


A 14 year-old male patient was referred to the endocrinology outpatient clinic for hypogonadism. He had an unremarkable family and personal history. He was born in China, and had lived in Spain with his biological family since the age of three.
This boy was referred by the urology outpatient clinic due to delayed genital development possibly due to hypergonadotropic hypogonadism with a total testosterone level of 3.7nmol/L (reference range: 9.9–27.8), FSH 18.3IU/L, and LH 13.3IU/L (1–25).
His family reported poor academic performance, difficulties in relations with other students, and progressive behavioral changes (attention deficit, irritability, impulse control problems) in the previous two years.
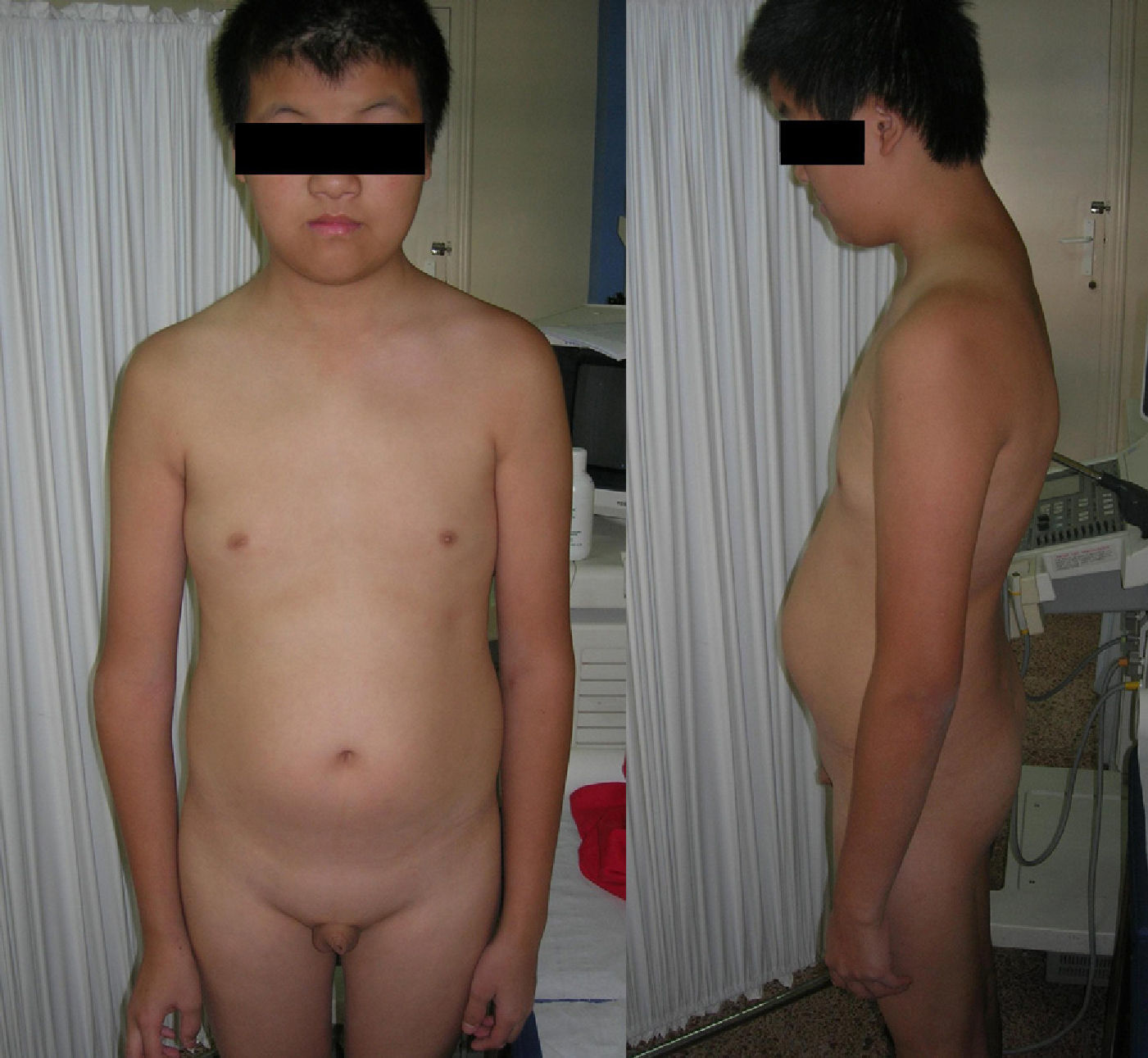
Physical examination found a weight of 50.5kg, a height of 169cm (97th percentile of height and 50th percentile of weight for Chinese adolescents of similar chronological age),1 and an arm span of 173cm. He had a longilinear body habitus, pectus excavatum, kyphoscoliotic pattern, prominent elbows, and increased abdominal fat. Low hair implantation, abnormal ears, hypertelorism, and epicanthus were also found. As regards secondary sexual characteristics, he had no pubic and axillary hair and showed infantile genitalia (2-mL testes, 3-cm penis). The rest of the examination was unremarkable (Fig. 1).

