


La presentación clínica más frecuente del hiperparatiroidismo primario (HP) es la hipercalcemia asintomática, siendo cada vez más infrecuente hacer el diagnóstico en presencia de manifestaciones óseas como la osteítis fibrosa quística (OFQ). La OFQ se presenta en menos del 5% de los pacientes con HP indicando una enfermedad más grave o de larga evolución. Se caracteriza por la aparición de dolor óseo junto con el hallazgo de alteraciones radiológicas específicas como incremento de la resorción ósea subperióstica en el tercio distal del radio y de las falanges medias, adelgazamiento distal clavicular, aspecto en «sal y pimienta» del cráneo, quistes óseos y tumores pardos en huesos largos. Los tumores pardos son el producto de la desmineralización ósea con activación de los osteoclastos, microhemorragias y microfracturas y reciben dicho nombre por la coloración típica secundaria a los depósitos de abundante hemosiderina. Histopatológicamente existe una combinación de actividad osteoclástica y osteoblástica con formación de quistes y numerosos macrófagos cargados con hemosiderina1. El diagnóstico diferencial de los tumores pardos incluye el granuloma reparativo de células gigantes y el tumor óseo de células gigantes (TCG).
Se describe el caso de un paciente con HP por un adenoma paratiroideo con tumores pardos que simulaban un TCG metastásico.
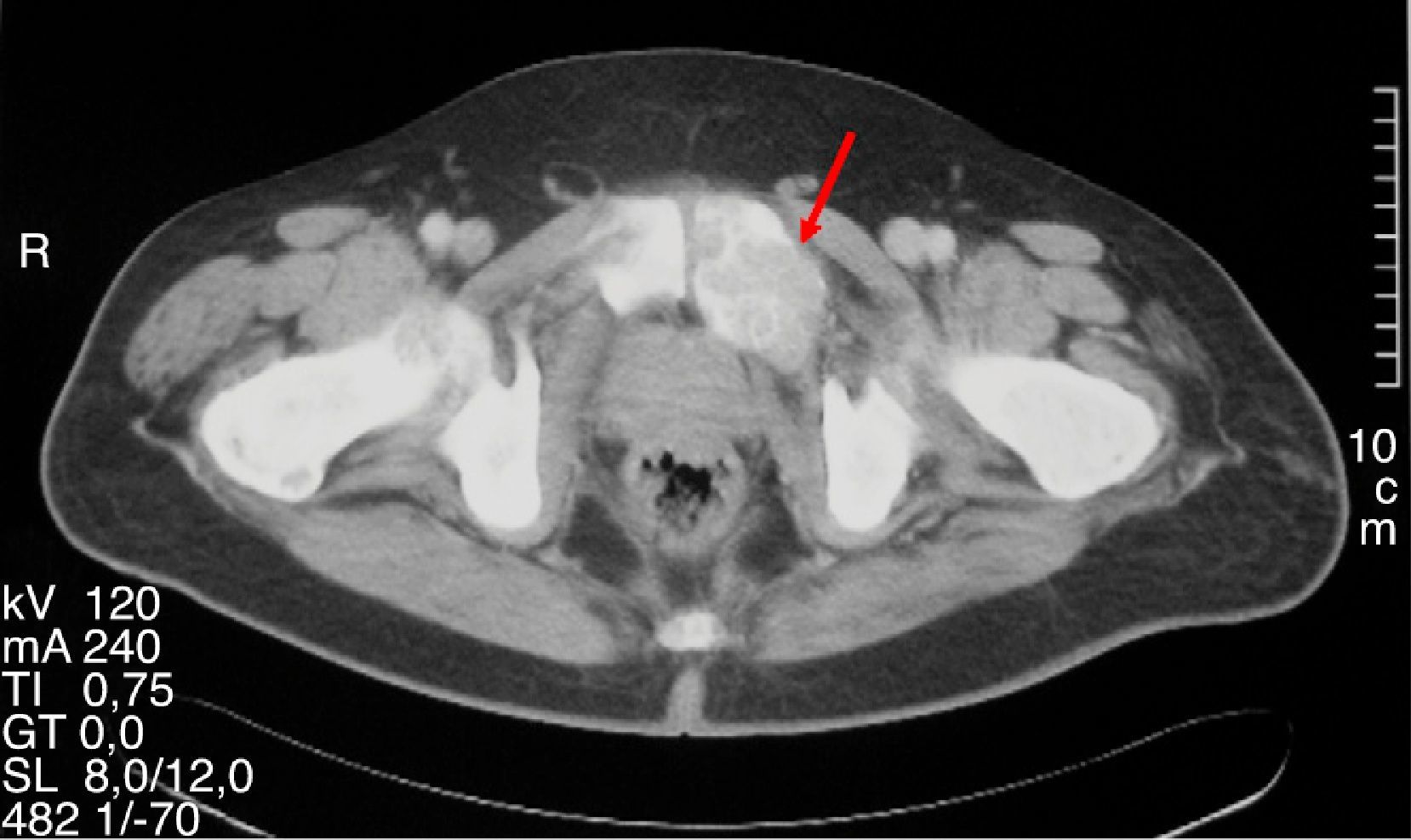
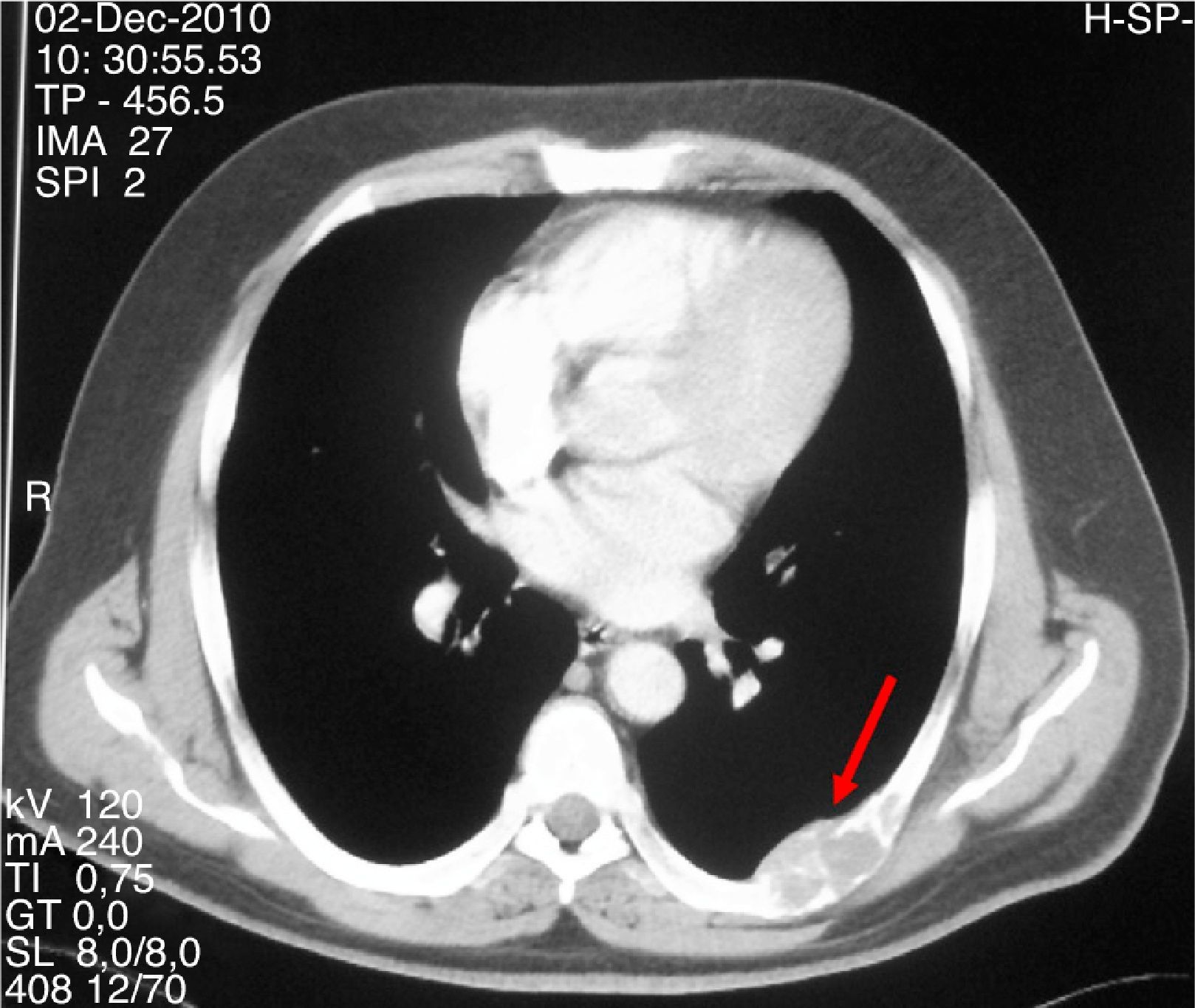
