


A pesar de que la apoplejía hipofisaria suele asentarse sobre un adenoma hipofisario, también puede aparecer sobre otros tipos de tumores menos frecuentes. El tumor teratoide/rabdoide atípico (TTRA) es un tumor maligno muy agresivo del sistema nervioso central, que habitualmente ocurre en niños menores de 3 años, con un pronóstico infausto a pesar del tratamiento quimio y/o radioterápico.
Presentamos el caso de una paciente de 43 años ingresada por sospecha de apoplejía hipofisaria por macroadenoma hipofisario. Refería cefalea de 3 meses de evolución. En las 2 semanas previas al ingreso presentó, de forma progresiva, malestar general, vómitos, debilidad de miembros inferiores, diplopía y, finalmente, ptosis palpebral. No existía clínica sugestiva de disfunción hormonal, poliuria ni polidipsia, y los ciclos menstruales estaban conservados. La exploración física destacaba un aceptable estado general con puntaje 15 en la escala de Glasgow; estaba normotensa y afebril, sin estigmas de hipercorticismo ni acromegalia. Presentaba paresia del III par izquierdo. Las exploraciones complementarias destacaban: hiponatremia grave (123mmol/l). TAC cerebral y RMN hipofisaria: tumoración selar de 20×23mm con crecimiento supraselar, compatible con macroadenoma hipofisario invasivo con sangrado subagudo. Campimetría: afectación de campo nasal y temporal superior e inferior en ambos ojos, más acentuada en ojo izquierdo. Perfil hormonal, compatible con hipopituitarismo parcial: prolactina 625,9μUI/ml (normal: 102-496); cortisol basal: 6,12μg/dl (normal: 6,2-19,4); TSH 0,29μUI/ml (normal: 0,27-4,2); FT4: 0,74ng/dl (normal: 0,93-1,7); FSH: 3,9mUI/ml (normal: 3,54-12,5); LH 0,3mUI/ml (normal: 2,4-12,6); HGH 0,45ng/ml (normal: 0-7), e IGF1 67ng/ml (normal: 100-310).
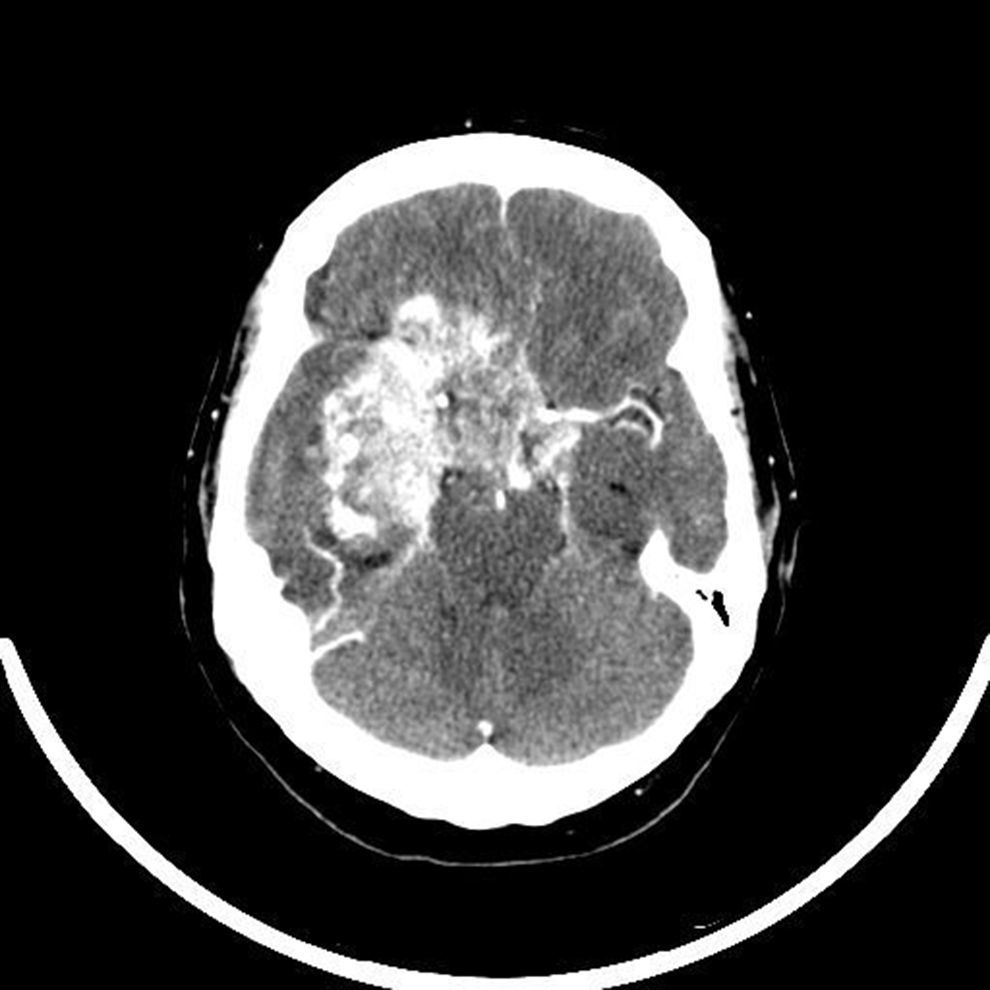
Diez días después de su ingreso se practicó resección parcial del tumor mediante abordaje transesfenoidal. En los días siguientes persistió la cefalea y desarrolló oscilaciones del nivel de conciencia. En una TAC craneal, sin contraste, realizada 5 días tras la intervención, se apreció una imagen ligeramente hiperdensa con respecto al parénquima cerebral, homogénea y redondeada en relación con lesión hipofisaria ya conocida, a pesar del tratamiento quirúrgico. Presentó una rápida progresión clínica, con desarrollo de incontinencia de esfínteres y amaurosis bilateral completa. En espera de los resultados histológicos, y ante la sospecha de tumoración maligna con crecimiento activo se consultó con oncología radioterápica para valorar tratamiento. En la TAC craneal realizada antes de iniciar la radioterapia (20 días tras la intervención) se observó una gran tumoración isodensa selar de 7,2cm de tamaño con infiltración de la región selar, seno cavernoso derecho, frontal y temporal derecho con efecto masa, y que realza intensamente con contraste intravenoso (fig. 1). El informe final de anatomía patológica confirmó la sospecha de malignidad: TTRA grado IV de la OMS. La determinación integrasa interactor 1 (INI1), marcador cuya ausencia a nivel nuclear en las células tumorales establece el diagnóstico definitivo de TTRA, resultó negativo en 2 ocasiones. Ante la rápida progresión de la enfermedad se mantuvo actitud conservadora, solo con medidas paliativas y tratamiento sustitutivo del hipopituitarismo persistente con glucocorticoides y tiroxina. Finalmente, la paciente falleció a los 35 días de ingreso.

