


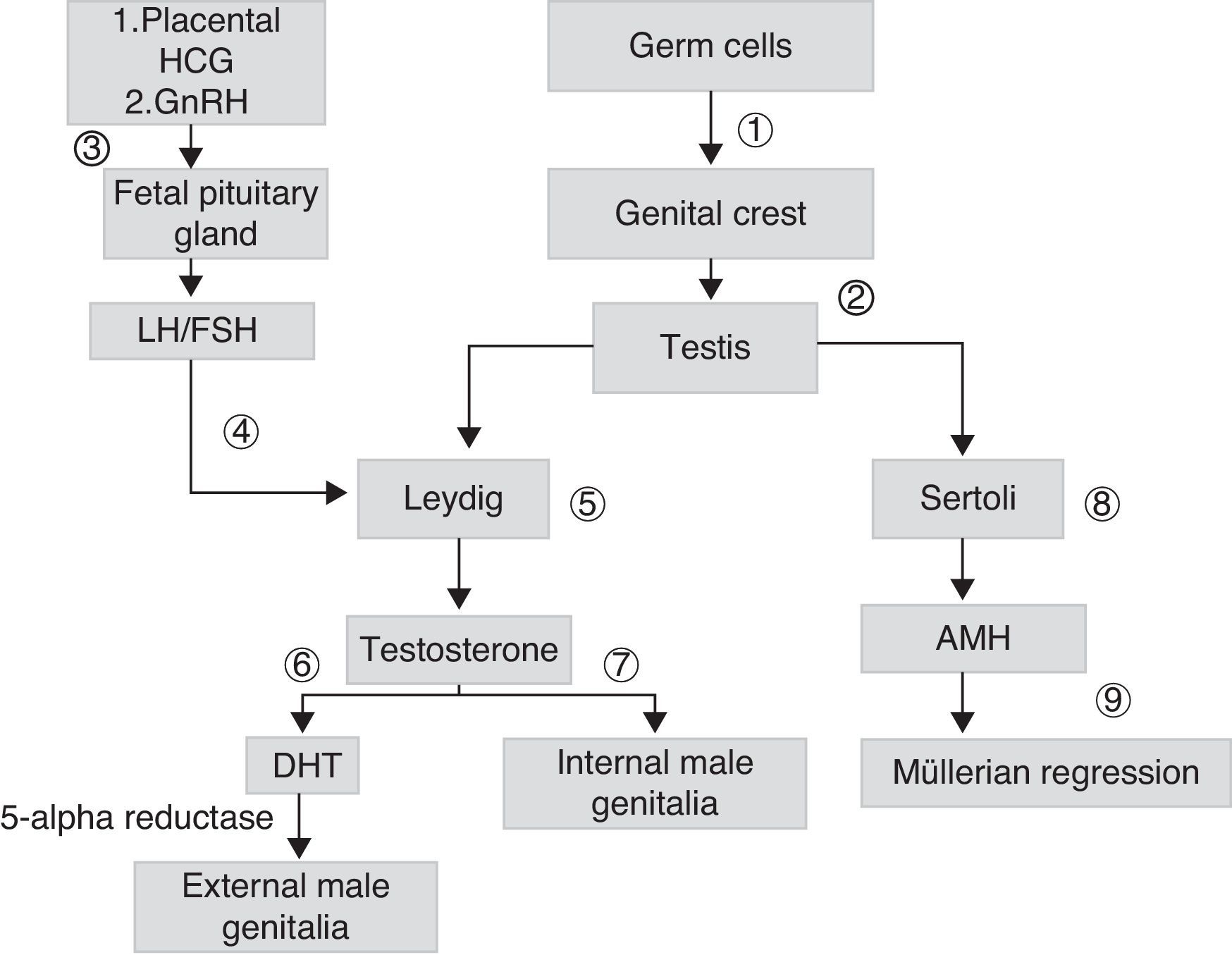
Sex differentiation is a process that starts early during embryogenesis. In males, the SRY gene, located in the short arm of chromosome Y, encodes for the testis-determining factor, which causes the gonad to differentiate into testis. Testicular androgen synthesis by Leydig cells starts from the eighth week of development. It is initially stimulated by placental chorionic gonadotropin (HCG), and subsequently by luteinizing hormone (LH). Fetal gonadotropins stimulate the testicular production of antimüllerian hormone (AMH), which stimulates the regression of müllerian ducts in males. Testosterone, whose action starts in the ninth week once the androgen receptor (AR) is present, allows for the stabilization of wolffian ducts and their differentiation into epididymis, deferent duct, and seminal vesicles. The conversion of testosterone into its most active metabolite, dehydrotestosterone (DHT), allows for the growth of the genital tubercle to form the penis and ventral raphe closure, so forming the penile urethra and scrotal raphe (Fig. 1).
