


Mutations in the DAX-1 gene (DAX1) (chromosome X gene 1 of dosage-sensitive sex reversal congenital adrenal hypoplasia; also called NROB1 gene [Nuclear receptor subfamily 0, group B, member 1] MIM ID *300473) are responsible for adrenal failure and hypogonadotropic hypogonadism in patients with congenital adrenal hypoplasia (OMIM# 300200). Mutations involve a loss of expression of the steroidogenic acute regulatory protein (StAR) and LHβ mediated by repression of the steroidogenic factor (1SF-1), as well as a decreased GnRH expression.1 More than 100 mutations in DAX1 have been reported to date.1 This clinical condition should be ruled out in patients with salt-losing syndromes once common causes of adrenal failure, such as defects in steroidogenesis (CYP21A2) and metabolic changes (adrenoleukodystrophy), have been ruled out. We report the case of two brothers with the mutation (R267P) and genotypic–phenotypic heterogenicity.
Case 1A 2-month-old male with vomiting and signs of dehydration and a poor weight curve for the previous 15 days. Laboratory tests showed hyponatremia and hyperkalemia, normal cortisol levels, and no elevation in steroidogenic precursors (Table 1). Primary hyperaldosteronism was diagnosed, and replacement therapy was started with mineralocorticoids and supplemental sodium chloride, with an adequate initial clinical and biochemical response. At 17 months of life, the patient was diagnosed with an associated glucocorticoid deficiency based on progressive hyperpigmentation, growth curve stability with weight at the 50th percentile (P) and P25 height, and low plasma cortisol levels. There was no family history of autoimmune disease, abortions or stillbirths, or neurological disease, and the patient had no siblings at the time of diagnosis. Tests for anti-adrenal antibodies and very long chain fatty acids were negative. The growth curve was normal under treatment with glucocorticoids 10mg/m2 SC/day and mineralocorticoid 0.175mg/day until progressive growth deceleration with delayed bone maturation started at 8 years. At 13 years and nine months, the patient showed no development of secondary sexual characteristics, a combined pituitary test with no growth hormone (GH) response to an insulin-induced hypoglycemia test (basal GH 0.5ng/mL; maximum peak [mp] 1ng/mL) and to a clonidine test with steroid impregnation (basal GH<0.2ng/mL; mp 0.2ng/mL); gonadotropin and androgen levels suggesting hypogonadotropic hypogonadism, with an abnormal HCG test (basal total testosterone 0.1ng/mL; mp 1ng/mL). Adrenal and hypothalamo–hypophyseal MRI was unremarkable. At 14 years, with a bone age of 10 years, treatment was started with testosterone propionate 50mg/month (4 doses), with no growth increase. At 14 years and 7 months, when the patient had a height of 142.6cm (cm/year, and a bone age of 10–11 years, treatment with recombinant growth hormone (rGH) was added. Growth velocity increased to 8cm/year in the first year and subsequently decreased to 6.5–4.7cm/year. At 20 years, the patient had a height of 170cm (P25; target height 167cm) and had adult secondary sexual characteristics. Treatment with rGH is currently continued for adult GH deficiency.
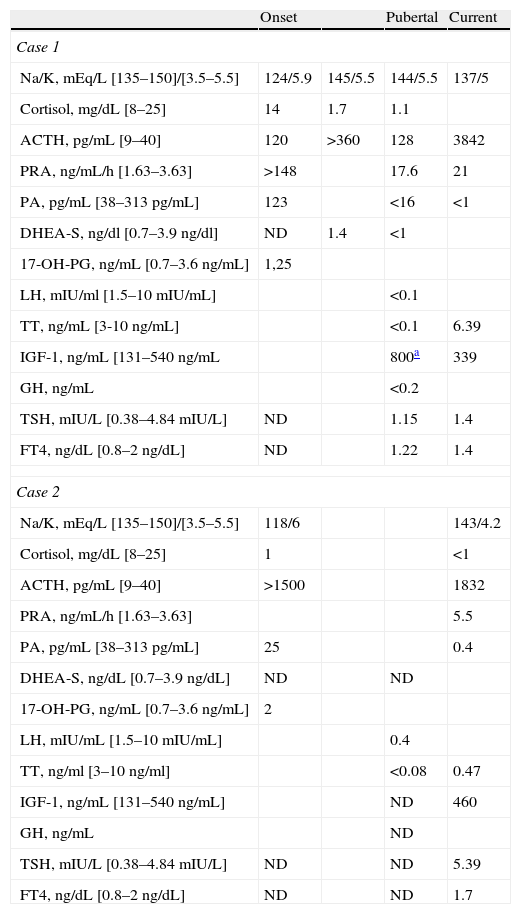
Basal biochemical and hormone measurements during follow-up.
