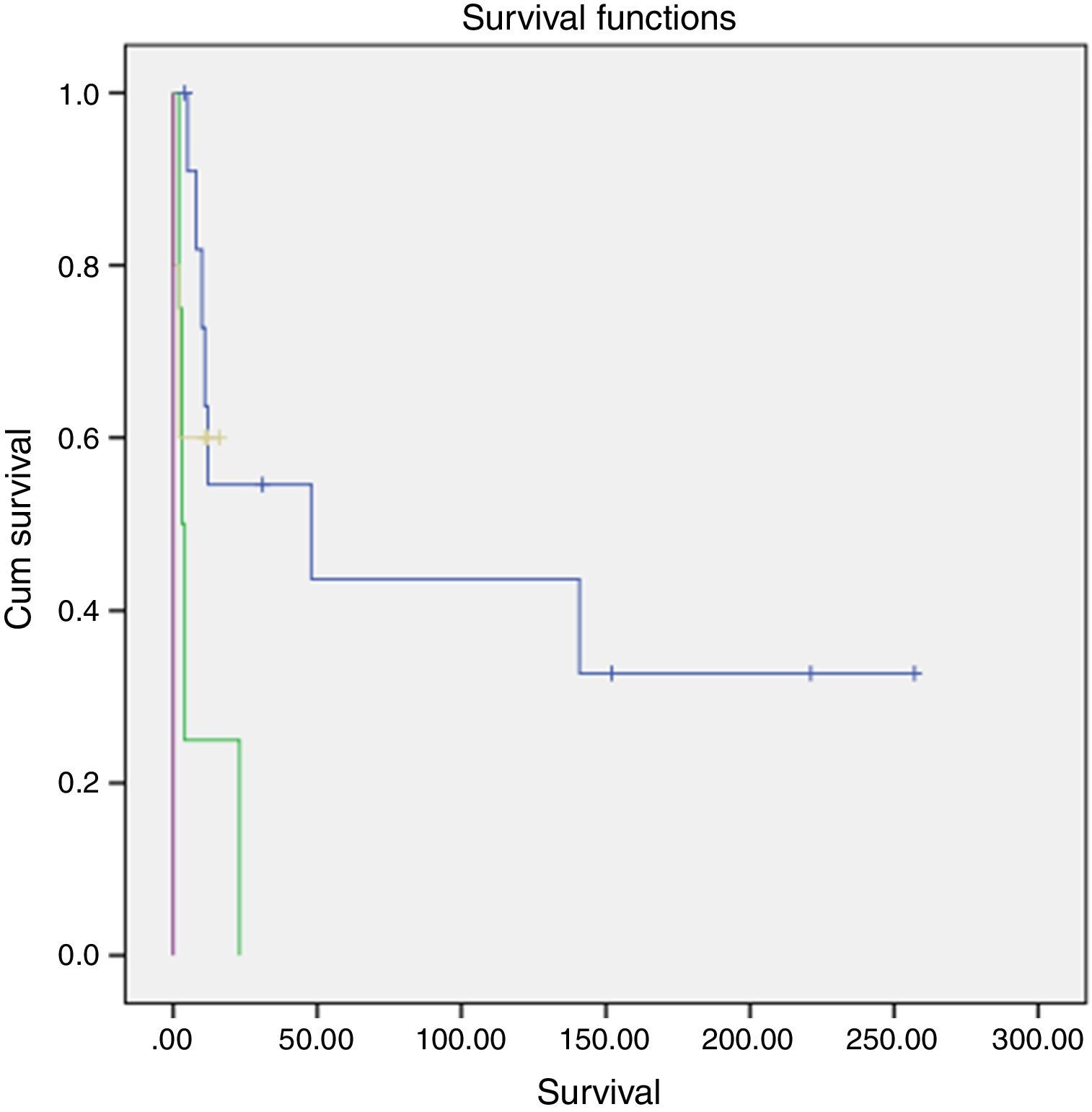
Survival and clinical evolutionThe median overall survival of our patients was 11 months (0–257 months), with a survival rate of 18.2% at 5 years. The survival time differed according to the stage of the disease and was significantly longer in patients diagnosed at stage II (p=0.002, log-rank test for) (Fig. 1).
 (blue: stage II, green: stage III, yellow: stage IV).")