Varón de 47 años que consultó por primera vez al Servicio de Traumatología de un hospital de Castilla-La Mancha en mayo de 2008 por dolor en la cadera y mano izquierda no asociado a traumatismo previo. Refería antecedentes personales de dislipemia, diabetes mellitus tipo 2, hipertensión arterial, obesidad grado i y cólico nefrítico con litiasis de oxalato cálcico. Como antecedentes familiares, 2 de sus hermanas habían sido diagnosticadas y operadas de HP por adenoma. Se realizó una radiografía simple de cadera y mano observando una imagen polilobulada quística que insuflaba y adelgazaba la cortical del tercer metacarpiano izquierdo y una lesión lítica supraacetabular y de la rama ilioisquiopubiana izquierda. En la TC de pelvis (noviembre 2008) se identificaron lesiones de gran tamaño en pala ilíaca, isquion, rama pubiana izquierda, ala sacra y cuello femoral derechos. Ante dichos hallazgos fue intervenido en julio y septiembre de 2009 realizando curetaje y relleno con injerto autólogo así como sustitutos óseos del tercer metacarpiano izquierdo y lesión supraacetabular izquierda. La anatomía patológica fue informada como TCG. Se realizaron controles con TC y RM de tórax y pelvis mostrando aumento de tamaño de las lesiones líticas polilobuladas y expansivas en pelvis (fig. 1), sacro, cuello femoral derecho y L5, con aparición de una nueva lesión en cabeza femoral derecha y 7.a arco costal izquierdo (fig. 