


Los tumores endocrinos son un grupo muy heterogéneo de neoplasias. Principalmente los tumores neuroendocrinos pueden presentarse agrupados en síndromes tumorales hereditarios, transmitidos con carácter autosómico dominante. En la actualidad están bien tipificados y se conocen las alteraciones genéticas responsables de los siguientes síndromes: MEN-1, MEN-2, síndrome de Von Hippel Lindau, neurofibromatosis tipo 1, esclerosis tuberosa, complejo de Carney, síndrome de hiperparatiroidismo y tumor de mandíbula, y síndrome de paraganglioma-feocromocitoma familiar1.
Presentamos el caso de una paciente con varios tumores de glándulas endocrinas, que a priori podría considerarse como un caso especial de síndrome tumoral endocrino.
Se trata de una mujer de 65 años remitida al Servicio de Endocrinología de Jaén por sospecha de hiperparatiroidismo primario e hirsutismo postmenopáusico. Sus antecedentes familiares incluían madre diabética tipo 2 y dos hermanos hipertensos. Entre sus antecedentes personales destacaban HTA, vitíligo, cólicos nefríticos y menopausia a los 51 años. Refería incremento del vello facial e hipertricosis en extremidades, caída de cabello frontoparietal y aumento de la libido desde 3 años antes. No existían datos de disfunción tiroidea ni cambios en el cuello.
En la exploración física objetivamos: peso 78kg, IMC 32kg/m2, tensión arterial 120/70mmHg, alopecia androgénica e hirsutismo grado 3 en la escala de Ferriman-Gallwey2 con hipertricosis muy marcada en extremidades.
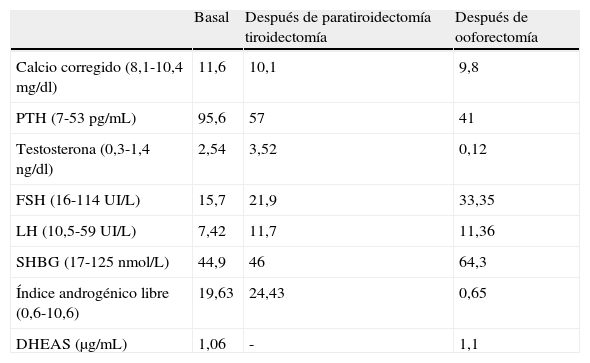
La analítica destacaba hipercalcemia con PTH elevada, así como testosterona por encima de la normalidad, sin otros datos patológicos (tabla 1). La gammagrafía con Tc-99m sestamibi mostró un depósito patológico en paratiroides superior derecha. La ecografía tiroidea mostró un nódulo de 2,4×2×1,5cm en istmo, de características benignas. Concluimos el estudio con ecografía transvaginal y TAC abdomino-pélvico sin hallazgos patológicos.
Datos analíticos antes y después de las cirugías (tiroidectomía y ooforectomía)
| Basal | Después de paratiroidectomía tiroidectomía | Después de ooforectomía | |
| Calcio corregido (8,1-10,4mg/dl) | 11,6 | 10,1 | 9,8 |
| PTH (7-53pg/mL) | 95,6 | 57 | 41 |
| Testosterona (0,3-1,4ng/dl) | 2,54 | 3,52 | 0,12 |
| FSH (16-114UI/L) | 15,7 | 21,9 | 33,35 |
| LH (10,5-59UI/L) | 7,42 | 11,7 | 11,36 |
| SHBG (17-125nmol/L) | 44,9 | 46 | 64,3 |
| Índice androgénico libre (0,6-10,6) | 19,63 | 24,43 | 0,65 |
| DHEAS (μg/mL) | 1,06 | - | 1,1 |
Ante el diagnóstico de hiperparatiroidismo primario se indicó paratiroidectomía. Durante la cirugía, se detectó un nódulo tiroideo sospechoso de malignidad, con biopsia intraoperatoria sugerente de carcinoma papilar tiroideo, que motivó una tiroidectomía total. Tras la cirugía, la paciente recibió ablación con I-131.
