


Symptomatic diabetes as a manifestation of another underlying condition is uncommon, and usually due to the use of drugs with hyperglycemic potential or to neoplastic processes in the pancreatic area. Among drugs, special mention should be made of high-dose steroids and, more recently and in young patients, atypical neuroleptics.1 The onset of cardinal symptoms of diabetes mellitus (DM) in a patient under 40 years of age, with no other organ-specific clinical signs or concomitant treatment suggests autoimmune diabetes (type 1A) as the first diagnostic possibility, while in elderly patients it should rule out an underlying neoplastic condition. We report the case of an apparent onset of type 1 DM in which subsequent evaluation showed an unexpected, reversible etiological diagnosis.
A 38-year-old male patient with unremarkable personal or family history was referred from the emergency room for hyperglycemia with ketosis. The patient reported urinary frequency and severe polydipsia for the previous three weeks, and the loss of 12kg in the previous year with no decreased appetite or other symptoms. The results of tests performed in the emergency room showed blood glucose 340mg/dL, ketone bodies in urine 50mg/dL, and pH 7.4, weight 56.5kg (BMI 17kg/m2), blood pressure 145/95mmHg, and heart rate 100 beats per minute, with no other physical examination findings. Basal-bolus insulin therapy (glargine and aspart) and diabetes education were started, and good glycemic control was achieved. Glycosylated hemoglobin at onset was 13.4%, with negative anti-IA2 (insulinoma antigen 2) and anti-GAD (glutamic acid decarboxylase) antibodies, and a basal C-reactive peptide level of 2.2ng/mL. Diagnosis of type 1A DM was therefore discarded, and additional tests were performed to rule out secondary causes.
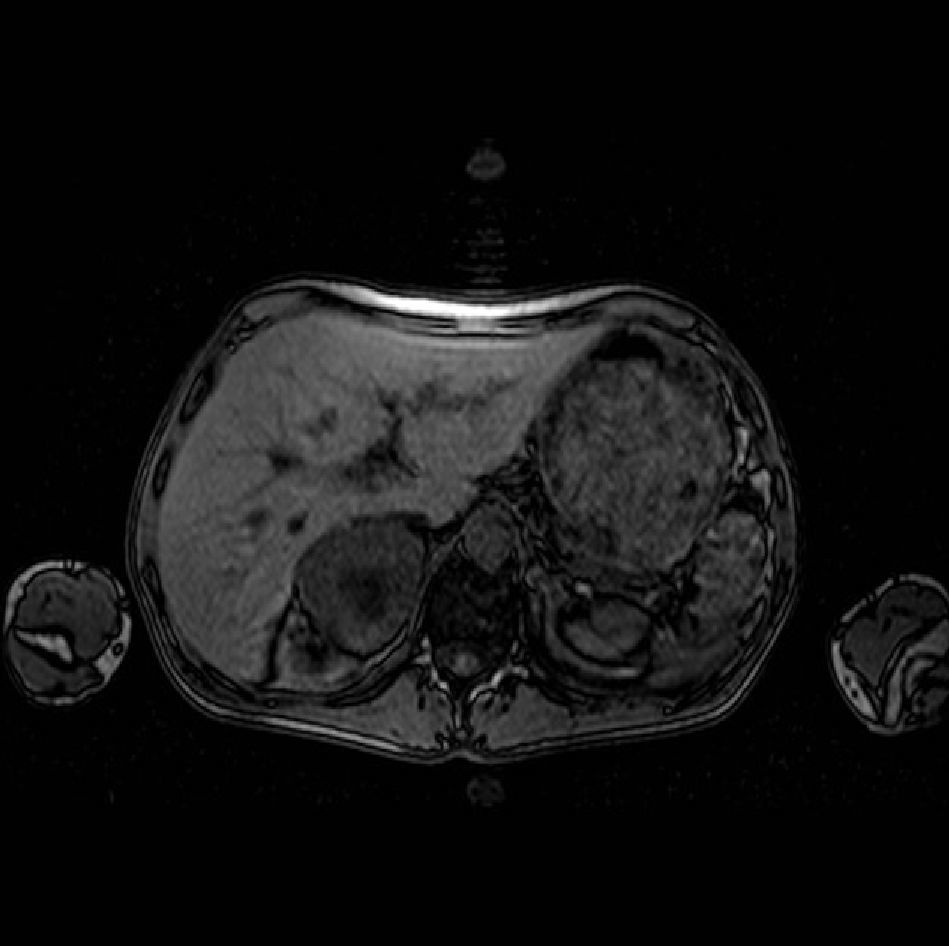
Levels of thyroid hormones, cortisol, calcitonin, calcium, and phosphorus were normal. Abdominal ultrasound revealed a 7-cm right adrenal mass, which magnetic resonance imaging confirmed to be a heterogeneous tumor with a necrotic center (Fig. 1). The results of measurements of catecholamines and their metabolites in 24-h urine were as follows: norepinephrine 767μg/24h (normal [N]: 12.1–85.5), epinephrine 270μg/24h (N: 1.7–22.4), dopamine 461μg/24h (N: 0–498), normetanephrines 3245μg/24h (N: 88–444), and metanephrines 2299μg/24h (N: 52–341). After preoperative preparation with phenoxybenzamine (no beta-blockers were required), laparoscopic adrenalectomy was successfully performed at four weeks. A pathological study confirmed a 7.5cm×5cm×5cm pheochromocytoma, 138g in weight, without capsular invasion. Insulin requirements dramatically decreased after surgery, and hypoglycemic therapy was not required after a few days. At the time of writing, the patient remains asymptomatic, euglycemic, and with normal blood pressure values He has recovered his normal weight and his hormone levels have remained normal.