2), cambios que fueron interpretados como progresión tumoral metastásica. Debido a la persistencia del dolor con imposibilidad total para la deambulación el paciente fue remitido a la Unidad de Tumores Óseos del Hospital Universitario La Paz, donde se revisaron las muestras de anatomía patológica concluyendo que las lesiones óseas eran altamente sugestivas de granulomas reparativos de células gigantes e histológicamente indistinguibles de tumores pardos, por lo que se sugirió descartar hiperparatiroidismo. El paciente fue derivado al Servicio de Endocrinología en noviembre del 2010 donde se amplió el estudio con analítica: calcio total 14mg/dl, calcio corregido 13,2mg/dl, calcio iónico 1,72mmol/L, fosfato 1,9mg/dl, magnesio 1,86mg/dl, calciuria 968,60mg/24 horas, creatinina 0,55mg/dl, PTHi 535pg/ml, vitamina D 13ng/ml. Se realizó TC corporal total que evidenció un nódulo de 1,5cm de diámetro en el lugar teórico de la paratiroides derecha y gammagrafía de paratiroides con 20 mCi de TC 99-sestamibi con hallazgos compatible con adenoma paratiroideo hiperfuncionante derecho. Asimismo, se descartó tanto bioquímica como morfológicamente la presencia de feocromocitoma asociado. Se realizó paratiroidectomía derecha cuya biopsia intraoperatoria fue informada como adenoma de paratiroides. El estudio histopatológico definitivo confirmó la presencia de un adenoma de paratiroides de 4,5 g de peso con dimensiones de 2,2×2 x 1,9cm.