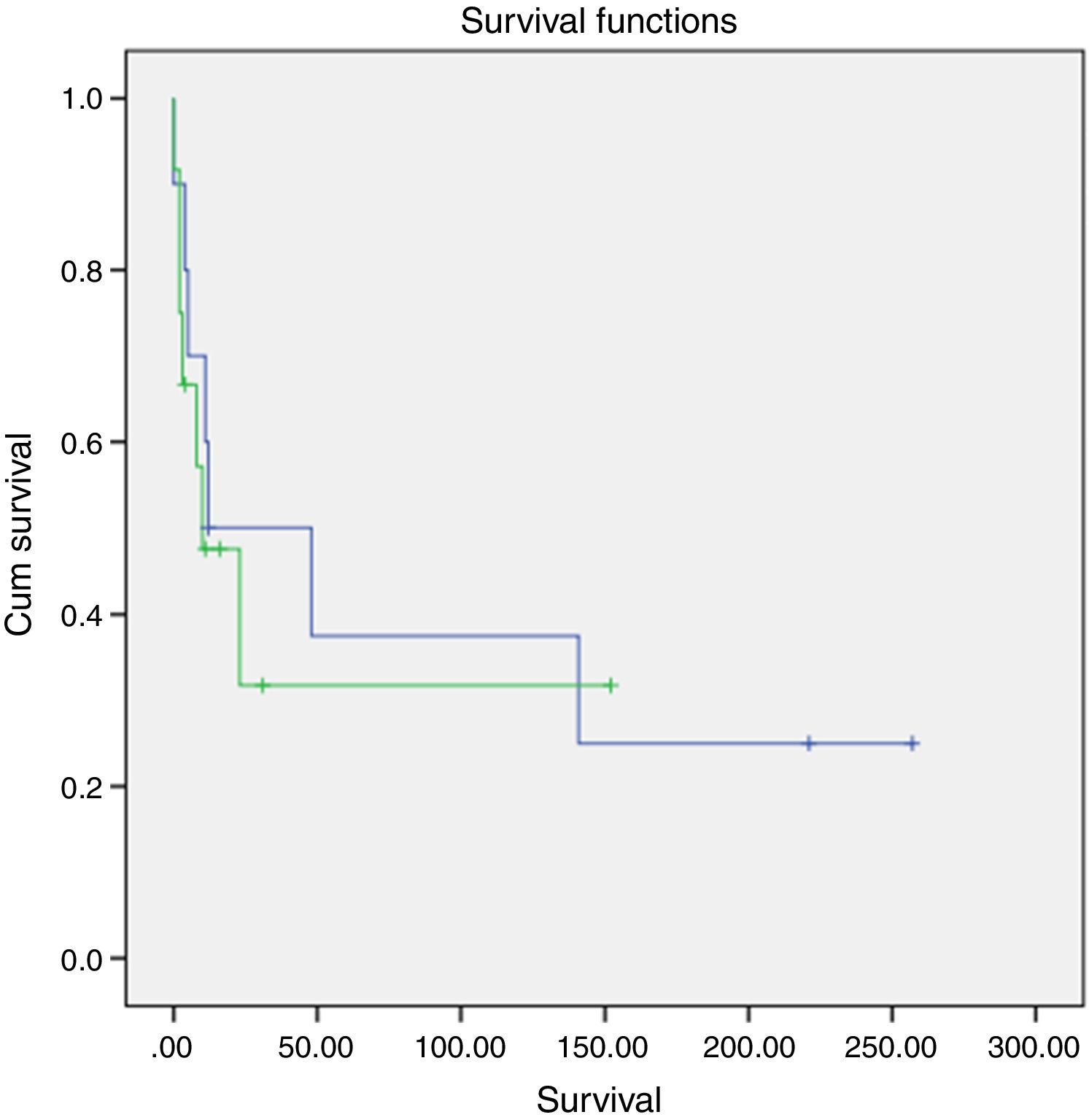
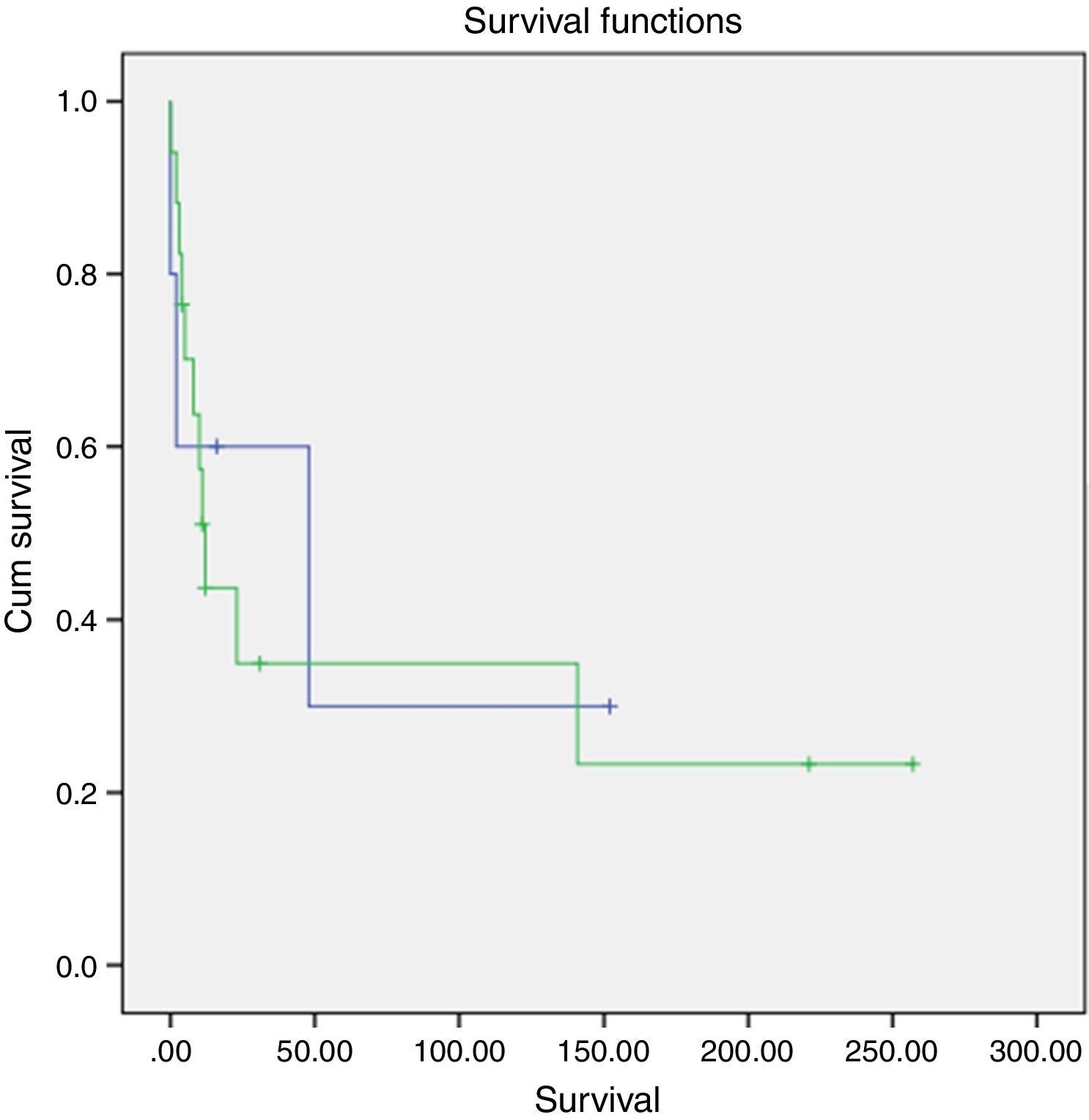
A slightly higher survival was also observed in patients diagnosed in younger ages and with nonfunctioning tumors (Figs. 2 and 3). However, this difference was not statically significant, probably conditioned by the limited number of patients in the sample (p>0.05).
. Log-rank test (p=0.692) (blue: <45 years; green: ≥45 years).")
. Log-rank test (p=0.834) (blue: functioning; green: non-functioning).")
The majority of our patients died due to progressive disease (54.5%); in three patients (13.6%) death was caused by the development of acute infectious complications; at present 6 (27.3%) patients are still alive, 4 (18.2%) of them with no evidence of disease. This subgroup of patients is composed exclusively by women aged 28–60 years (median 37 years) in whom ACC had been found in stage II at diagnosis. Two patients had nonfunctioning tumors, one had isolated Cushing's syndrome and another had hyperandrogenism. In all these patients a complete surgical removal (R0) of the adrenal lesion had been reported, and there have been no recurrence or distant metastasis identified during the follow-up. Currently, they present an overall median survival of 186 months (31–257).