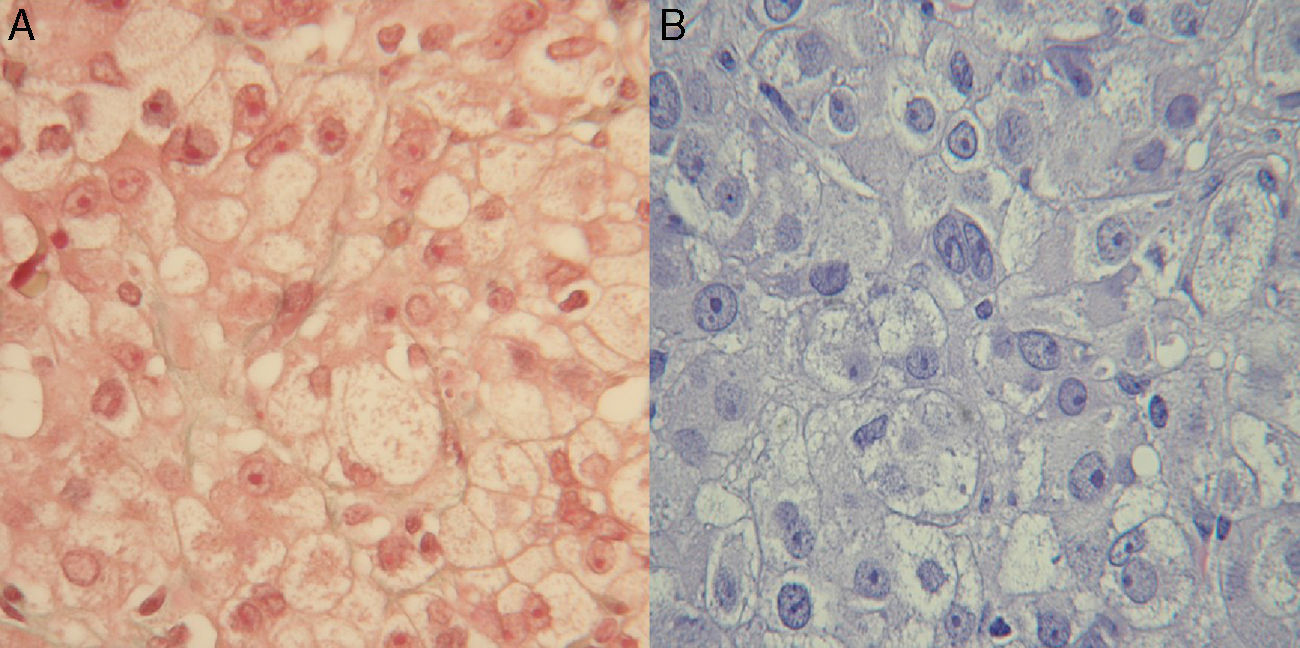
En revisiones posteriores la clínica de hiperandrogenismo persistía, e incluso empeoró. Ante la sospecha de un origen ovárico, y dado que las pruebas de imagen habían sido negativas, se remitió a Ginecología para una laparoscopia exploradora. Se realizó una ooforectomía bilateral, con diagnóstico histológico de tumor de células esteroides en ovario derecho, y de hipertecosis y quistes foliculares en ovario izquierdo (fig. 1).
 Tricómico. B) H & E.")
Tras la ooforectomía, la paciente mejoró de forma manifiesta del hiperandrogenismo, con recuperación parcial del cabello y disminución del hirsutismo. En el estudio hormonal realizado 3 meses después los parámetros bioquímicos y hormonales se habían normalizado (tabla 1).
Dos años después de la ooforectomía, la paciente fue derivada nuevamente a Endocrinología por clínica cardinal de diabetes y pérdida progresiva de peso (10kg en 4 meses), con hiperglucemia de 342mg/dl acompañada de glucosuria y cetonuria, por lo que se inició insulinoterapia en su centro de salud. Su estudio mostró HbA1c 12,1%, anticuerpos anti GAD y anti IA2 negativos y péptido C 1,4ng/dl.
En poco tiempo mejoró el control metabólico y la clínica, la dosis de insulina se fue reduciendo hasta suspenderla al cabo de un año, y actualmente se mantiene en tratamiento con antidiabéticos orales con excelente control metabólico (último valor de HbA1c: 6,2%). Si bien inicialmente se pensó en una diabetes subtipo LADA por la severidad clínica de la hiperglucemia, la posterior evolución y los antecedentes familiares orientaron el diagnóstico hacia una diabetes mellitus tipo 2.
En conjunto, esta paciente fue diagnosticada de hiperparatiroidismo primario (HPP), carcinoma papilar de tiroides, tumor ovárico de células esteroides y diabetes mellitus. Estas patologías pueden darse con carácter esporádico, pero llama la atención que confluyan todas ellas fuera del contexto clínico de alguno de los síndromes tumorales endocrinos descritos hasta el momento.
Es conocida la asociación entre HPP y cáncer de tiroides: de tipo medular en el caso de los síndromes MEN I y IIA, y de tipo diferenciado fuera de estos síndromes. Existen varios casos publicados en los que durante la cirugía del HPP se descubre un carcinoma papilar de tiroides, estimándose la incidencia de HPP combinado con cáncer tiroideo no medular entre el 2-13%3. Permanece la controversia sobre si ambas patologías se diagnostican de forma coincidente o son causadas por factores de riesgo y/o cambios genéticos comunes. Se ha descrito la mutación de un protooncogén, responsable de la aparición simultánea de carcinoma papilar y medular de tiroides junto con HPP4. Por todo ello, se recomienda que ante un HPP se realice una ecografía cervical para descartar nódulos tiroideos subsidiarios de estudio por sospecha de carcinoma de tiroides no medular.
Nuestra paciente presentó concomitantemente un cuadro de hiperandrogenismo, que dada su obesidad y el antecedente de HPP podría haber estado relacionado con el síndrome de insulinorresistencia presente en ambas situaciones5,6. Por otra parte, está descrita la presencia de tumores hipofisarios productores de gonadotropinas asociados al MEN I, que puede ocasionar hiperestimulación ovárica7,8. Sin embargo, en nuestro caso el hallazgo de gonadotropinas anormalmente bajas para la edad y estado de menopausia de la paciente orientaron el diagnóstico hacia la patología ovárica.
El tumor ovárico extirpado, llamado tumor de células esteroides, de Grawitz o corticosuprarrenaloma, se denomina así por contener residuos de corteza adrenal en su histología9. Aparece típicamente en mujeres adultas, en especial tras la menopausia. En el 70-80% de los casos el tumor tiene actividad androgénica y es virilizante; un pequeño porcentaje puede secretar estrógenos, y se han descrito cuadros de hipercorticismo, incluida la aparición de diabetes10, si bien en nuestro caso esta se desarrolló 2 años después de la ooforectomía.
Nuestra pregunta es hasta qué punto estos procesos son independientes entre sí y se asocian de forma fortuita, o bien si existe algún factor aún no identificado responsable de cierta predisposición al desarrollo de este tipo de endocrinopatía.
