


Most neoplasms growing in the sella turcica are pituitary adenomas. The remaining neoplasms are benign tumors (craniopharyngiomas and meningiomas), malignant tumors (primary germ cell tumors, chordomas, lymphomas, and pituitary adenocarcinomas, or metastatic disease, mainly from breast and lung cancer), and other uncommon conditions (cysts, abscesses, arteriovenous fistulas, lymphocytic hypophysitis). Schwannoma accounts for up to 8% of primary brain tumors,1 and is usually associated with the cranial nerves and shows a special predilection for the vestibular portion of the 8th cranial nerve at the cerebellopontine angle, while intrasellar localization is exceptional.
We report here the 17th case of intrasellar schwannoma published to date since the one reported by Perone et al.,2 which mimicked a non-secreting pituitary adenoma both clinically and radiographically.
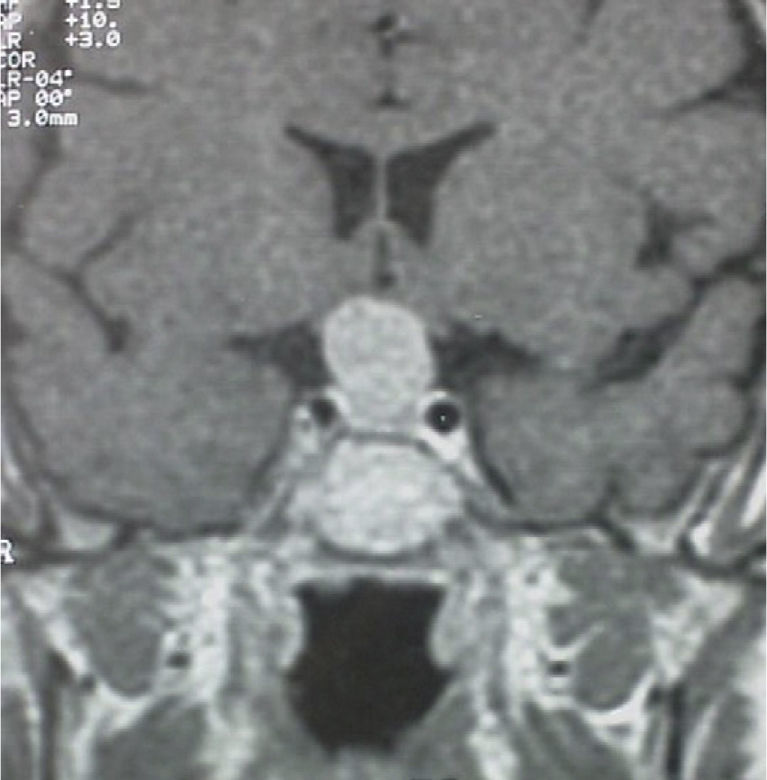
A 56-year-old female patient attended our clinic for a pituitary mass. Her family history was unremarkable. Her personal history included migraine and Ménière syndrome being monitored by the otolaryngology department, which had requested magnetic resonance imaging (MRI) of the brain that revealed a pituitary tumor, and cholecystectomy for cholelithiasis. The patient had had two uneventful pregnancies and menopause at 50 years of age. She reported no visual disturbances or clinical signs suggesting pituitary hypofunction or hyperfunction. Physical examination revealed blood pressure values of 110/70mmHg, heart rate of 78 beats per minute, height of 159cm, and weight of 62kg, with a body mass index of 24.5kg/m2. Patient hydration and color were normal. She had no acromegaloid traits or features suggesting Cushing's syndrome. No goiter was palpated. Cardiopulmonary auscultation and basic abdominal examination were normal, and there were no lower limb edema or signs of thrombosis. No marked visual field defects were found in confrontation campimetry. There was no galactorrhea, either spontaneous or on nipple expression. MRI of the head (Fig. 1) revealed a pituitary macroadenoma homogeneously enhanced following contrast injection and slightly compressing optic chiasma. No space-occupying lesions were identified in the cerebellopontine angles, and there were no changes in the 7th and 8th cranial nerves in both sides. Campimetry demonstrated bitemporal hemianopsia.