Based on these findings, laboratory tests were requested, including a hormone profile, X-rays of the left hand, and karyotype. The patient was also referred to the infantile mental health unit (USMI). Results of supplemental tests were as follows: total testosterone, 5.5nmol/L; sex hormone-binding globulin (SHBG), 29.9nmol/L (NV 10–80); LH, 29.6IU/L; FSH, 37.3IU/L; 17-beta-estradiol, 24pmol/L (20–1800); TSH, 3.93μU/mL (0.4–4); total cholesterol, 106mg/dL (150–200); and triglycerides, 58mg/dL (70–170). Basic blood chemistry, liver tests, and complete blood count were normal. Bone age was 13 and a half years. Karyotype showed polysomy 48,XXXY. The patient was assessed by the clinical psychology team of USMI and reported to have mild mental retardation (intelligence quotient (IQ) of 62 with marked retardation in the speech area) and difficulties in cultural adaptation.
A diagnosis of partial hypergonadotropic hypogonadism due to Klinefelter-like syndrome secondary to aneuploidy 48,XXXY was therefore made. Treatment was started with testosterone cypionate 50mg every 4 weeks by the intramuscular route, with three-monthly dose titration based on total testosterone and gonadotropin (FSH and LH) levels. Drug treatment was well tolerated and had no influence on behavior. Based on recommendations by the USMI, the patient entered a specific support and follow-up program at school.
Klinefelter syndrome encompasses a group of disorders characterized by the presence of at least one X chromosome additional to the normal male karyotype, 46,XY. The classical form is a karyotype 47,XXY, but there are other much more uncommon variants, such as those caused by aneuploidies 48,XXYY, 49,XXXXY, and 48,XXXY.2 The 47,XXY is the most common chromosome abnormality in humans, with an incidence of one case per 650 males born. The incidence of the 48,XXXY variant is very low and is estimated at approximately 1:50,000 males born.3
While patients with these polysomies share common clinical characteristics, there are differential traits between the different forms reported. This syndrome is traditionally characterized by tall height, narrow shoulders, gynecomastia, decreased testicular size and penis length, facial dysmorphism, hypergonadotropic hypogonadism, and mental retardation, among other changes. These traits vary depending on the underlying chromosome abnormality and specifically on the number of surplus X chromosomes. Thus, patients with polysomy 48,XXXY have higher mean heights as compared to those with polysomy 47,XXY (190cm versus 179–188cm) and are more likely to have congenital malformations such as radioulnar synostoses or clinodactyly.3,4 Facial and body dysmorphism (hypertelorism, epicanthus, narrow lid opening, low hairline implantation, flat feet, joint hyperextensibility and hyperlaxity) are also more common in polysomy 48,XXXY. Decreased testicular volume (less than 3mL and usually lower than 1.5mL) is a constant trait, and is also more likely in patients with polysomy 48,XXXY.3 Other frequent changes include metabolic syndrome, type 2 diabetes mellitus, osteoporosis, and breast cancer. It is not known whether the risk of these changes is greater in variants of Klinefelter syndrome.5
As regards the cognitive sphere, significant intellectual disability is uncommon in patients with polysomy 47,XXY, in whom mean IQ ranges from 89 and 102. By contrast, more than 50% of patients with aneuploidy 48,XXXY have a variable mental retardation (IQ 40–75). Learning, speech, and motor retardations are more common in variant 48,XXXY, but also appear in the classical form.2,6–8 An immature, passive, cooperative, and not particularly aggressive behavior has been reported in patients with the 48,XXXY variant.2
In children and adolescents, Klinefelter syndrome should be suspected in the presence of any of the following clinical signs: retarded development, reduced testicular size, gynecomastia, longilinear body habitus, deficient verbal communication, academic problems, difficulties in psychosocial relations and/or behavioral changes. A karyotype is indispensable for confirmation of the diagnosis.2
When Klinefelter syndrome is suspected at the first visit of a patient, a complete physical examination is recommended. Sex hormone (estrogens, testosterone, SHBG, FSH, and LH) and routine laboratory tests (plasma glucose, lipid profile, thyroid hormones, and complete blood count) should be performed. An oriented history focusing on the practice of physical exercise, sexuality, and libido should be collected.5 Patients should be monitored every three months and, once replacement therapy is stabilized, once a year. Because of the increased risk of developing osteoporosis and hypoparathyroidism, regular monitoring of vitamin D, calcium, phosphorus, and bone densitometry is recommended.5
Most of these patients are not diagnosed in childhood. However, the lack of development of secondary sexual characteristics with puberty demonstrates the existence of an underlying disease. Testosterone replacement therapy is aimed at promoting the development and maintenance of secondary sexual characteristics, improving sexual function, and increasing muscle mass and strength, as well as preventing excess height. This treatment has been reported as possibly having positive effects on character, behavior, and concentration.5 An additional advantage is based on its preventive effect upon the development of osteoporosis, obesity, metabolic syndrome, and diabetes. Replacement therapy will not increase testicular size in any case.2,5,9
In prepubertal males with Klinefelter syndrome and evidence of androgen deficiency, testosterone replacement therapy should be started at about 12 years of bone age. To avoid excessively rapid virilization and bone maturation, treatment should be started with parenteral preparations at low doses (e.g. testosterone cypionate or enantate 50mg every 4 weeks), and replacement doses should be increased gradually. The dosage should initially be adjusted every three months, and once both gonadotropin and testosterone levels are within the normal range, adjustments may be made annually.5,9
Klinefelter syndrome is a common endocrine disease in its classical form, but there are rare variants of the syndrome with differential characteristics. Early diagnosis allows for the identifying of difficulties in the educational and speech areas and for starting hormone replacement therapy at the appropriate time, thus improving substantially the quality of life of these patients.5
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Romero Lluch A, et al. Alteraciones endocrinológicas y psicológicas en la polisomía 48,XXXY. Endocrinol Nutr. 2012;59:396–8.
