


Although pituitary apoplexy usually occurs in a pituitary adenoma, it may also occur in other less common types of tumors. The atypical teratoid/rhabdoid tumor (ATRT) is a very aggressive malignant tumor of the central nervous system usually occurring in children under three years of age and has a poor prognosis despite chemotherapy and/or radiotherapy.
We report the case of a 43-year-old female patient admitted to hospital for suspected pituitary apoplexy due to pituitary macroadenoma. She complained of headaches over the previous three months. In the two weeks prior to admission, the patient progressively experienced malaise, vomiting, lower limb weakness, diplopia and, finally, eyelid ptosis. There were no clinical signs suggesting hormone dysfunction, urinary frequency, or polydipsia, and her menstrual cycles were maintained. A physical examination showed an acceptable general condition with a score of 15 on the Glasgow scale. The patient had normal blood pressure and no fever or signs of hypercorticism or acromegaly. Paresis of the third left cranial nerve was found. Supplemental tests revealed severe hyponatremia (123mmol/L). CT of the brain and pituitary MRI: 20×23mm sellar tumor with suprasellar growth, consistent with an invasive pituitary adenoma with subacute bleeding. Campimetry: superior and inferior nasal and temporal field involvement, especially marked in the left eye. A hormone profile was consistent with partial hypopituitarism: prolactin 625.9μIU/mL (normal: 102–496); basal cortisol: 6.12μg/dL (normal: 6.2–19.4); TSH 0.29μIU/mL (normal: 0.27–4.2); FT4: 0.74ng/dL (normal: 0.93–1.7); FSH: 3.9mIU/mL (normal: 3.54–12.5); LH 0.3mIU/mL (normal: 2.4–12.6); HGH 0.45ng/mL (normal: 0–7), and IGF-1 67ng/mL (normal: 100–310).
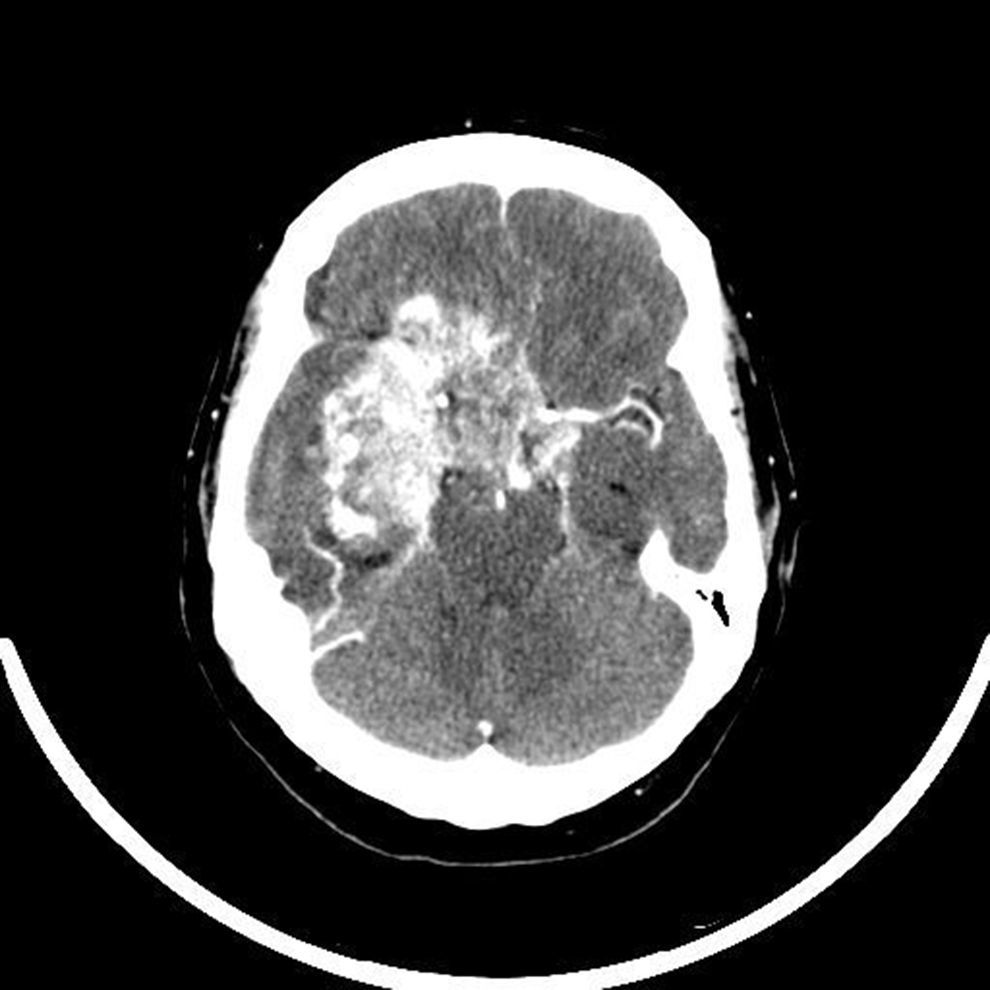
Ten days after admission, partial tumor resection was performed using a transsphenoidal approach. Over the following days, the headaches persisted and the patient experienced fluctuations in consciousness. A CT scan of the head with no contrast, performed five days after surgery, showed a slightly hyperdense image as compared to the brain parenchyma, homogeneous and rounded and related to the already known pituitary lesion, despite surgery. Rapid clinical progression occurred, with the development of sphincter incontinence and complete bilateral amaurosis. While waiting for the histological results and because of a suspected actively growing malignant tumor, radiotherapeutic oncology was consulted to assess treatment. A CT scan performed before the start of radiotherapy (20 days after surgery) showed a big sellar isodense tumor, 7.2cm in size that had infiltrated the sellar region and right cavernous, frontal and right temporal sinuses with a mass effect, strongly enhanced with intravenous contrast (Fig. 1). The final pathological report confirmed the suspected malignancy: ATRT grade IV of the WHO. Measurement of integrase interactor 1 (INI1), a marker whose absence in tumor cell nuclei establishes the final diagnosis of ATRT, was negative twice. Because of rapid disease progression, a conservative approach was adopted. Only palliative measures and replacement therapy with glucocorticoids and thyroxine for persistent hypopituitarism were used. Finally, the patient died 35 days after admission.