, form primary sex cords originating testes (2). Leydig cells are stimulated by placental chorionic gonadotropin (HCG) and later by luteinizing hormone (LH) (4), which depends on the gonadotropin releasing hormone (GnHRH) secreted by fetal hypothalamus (3), which stimulates the production of gonadotropins. These stimulate the production of androgens (7) and antimüllerian hormone (AMH) (8), which allows for the regression of müllerian ducts (9). The testosterone initially secreted (7) allows for the development of internal male genitalia. The conversion of testosterone into dehydrotestosterone (DHT) allows for the formation of external male genitalia (6).")
Scheme of embryonic sexual development: germ cells migrate to genital crests (1), form primary sex cords originating testes (2). Leydig cells are stimulated by placental chorionic gonadotropin (HCG) and later by luteinizing hormone (LH) (4), which depends on the gonadotropin releasing hormone (GnHRH) secreted by fetal hypothalamus (3), which stimulates the production of gonadotropins. These stimulate the production of androgens (7) and antimüllerian hormone (AMH) (8), which allows for the regression of müllerian ducts (9). The testosterone initially secreted (7) allows for the development of internal male genitalia. The conversion of testosterone into dehydrotestosterone (DHT) allows for the formation of external male genitalia (6).
Complete androgen insensitivity syndrome (CAIS) is an XY sexual development disorder where AR function loss exists. CAIS occurs in subjects with the 46 XY karyotype with external genitalia of the female phenotype.
We report the case of a term newborn delivered after a physiological pregnancy. She was the first daughter of healthy, non-consanguineous parents. Birth weight was 3420g and birth length 50cm, while the Apgar score was 9–10. Physical examination disclosed normal external female genitalia and two 1-mm masses in both inguinal canals, which led to inguinal hernia being suspected. Abdominal ultrasound examination revealed a persistent peritoneo-vaginal process in both inguinal regions with herniation of abdominal contents and two homogeneous ovoid formations 1cm in diameter with Doppler flow inside, with no follicles suggesting gonads. At two days of life, LH and follicle-stimulating hormone (FSH) levels were less than 0.1IU/L (reference values (RV): 0.02–7.0mIU/L), testosterone was 1.45nmol/L (RV: 0.42–0.72nmol/L), and the estradiol level was <73.4pmol/L (markedly elevated values were to be expected).