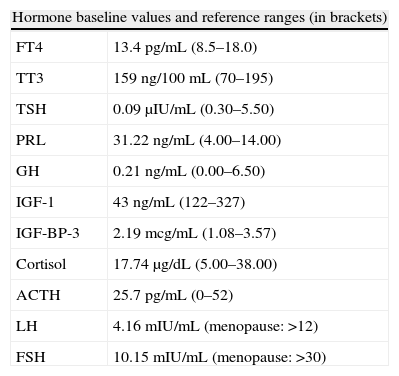
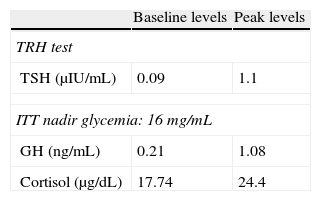
Pituitary hormone tests at baseline (Table 1) and after stimulation with thyrotropin-releasing hormone (TRH) and insulin (ITT) (Table 2) showed a preserved adrenal axis, mild hyperprolactinemia, gonadotropin levels inappropriately low for postmenopausal status, low levels of insulin-like growth factor type 1 (IGF-1) with no growth hormone (GH) response to insulin-induced hypoglycemia and low thyroid-stimulating hormone (TSH) levels, with poor response to stimulation with TRH but with normal free circulating tetraiodothyronine (T4) levels. A non-functioning pituitary adenoma with secondary TSH, growth hormone, and gonadotropin deficiency with mild hyperprolactinemia secondary to brainstem compression was diagnosed.
Pituitary function tests at baseline and after stimulation.
| Hormone baseline values and reference ranges (in brackets) | |
| FT4 | 13.4pg/mL (8.5–18.0) |
| TT3 | 159ng/100mL (70–195) |
| TSH | 0.09μIU/mL (0.30–5.50) |
| PRL | 31.22ng/mL (4.00–14.00) |
| GH | 0.21ng/mL (0.00–6.50) |
| IGF-1 | 43ng/mL (122–327) |
| IGF-BP-3 | 2.19mcg/mL (1.08–3.57) |
| Cortisol | 17.74μg/dL (5.00–38.00) |
| ACTH | 25.7pg/mL (0–52) |
| LH | 4.16mIU/mL (menopause: >12) |
| FSH | 10.15mIU/mL (menopause: >30) |
ACTH: corticotropin; FSH: follicle-stimulating hormone; GH: somatotropin; IGF-BP-3: insulin-like growth factor receptor binding protein-3; IGF-1: insulin-like growth factor-1; LH: luteinizing hormone; PRL: prolactin; FT4: free thyroxine; TSH: thyroid-stimulating hormone; TT3: total triiodothyronine.
Results of thyroid-stimulating hormone (TSH) stimulation with thyrotropin-releasing hormone (TRH) and somatotropin (GH) and cortisol stimulation test with insulin (ITT).
| Baseline levels | Peak levels | |
| TRH test | ||
| TSH (μIU/mL) | 0.09 | 1.1 |
| ITT nadir glycemia: 16mg/mL | ||
| GH (ng/mL) | 0.21 | 1.08 |
| Cortisol (μg/dL) | 17.74 | 24.4 |
The patient was referred to Neurosurgery for tumor resection, which was performed by a transsphenoidal approach. Intraoperative study suggested a pituitary adenoma, but final study of paraffin-embedded tissue showed pituitary tissue with a trabecular and acinar pattern without atypia, with variable expression of GH, prolactin (PRL) and, to a lesser extent, TSH, follicle-stimulating hormone (FSH), and adrenocorticotropic hormone (ACTH). Final diagnosis was of non-neoplastic pituitary tissue, consistent with diffuse pituitary hyperplasia, predominantly of GH and PRL secretory cells.
A second MRI of the brain suggested persistent tumor with intrasellar and suprasellar growth which compressed optic chiasma without invading the cavernous sinuses. The mass, 1.5cm in maximum size, was homogeneous in both structure and uptake. These findings were consistent with macroadenoma.
A second surgical procedure consisting of radical resection was performed through a right frontal craniotomy. Pathological tumor study showed an immunophenotype positive for vimentin and protein S-100 and negative for actin, desmin, cytokeratins, and EMA diagnostic of neurinoma/schwannoma.
The patient experienced anterior and posterior postoperative panhypopituitarism, and was therefore prescribed replacement therapy with hydrocortisone, levothyroxine, growth hormone, and desmopressin. The initially affected visual field mostly recovered, with persistence of some small peripheral defects (more marked in the left than in the right eye).
Patient course has been excellent to date, after ten years of follow-up.