Durante el postoperatorio el paciente presentó hipocalcemia sintomática requiriendo tratamiento con calcitriol y calcio con el que permanece en la actualidad. El paciente continúa seguimiento en el Servicio de Endocrinología, refiriendo importante mejoría sintomática y logrando deambulación con muletas. El estudio genético no encontró mutaciones en el gen MEN-1.
El TCG es un tumor altamente vascularizado que se localiza en las metáfisis o epífisis de los huesos largos o bien en la pelvis, sacro o vértebras2. La apariencia radiológica e histológica de los tumores pardos típicos de la OFQ puede simular estrechamente un TCG, como en el caso de nuestro paciente, siendo las manifestaciones clínicas y los resultados de laboratorio (PTHi) los que permiten hacer la diferenciación. Algunos autores3,4 han reportado casos de OFQ donde la primera sospecha, basada en la clínica y las imágenes radiológicas, fue la existencia de una enfermedad ósea metastásica secundaria. Sin embargo, en nuestro paciente el diagnóstico de un tumor óseo primario metastásico se había establecido en base a los hallazgos histológicos, mientras que la historia familiar, no especificada en los casos referidos, era congruente con un HP.
Por otro lado, el déficit de vitamina D se detecta frecuentemente en pacientes con HP3,5 y se asocia con una exacerbación de la presentación bioquímica y fenotípica de la enfermedad (mayores concentraciones séricas de PTH, grandes adenomas paratiroideos y mayor riesgo de fracturas), hecho que podría haber contribuido a la florida clínica de nuestro paciente.
Es conocido que las formas familiares de hiperparatiroidismo son infrecuentes (5%), siendo las causas más frecuentes los síndromes de neoplasia endocrina múltiple (MEN) tipo 1 y 2A, el síndrome hiperparatiroidismo-tumor mandibular (HTP-JT) y el hiperparatirodismo familiar aislado (FIHP)6. En el MEN 1 el hiperparatiroidismo es la presentación más precoz y frecuente (>90%), mientras que en el MEN 2A tiene una presentación tardía con una penetrancia baja. A pesar de que en nuestro paciente el estudio genético para MEN 1 fue negativo, hay que recordar que hasta en un 30% de los casos analizados el resultado puede ser un falso negativo, bien producto de patrones mutacionales en los que estén implicados diferentes regiones del gen o de mutaciones en genes todavía no conocidos que afecten la transcripción o acción de la menina7. Este hecho, asociado a la probable aparición asincrónica de distintos aspectos del MEN-1, hace necesario un seguimiento continuo. El MEN 2A es poco probable al no presentar compromiso neoplásico tiroideo ni feocromocitoma. En el diagnóstico diferencial se debería incluir también el HTP-JT por la gran afectación ósea y el gran tamaño del adenoma. El hallazgo final de un carcinoma paratiroideo hubiera apoyado este diagnóstico ya que su aparición es frecuente en el HTP-JT8. No obstante, la ausencia de lesiones fibro-óseas mandibulares o maxilares y renales lo hace poco factible. Por último, aunque el FIHP pudiera constituir en algunos casos una variante de otros síndromes hiperparatiroideos, no se puede excluir la posibilidad de que mutaciones en genes localizados en loci aún no identificados, diferentes a las descritas en los MEN 1 y 2, y en el HTP-JT, puedan causar este síndrome.
El caso aquí descrito es de interés al ilustrar la importancia de evaluar el metabolismo fosfo-cálcico y la función paratiroidea en todos los pacientes con lesiones óseas, sospechar la existencia de un posible HP en caso de lesiones sugestivas, e indagar acerca del probable componente genético subyacente, a través de la realización de una historia familiar y personal detallada.