La apoplejía hipofisaria se define como un fenómeno, isquémico o hemorrágico, que suele producirse sobre un adenoma hipofisario, aunque también puede asentar sobre otro tipo de tumores e incluso en una glándula hipofisaria normal1. En nuestra paciente, la presentación clínica era muy sugestiva y las pruebas de imagen iniciales mostraban una tumoración compatible con adenoma hipofisario, que es la lesión hipofisaria más frecuente en adultos. Sin embargo la evolución desfavorable y rápidamente progresiva orientaba a un proceso maligno, cuyo diagnóstico final fue TTRA.
El TTRA del sistema nervioso central, habitualmente afecta a niños de temprana edad, siendo el 94% de los pacientes descritos menores de 5 años2,3. Las características radiológicas no son específicas de este tipo de tumor. Histológicamente se compone de células rabdoides en su totalidad o combinadas con zonas indistinguibles de tumor neuroectodérmico primitivo (PNET, por sus siglas en inglés) y/o con componente mesenquimal y/o epitelial4,5. El TTRA es el único tumor del sistema nervioso central en el que se ha encontrado una alteración patognomónica a nivel de un gen supresor, el gen INI1/hSNF5. La pérdida de expresión de INI1 se considera diagnóstico de TTRA, por lo que una adecuada evaluación inmunohistoquímica es fundamental en la evaluación de esta variante tumoral.
Los protocolos de tratamiento en adultos se han extrapolado de la literatura pediátrica, siendo indicada la resección quirúrgica agresiva seguida de tratamiento multidisciplinar. En adultos, el uso de radioterapia craneoespinal es un tratamiento estándar, asociada a distintos regímenes de quimioterapia, aunque este no está bien definido para adultos.
El TTRA se caracteriza por un comportamiento agresivo en la mayoría de los niños, que habitualmente fallecen en los primeros 7 meses tras el diagnóstico, a pesar del tratamiento intensivo. En la revisión bibliográfica realizada en adultos, solo 9 casos se han producido en la región selar o supraselar. La supervivencia es variable en los diferentes casos publicados en la literatura, con un promedio de 26 meses (rango: 2 semanas/17 años)3,6,7; en nuestro caso la evolución fue fulminante, de tan solo 35 días desde el diagnóstico, siendo la forma de presentación más agresiva y de más rápida evolución de entre aquellos que han asentado en la silla turca8,9.
A modo de conclusión, recordamos que la apoplejía hipofisaria asienta con frecuencia sobre adenomas hipofisarios, si bien siempre deben tenerse en cuenta otros tumores primarios que afectan a la silla turca. El TTRA es un tumor del sistema nervioso central, excepcional en adultos y en localización hipofisaria, lo que puede dificultar el diagnóstico. El diagnóstico requiere una adecuada evaluación inmunohistoquímica, ya que la pérdida de expresión de INI1 es patognomónica de este tipo de tumor. Debido a su escasa incidencia, no existen protocolos de manejo terapéutico específicos para la población adulta, siendo el pronóstico infausto en un tiempo variable. Nuestro caso pone de manifiesto la rápida progresión de la enfermedad, y se trata de uno de los casos de TTRA en adultos con un comportamiento más agresivo y menor supervivencia desde el diagnóstico, según lo publicado hasta ahora en la literatura.