DiscussionOnly 3 (13.6%) patients of our series were diagnosed incidentally, most of the patients had nonspecific complaints related to the presence of hormonal hypersecretion, which may have contributed to an earlier diagnosis, and half of the patients were diagnosed at stage II. The frequency of functioning tumors was similar to that reported in the current literature, which describes the presence of isolated Cushing's syndrome in about 45%, Cushing's syndrome combined with hyperandrogenism in 25% and isolated hyperandrogenism in less than 10% of the cases of ACC.10,11 ACC is usually characterized by multiple defects of steroid biosynthesis enzymes, leading to the secretion of steroid precursors typical of adrenal enzymatic blocks. In general, an isolated excess of a single steroid hormone constitutes a rare event.4 In our series, all the patients with hyperandrogenism presented hypersecretion of multiple hormones and precursors.
Surgical treatment, through laparotomy, was the first therapeutic option for almost all the patients, only two cases did not gather surgical conditions. However, the best surgical approach still remains in debate.12 The preoperative evidence of locally advanced disease unequivocally requires a laparotomy approach. However in the presence of a well-defined lesion under 10cm, some authors consider that it can be treated through a laparoscopic intervention,13 while others are convinced that such approach may increase the risk of recurrence.14 Regardless the technique used, the primary objective is to achieve a complete tumor removal that potentially cures the patient.11 In fact, the total removal of the lesion, with disease-free margins (R0), represents the most important prognostic factor.15 During an early stage of the disease an isolated adrenalectomy may be sufficient; however, the presence of invasion of adjacent structures implies the removal of all involved organs. As ACC usually spread through the lymphatic system, the routine performance of lymphadenectomy can be also applied. Unfortunately it was not performed by routine in our series; this fact may have probably contributed to a worse prognosis of our patients. According to the German ACC Study Group, this procedure allows a significant reduction in the risk of recurrence and death caused by this disease (hazard ratio: 0.54).16 Finally, the surgeon's expertise must also be taken into consideration. It has been shown that centers performing a large number of adrenalectomies per year obtain significantly higher survival rates and lower rates of local recurrence. In our study all patients were operated by experienced surgeons who met the current recommendations, performing more than 20 adrenalectomies per year.17 This point may have contributed to the high percentage of R0 in our sample (90%), that had important repercussion on patient's prognosis.
While prior or intraoperative identification of local invasion or distant metastasis establishes the diagnosis of ACC, the absence of these findings relies on postoperative histopathological analysis. The histopathological study of adrenal tumors can be challenging, with an error rate of up to 13%.9 Currently, the Weiss histopathological criteria constitute the most used system to assess the likelihood of malignancy.18 Despite a relatively high inter-observer variability it still represents a simple and reliable tool. In our center its systematic use is relatively recent, and only the performance of future analysis may assess its impact on the diagnosis and prognosis of these patients. Several immunohistochemical markers have been reported to be useful for the characterization of resected adrenocortical neoplasms. These include the Ki67 proliferative index, p53, insulin-like growth factor (IGF)-II, cyclin E, β-catenin, and steroidogenic factor-1 (SF1).7 Tumor staining with Ki-67 has been utilized to help differentiate benign form malignant adrenal tumors. A cut-off value between adenoma and ACC has been found to vary from 1.5% to 4%.5 A recent study of 17 patients revealed that Ki-67 index of 7% or more was associated with significantly shortened disease-free survival.6
In our study mitotane was the only adjuvant therapy used, and was prescribed to about one-fourth (22.7%) of the patients. This drug has been used for more than 50 years, and continues to be the key element of medical treatment of ACC.19 Nevertheless, its molecular mechanisms of action are not fully understood yet. According to recent data it probably exerts effects through the down-regulation of the mitochondrial respiratory chain.20 Some authors have showed that mitotane treatment allows prolonging patients’ survival. One of these studies demonstrated a significantly longer survival in patients receiving adjuvant treatment with mitotane compared with those patients who did not receive it (10:25 vs. 42 months and 110 vs. 52 and 67 months respectively).21 In another study, patients in stage II treated with mitotane after surgery had a better prognosis for this stage.22 However, the rarity of this disease, the use in some series of insufficient drug dose, and the large inter-individual variability of mitotane metabolism (key aspect of its therapeutic action) did not allow obtaining definite and consistent conclusions from all published studies.23 In our study, the adjuvant mitotane treatment also seems to be associated with an improvement of survival of patients in stage III (13.5 vs. 2.5 months), but the small number of cases limits the achievement of definitive conclusions. Other treatment options include adjuvant chemotherapy or radiotherapy (RT). According to results of the FIRM-ACT study, mitotane combined with chemotherapy (EDP) may have benefits as first-line therapy in patients with ACC diagnosed in an advanced stage.24 Tumor bed RT may be considered when there is high risk of local recurrence (R1) or in case of symptomatic lesions.19 None of our patients received adjuvant RT, and chemotherapy (EDP) was only added in two patients with recurrent or metastatic disease.