| Onset | Pubertal | Current | ||
| Case 1 | ||||
| Na/K, mEq/L [135–150]/[3.5–5.5] | 124/5.9 | 145/5.5 | 144/5.5 | 137/5 |
| Cortisol, mg/dL [8–25] | 14 | 1.7 | 1.1 | |
| ACTH, pg/mL [9–40] | 120 | >360 | 128 | 3842 |
| PRA, ng/mL/h [1.63–3.63] | >148 | 17.6 | 21 | |
| PA, pg/mL [38–313pg/mL] | 123 | <16 | <1 | |
| DHEA-S, ng/dl [0.7–3.9ng/dl] | ND | 1.4 | <1 | |
| 17-OH-PG, ng/mL [0.7–3.6ng/mL] | 1,25 | |||
| LH, mIU/ml [1.5–10mIU/mL] | <0.1 | |||
| TT, ng/mL [3-10 ng/mL] | <0.1 | 6.39 | ||
| IGF-1, ng/mL [131–540ng/mL | 800a | 339 | ||
| GH, ng/mL | <0.2 | |||
| TSH, mIU/L [0.38–4.84mIU/L] | ND | 1.15 | 1.4 | |
| FT4, ng/dL [0.8–2ng/dL] | ND | 1.22 | 1.4 | |
| Case 2 | ||||
| Na/K, mEq/L [135–150]/[3.5–5.5] | 118/6 | 143/4.2 | ||
| Cortisol, mg/dL [8–25] | 1 | <1 | ||
| ACTH, pg/mL [9–40] | >1500 | 1832 | ||
| PRA, ng/mL/h [1.63–3.63] | 5.5 | |||
| PA, pg/mL [38–313pg/mL] | 25 | 0.4 | ||
| DHEA-S, ng/dL [0.7–3.9ng/dL] | ND | ND | ||
| 17-OH-PG, ng/mL [0.7–3.6ng/mL] | 2 | |||
| LH, mIU/mL [1.5–10mIU/mL] | 0.4 | |||
| TT, ng/ml [3–10ng/ml] | <0.08 | 0.47 | ||
| IGF-1, ng/mL [131–540ng/mL] | ND | 460 | ||
| GH, ng/mL | ND | |||
| TSH, mIU/L [0.38–4.84mIU/L] | ND | ND | 5.39 | |
| FT4, ng/dL [0.8–2ng/dL] | ND | ND | 1.7 | |
ACTH: adrenocorticotropic hormone; PA: plasma aldosterone; PRA: plasma renin activity; DHEA-S. dehydroepiandrosterone sulphate; 17-OH-PG: progesterone; IGF-1: insulin-like growth factor; LH: luteinizing hormone; ND: no data available; FT4: free T4; TSH: thyroid-stimulating hormone; TT: total testosterone.