This case, interesting because of the rarity of the condition, shares the clinical and radiographic characteristics of all other similar cases reported. As reviewed by Perez et al.,3 most patients have visual field defects caused by extrasellar tumor extension with modest secondary hyperprolactinemia, and half of the patients have evidence of hypopituitarism. In MRI, schwannomas have a hypointense, dull grey appearance in T1-weighted images and are usually hyperintense in T2-weighted images, with homogeneous enhancement after contrast administration. Most of them show suprasellar extension. They are usually slowly growing, indolent tumors. However, malignant transformation may occur.4 Surgery should be performed, initially through a transsphenoidal approach, but complete resection is uncommon due to its harder consistency and greater vascularization. Other approaches have therefore been proposed.5,6
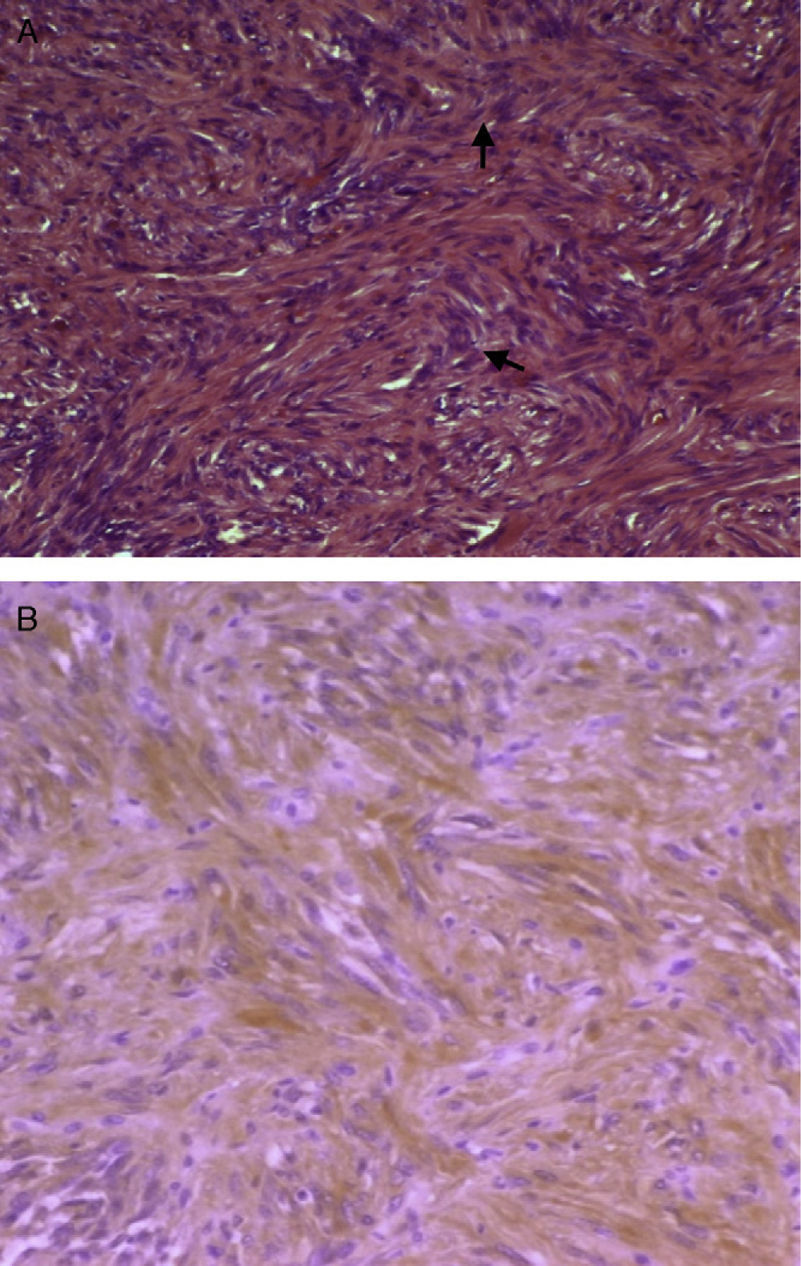
Macroscopically, schwannoma, also called neurinoma, neurilemoma, neuroma, or neurolemoma, is well-circumscribed and pseudoencapsulated and shows, as previously discussed, a marked vascularization that makes it more prone to bleeding. Microscopically, an alternating pattern between areas with type A and B Antoni patterns is characteristic. Areas with type A Antoni pattern consist of compact fusiform cells of ill-defined cytoplasmic margins, whose nuclei are arranged as a palisade leaving a lighter center, forming “Verocay bodies” (Fig. 2A). Areas with type B Antoni pattern are less cellular and may show microcystic changes, inflammatory cells, and collagen fibers, as well as large vascular spaces. In immunohistochemistry, cells are positive for protein S-100 (a marker of nerve sheath tumors and melanoma) (Fig. 2B). The reported tumor was positive for vimentin, a marker of meningioma, among other tumors, and for protein S-100. Meningioma is negative for S-100 and positive for EMA. Histomorphological data (absence of pigment) did not suggest melanocytoma either, although this is positive for S-100. The two main alternative diagnoses to be considered, fibrous meningioma and melanocytoma, were therefore ruled out.