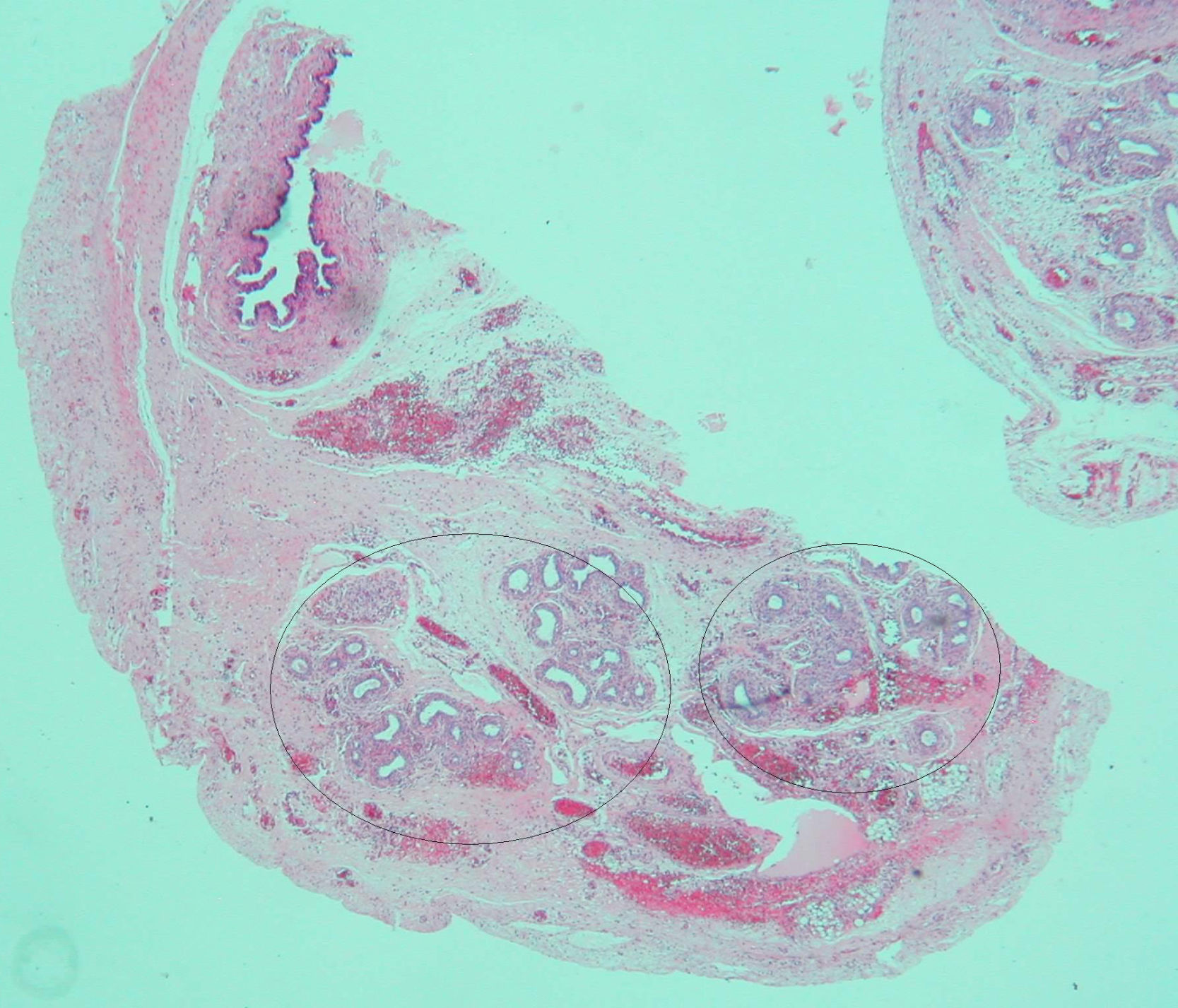
Based on the suspected presence of gonads in the inguinal canal, the patient underwent surgery on her third day of life. Intraoperative vaginoscopy showed no uterine cervix. A gonadal biopsy revealed a fragment of testicular parenchyma consisting of solid cords with Sertoli cells and spermatogonia. Leydig cells were not identified. Left gonad biopsy showed a high number of spermatogonia, a hydatid with a cystic aspect, and many tubular structures consistent with epididymis. A duct consistent with a müllerian remnant was found in the vicinity (Fig. 2). Both testes were replaced into the abdomen.
 which could correspond to müllerian remnants.")