A male diagnosed at 18 months with adrenal insufficiency due to salt-losing syndrome (Table 1). Two weeks before diagnosis, after receiving the oral DTP and polio vaccine, the patient showed a low mood and progressive anorexia, with abdominal pain at night and foul-smelling soft stools. Weight and height gain was adequate until 16–17 months of age, when a flat weight curve, weight<P3, height between P25 and P50, and hyperpigmentation were seen. An 8-year-old brother had been diagnosed with primary adrenal insufficiency (case 1). Tests for anti-adrenal antibodies and very long chain fatty acids were negative. The patient had a normal weight and height evolution under treatment with glucocorticoids at 10mg/m2 SC/day and mineralocorticoid at 0.125mg/day, and received oral NaCl supplements in the first year following diagnosis. At 12 years, a gradual decrease in growth velocity was seen, as well as no progression of pubertal development. At 14 years, a combined pituitary test showed a somatotropic axis with no changes and hypogonadotropic hypogonadism (basal and mp LH<0.07mIU/mL, basal and mp FSH 0.6/0.8IU/L, basal total testosterone <0.08ng/mL). Testosterone propionate (50mg/month) was started for pubertal induction, leading to increased growth and bone maturation and progressive development of secondary sexual characteristics. At 17 years of age, the patient had a height of 165.5cm (P15) and a bone age of 14.5 years, with a persistent growth velocity of 6cm/year.
A molecular study of both patients and their parents by direct DAX1 sequencing using various pairs of primers for exon 1 amplification showed that both the index case (case 1) and his younger brother had in hemizygosis a change in position 800 (c.800G>C), which caused substitution at protein level p.Arg267Pro (NR0B1.0004, ARG267PRO). A DNA study of the mother confirmed the presence of the same mutation in heterozygosis. No mutation was found in the father.
Chromosome X-linked heredity is usually recessive for women. In the reported case, the mother had the mutation in heterozygosis and was therefore a carrier, because the normal dominant allele prevented the expression of the affected gene. The XY sons had the mutation in hemizygosis and were therefore affected by the condition.
There is a phenotypic heterogenicity associated with DAX1 mutations. The lack of genotype–phenotype correlation in some mutations is presumably caused by the influence of other modifying genes, which leads to significant within-family variations in age at onset and expression.2 Onset usually occurs at an earlier age in the younger brother. Family history usually reveals the presence of unexplained deaths of children during infancy or siblings with congenital adrenal hypoplasia. The form of presentation is, in most cases, a salt-losing syndrome with hyponatremia, hyperkalemia, and metabolic acidosis in the first months of life, preceded by the loss of a channel of growth. Subjects may erroneously be diagnosed with CYP21A2 deficiency, isolated hypoaldosteronism, or pseudohypoaldosteronism.2 While basal cortisol at diagnosis may vary, ACTH is invariably elevated (case 1) and there is an inadequate cortisol elevation in response to the ACTH stimulation test. Aldosterone deficiency precedes hypocortisolism in most patients (case 1).3 There have been reports of cases of transient isosexual precocity in infancy and childhood with high testosterone levels for age, penile elongation, sometimes associated with testicular enlargement, and no other signs of sexual development; some mechanisms proposed for this phenomenon implicate the NR0B1 gene in the prepubertal control mechanism of the gonadal axis, ACTH-mediated stimulus of testicular steroidogenesis, or autonomous hyperplasia of Leydig cells.2 In the pubertal period, these patients will require testosterone replacement for the development of secondary sexual characteristics. Since DAX1 abnormalities may affect testicular development and spermatogenesis, fertility treatment using pulsatile GnRH and gonadotropins is often ineffective.4 Mental disability (motor, speech, and social behavior) may also occur. To our knowledge, there are no cases reported in the literature of an association between DAX1 and GH deficiency as seen in case 1.
DAX1 mutations account for 58% of cases of primary adrenal insufficiency of “unknown etiology” in children (newborn-13 years) in whom autoimmune causes, deficient steroidogenesis, or metabolic causes have been ruled out.5 Although it does not change the therapeutic strategy, molecular diagnosis allows for genetic counseling to relatives and is warranted in children with the onset of a salt-losing syndrome of unknown etiology, with or without associated cortisol deficiency. High clinical suspicion is required to prevent erroneous diagnosis and to allow for an adequate therapeutic approach.
Please, cite this article as: Sánchez-Pacheco M, et al. Hipoplasia adrenal congénita e hipogonadismo hipogonadotropo: variabilidad fenotípica de la mutación R267P del gen DAX-1. Endocrinol Nutr. 2012;59:140–2.