 Micrograph showing Antoni A areas consisting of compact fusiform cells (H&E, 100×). (B) Immunohistochemistry positive for protein S-100, with dominant cytoplasmic pattern, of a brown color due to the chromogen used (diaminobenzidine) (protein S-100, 100×). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)")
(A) Micrograph showing Antoni A areas consisting of compact fusiform cells (H&E, 100×). (B) Immunohistochemistry positive for protein S-100, with dominant cytoplasmic pattern, of a brown color due to the chromogen used (diaminobenzidine) (protein S-100, 100×). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
As regards the endocrinological aspects, the existence of secondary hypothyroidism with FT4 levels of 13pg/mL is questionable despite the poor TSH response to a TRH stimulation test. Response is also decreased in hyperthyroidism, but our patient had no thyroid pathology or consistent symptoms.
Pathological study revealed diffuse pituitary hyperplasia at the expense of somatotroph and lactotroph cells, which agrees with the slight increase of PRL in blood (if we leave aside potential pituitary stalk compression), but is inconsistent with low IGF-1 levels and GH response during ITT. A second GH stimulation test could have been performed, but the patient refused it. Surgery itself may modify the initial behavior, and we functionally assumed a GH deficiency which was subsequently not confirmed by immunohistochemistry. These discrepancies between biochemical behavior and immunohistochemical results are frequently found in clinical practice.
The first pathological diagnosis was conditioned by a probable biopsy bias, possibly corresponding to perilesional hyperplastic pituitary tissue leading to an intraoperative diagnostic judgment of probable adenoma which was not subsequently confirmed. Repeat surgery due to the persistence of the mass in MRI was diagnostic of schwannoma. Alternatively, the patient could also have had an early collision sellar lesion. Koutourousiou et al.7 reported eight cases of pituitary adenomas occurring concomitantly with a second sellar lesion (whether of adenomatous origin or not), the so-called “collision sellar lesions”, in their series of 548 patients undergoing surgery for pituitary adenoma through the transsphenoidal approach. The cohort included five women and three men with a mean age of 48 years (range, 38–76 years). All patients underwent transsphenoidal surgery because of a preoperative diagnosis of pituitary adenoma with no suspected dual lesion based on clinical or radiographic evidence. There was a single case of intrasellar schwannoma coexistent with a GH-secreting adenoma which was subsequently reported.8 This was a 38-year-old male patient with headache and decreased libido who was diagnosed with acromegaly. MRI revealed a 19-mm pituitary tumor with suprasellar extension. Histological study showed adenoma immunohistochemically positive for GH and a pituitary schwannoma at the periphery. Patient follow-up for two years showed no tumor recurrence. The authors concluded in their review that preoperative diagnosis of a dual lesion is very difficult because clinical and radiographic signs characteristic of pituitary adenoma occur in most cases. Final diagnosis is made based on histological study. The pathogenetic mechanism of these dual lesions is currently unknown.
Finally, a reference should be made to the question of the origin of intrasellar schwannomas, because Schwann cells are not located in the central nervous system. Maartens et al.9 differentiate the reported cases into true intrasellar schwannomas and parasellar schwannomas (which are a majority) based on the Russel and Rubinstein10 hypothesis that neurinomas grow from Schwann cells located in nervous plexuses close to the sella turcica. Other proposed explanations include that they are perivascular Schwann cells11 or ectopic Schwann cells10; are derived from pluripotential cells12; or are migrations of cells from the neural crest which form foci of Schwann cells (schwannosis) in parenchyma of the central nervous system.11 In the reported case, the origin was even more uncertain, if possible, because the (chance?) coexistence of GH and PRL cell hyperplasia had to be explained.
To sum up, the remote possibility of an intrasellar schwannoma should be considered in differential diagnosis of pituitary lesions, and immunohistochemistry is determinant in the final diagnosis of these rare sellar masses. The few cases reported to date do not allow the profile of sellar schwannoma to be established with certainty.
Please cite this article as: Boj Carceller D, et al. Schwannoma intraselar: un caso excepcional de incidentaloma hipofisario. Endocrinol Nutr. 2012;59:336–8.