Pituitary apoplexy is defined as an ischemic or hemorrhagic event usually occurring in a pituitary adenoma, although it may also occur in other types of tumor and even in a normal pituitary gland.1 In our patient, the clinical presentation was highly suggestive, and initial imaging tests showed a tumor consistent with pituitary adenoma, the pituitary lesion most common in adults. However, the unfavorable and rapidly progressive course suggested a malignant condition, finally diagnosed as ATRT.
ATRT of the central nervous system usually affects young children, and 94% of the patients reported have been younger than 5 years.2,3 Radiographic characteristics are not specific of this type of tumor. Histolologically, ATRT consists of rhabdoid cells, alone or combined with indistinguishable areas of primitive neuroectodermal tumor (PNET) and/or with a mesenchymal and/or epithelial component.4,5 ATRT is the only tumor of the central nervous system in which a pathognomonic change has been found in a suppressor gene, INI1/hSNF5. The loss of INI1 expression is considered diagnostic of ATRT, and adequate immunohistochemical assessment is therefore essential in order to evaluate this tumor variant.
Treatment protocols for adults have been extrapolated from the pediatric literature, and aggressive surgical resection followed by multidisciplinary treatment is indicated. In adults, craniospinal radiotherapy is a standard treatment, associated with different chemotherapy regimens, although this is not well defined for adults.
ATRT is characterized by aggressive behavior in the majority of children, who usually die within seven months of diagnosis despite intensive treatment. In the literature review performed in adults, only nine cases have occurred in the sellar or suprasellar region. Survival is variable in the different cases reported in the literature, with a mean of 26 months (range, 2 weeks/17 years)3,6,7; in the reported case, the course was fulminant, as the patient died only 35 days after diagnosis, and the form of presentation was the most aggressive and most rapidly progressive among all the tumors occurring in the sella turcica.8,9
In conclusion, it should be borne in mind that pituitary apoplexy commonly occurs in pituitary adenomas, although other primary tumors affecting the sella turcica should always be taken into consideration. ATRT is a tumor of the central nervous system which is exceptional in adults and in a pituitary location, which may make diagnosis difficult. Diagnosis requires adequate immunohistochemical assessment, because the loss of INI1 expression is pathognomonic of this type of tumor. Because of the low incidence of ATRT, there are no specific therapeutic management protocols for the adult population, and the tumor is associated with a gloomy prognosis within a variable time period. The case reported demonstrates the rapid progression of the disease, and is one of the cases of ATRT in adults with the most aggressive behavior and shortest survival time among the diagnoses reported to date in the literature.
Please cite this article as: Larrán-Escandón L, Mateo-Gavira I, Vilchez-López FJ, Gómez Cárdenas E, Aguilar Diosdado M. Apoplejía hipofisaria como forma de presentación de un tumor teratoide/rabdoide atípico en el adulto. Endocrinol Nutr. 2016;63:364–365.