Pheochromocytoma is a catecholamine-secreting tumor derived from the enterochromaffin cells of the adrenal medulla. It is an uncommon tumor, diagnosed in approximately 1–2 cases per 100,000 inhabitants a year,2 although its prevalence is greater (0.05–0.1%) in autopsy studies.3,4 Clinical presentation widely ranges from asymptomatic patients to severe hypertensive crises.5 Although the classical triad of palpitations, headache, and sweating is highly specific for diagnosis, most patients have sustained or have paroxysmal arterial hypertension5 with no other associated clinical signs. The prevalence of any change in carbohydrate metabolism in patients with pheochromocytoma is highly variable, according to different studies. In a series of 60 patients, 24% had DM and a clear relationship was seen between urinary catecholamine and blood glucose levels.6 In another series of 191 patients, its prevalence was a little higher than 35%.7 By contrast, in a study conducted on 1093 diabetic patients with abdominal ultrasound and glucagon tests, pheochromocytoma was diagnosed in 0.96/1000 patients,8 a prevalence similar to that found in the hypertensive population (1/1000).2 Although hypoglycemia secondary to pheochromocytoma is not an uncommon finding, its onset with cardinal clinical signs is rare, and only four patients with ketoacidosis have been reported in the literature.9 Although our patient had no acidosis, hyperglycemia and ketosis were very significant and, because of the patient's age, suggested the onset of classical type 1 diabetes. The absence of the characteristic clinical signs of pheochromocytoma, except for hypertension, delayed final diagnosis.
In pheochromocytoma, carbohydrate changes are due to multiple factors. There is, on the one hand, decreased insulin secretion, increased glucagon levels, and stimulation of glucogenolysis secondary to increased norepinephrine levels, and on the other hand, decreased peripheral glucose uptake and increased hepatic gluconeogenesis secondary to excess epinephrine. After tumor resection, a majority of patients do not require hypoglycemic treatment.10
In conclusion, the variable presentation of these tumors should be emphasized. In this case, the only sign was gradual weight loss until the onset of the cardinal symptoms of diabetes mellitus. Pheochromocytoma should therefore be considered in young patients with arterial hypertension and non-autoimmune diabetes mellitus.7,8
Conflict of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Egaña Zunzunegui N, et al. Debut de diabetes mellitus en paciente joven: ¿diabetes tipo 1? Endocrinol Nutr. 2012;59:275–6.