In 9 (40.9%) patients the treatment with mitotane was only prescribed after recurrence or development of metastasis. As expected in these circumstances the results are even less favorable. In most cases, metastatic disease conditions death within about 1 year, but there are reports stating a long-term response of some patients.25 It is empirically acceptable, but it is not scientifically proven, that the association of chemotherapy (particularly EDP) with mitotane determines a better clinical outcome than the use of mitotane alone. However the results obtained with chemotherapy have been disappointing.24 As time to recurrence is inversely proportional to probability of death, it should influence the therapeutic choice; it is proposed that patients with early recurrence (within 12 months) should receive more aggressive medical therapy.19 In our series, neither treatment with mitotane (alone or in combination with chemotherapy) nor surgical removal of metastatic lesions conditioned significant improvement in survival time of the patients with progressive disease. Nevertheless in almost all of our patients the recurrence of the disease was an early event, aspect that may indicate the presence of a more aggressive tumor.
The optimal dose and duration of medical treatment with mitotane, either adjuvant or after recurrence, is not defined yet. The therapeutic goal is to attain mitotane serum levels between 14 and 20mg/L. Significantly longer survival time has been observed in those patients achieving these levels (24 vs. 18 months, death hazard ratio 0.52, 95% CI: 0.28–0.97).26 Similarly to findings reported by others authors,27 our patients received widely varying doses of the drug, with 70% of them achieving therapeutic levels of mitotane. This can be explained not only by the large inter-individual variability in drug metabolism, so that only 38% of plasma levels directly depends on the mitotane dose,28 but also by the development of side effects that were reported by our patients. Although the tight laboratory surveillance of mitotane serum levels has avoided the use of supra-therapeutic doses, the scrupulous compliance of the prescribed dosage was difficult to assess.
The global survival rate of our sample was very low. However, there were some particular cases presenting disease remission for over 10 years. The diagnosis of ACC at an early stage, when allowing a complete resection of the tumor, seems to have been the determining cause of this favorable course. The staging system was created to differentiate cohorts of patients according to their prognosis (5-year survival of 81%, 61%, 50% and 13% in stages I, II, III and IV, respectively).29 However, recent data suggest that the survival rate has improved over the last years in all stages. The reasons are not totally clear, but the improvement of surgical techniques with a complete tumor removal even for advanced disease stages, or the widespread use of adjunctive therapy with mitotane may explain, at least in part, this evolution. On the other hand, there also seems to exist a great heterogeneity of ACC itself, and the disease progression can be independent of the stage.30 Long survival have been reported even in some patients with recurrent or metastatic ACC.31
Survival analysis was performed in our sample in order to assess other possible determinants of prognosis. In accordance with reports by other authors,32 patients with ACC diagnosed at younger age presented higher survival rates compared to older ones. In agreement with data from literature,32,33 hormonal hypersecretion, particularly hypercortisolism, seems to have adversely affected the outcome of our patients. Initially some authors affirmed that nonfunctioning ACC, more common in older adults, tended to progress faster.34 However, later studies demonstrated that, regardless the age, the presence of cortisol hypersecretion conditions a shorter survival (hazard ratio 3.9).32 It remains unclear whether cortisol production only reflects the biological aspect of the tumor, or if it directly affects patient's survival (facilitating cell proliferation, suppression of anti-tumor mechanisms, systemic deleterious effects, etc.).33 There are other prognostic factors, particularly related to the histological and molecular tumor characteristics, that were not analyzed in this study. Over the past few years our knowledge about the molecular events underlying ACC pathogenesis has improved, and in the future molecular markers may help us to define the risk of recurrence and to guide therapeutic decisions. These elements, combined with the clinical aspects known through the publication of series such as this, may enable an even more specific and effective therapeutic approach.
There are some limitations to this study, mainly the retrospective nature of our analysis based on a limited number of patients. However, the authors report the experience of a single center, allowing the access to individual clinical records along the whole follow-up time, as well as to histopathological confirmation of ACC in all patients.
In conclusion, despite the global poor prognosis, our study shows that ACC patients can sporadically achieve a long survival, and that some patients can even be cured, particularly if the tumor is detected in an early stage and if they are subjected to a complete surgical removal. According to our data, diagnosis at younger age, the presence of nonfunctioning tumors and the use of mitotane as adjuvant treatment allows an improvement in the prognosis of these patients.
FundingNone.
Conflict of interestsThe authors declare that they have no competing interests.