The karyotype was 46 XY. The molecular study of the AR gene found a base change in exon 5, an arginine by glutamine replacement at codon 752 (R752X). This mutation had previously been reported in the literature in a patient with CAIS,1 and therefore confirmed the diagnosis. The molecular study of the mother was positive for the same mutation, but she had no phenotypic changes.
CAIS is an X-linked autosomal recessive disease caused by a mutation in AR that may be inherited (70%) or occur de novo (30%). The gene is located in the chromosomal region Xq11-12 and consists of eight exons encoding for a 919 amino acid protein. It has four functional domains: NTD (N-terminal transactivation domain), encoded in exon 1; DBD (DNA binding domain), encoded in exons 2 and 3; a hinge region; and LBD (ligand binding domain), encoded by exons 4–8.2 AR is located in cell cytoplasm bound to protein, and upon ligand binding translocates to the nucleus, where it is bound to DNA. The same mutation may cause different phenotypes in the same affected family.3
Prevalence is estimated at 2–5/100,000.4 Clinically, CAIS occurs in individuals with karyotype 46 XY and normal external female genitalia, blind-ending vagina, and intra-abdominal testes. Leydig cells secrete testosterone in adequate amounts for a male of that age, which is normally converted into dehydrotestosterone (DHT) through 5-alpha reductase, but the effect of DHT is virtually nil because of the presence of non-functioning AR. AMH production by Sertoli cells and the synthesis of its receptor AMHR2 is not affected by AR impairment. This allows for a normal action of AMH during the embryonic period, explaining the regression of müllerian structures (upper third of vagina, uterus, and ovaries) in patients with CAIS.
CAIS should be suspected in two instances: firstly, in a phenotypically female newborn with bilateral inguinal hernia, because this is uncommon in females. One to 25% of these lesions correspond to CAIS.5 The second presentation form occurs in a female adolescent with adequate breast development and a little or no pubic and/or axillary hair and with primary amenorrhea, in whom the absence of an uterus and ovaries is shown.
LH and testosterone are normal or in the upper limit during the first three months of life. In patients with preserved gonads, reactivation of the hypothalamic-pituitary axis during puberty induces Leydig cell activity, increasing intratesticular androgens and allowing for the arrest of Sertoli cell proliferation. AMH production is maintained due to AR insensitivity in the nucleus of Sertoli cells.6 Testosterone and LH remain elevated due to androgen resistance at hypothalamic-pituitary level. Testosterone is aromatized peripherally to estrogens, which explains the normal female breast development seen in these cases.7 Gonad preservation until puberty appears to be safe, because the risk of gonadoblastoma or other tumors only increases subsequently.8
The presence of müllerian remnants has sporadically been reported and has no clinical consequences. Various hypotheses have been suggested regarding their persistence, including (1) the presence of defects in AMH secretion or function, (2) the absence of a müllerian tissue response to the action of AMH due to the presence of high estrogen levels (secondary to peripheral testosterone conversion), or (3) early testicular descent, which places müllerian structures outside the range of action of AMH during embryogenesis.9 These hypotheses were evaluated in a 20-week fetus with prenatal diagnosis of CAIS and the presence of müllerian remnants. Three events were seen: first, increased AMH expression by Sertoli cells; second, decreased expression of peritubular mesenchymal cells positive for the AMH receptor (AMHR2); and third, Leydig cell hyperplasia. These findings suggest a new hypothesis with regard to AMH action during the embryonic period, based on prenatal resistance to AMH.10
In conclusion, the presence of minor müllerian structures in cases of CAIS may be more common than previously reported, but this should not affect diagnosis, and does not modify either its course or its treatment.
We thank Dr. Alejandro Martínez-Aguayo for his critical review of the manuscript.
Please cite this article as: Grob F, et al. Síndrome de insensibilidad completa a andrógenos con persistencia de restos mullerianos. Descripción de un caso. Endocrinol Nutr. 2013;60:216–8.
