


Multiple endocrine neoplasia type 1 (MEN1) is a rare inherited disease characterized by hyperplastic and neoplastic disorders mainly affecting parathyroids, anterior pituitary and gastroenteropancreatic endocrine tissues.1 In most subjects with MEN1 phenotype a heterozygous germline mutation in the MEN1 gene is identified. In addition, somatic mutations and large deletions of the MEN1 locus and its surrounding regions in chromosome 11, known as loss of heterozygosity (LOH), are found in MEN1 associated tumors.2 We hereby present a MEN1 patient who developed a thyroid neuroendocrine carcinoma, and analyze the potential involvement of MEN1 gene in the development of this tumor.
A 44 year-old Caucasian woman with MEN1 familial background was identified as a carrier of the mutation c.549G>A (p.Trp183*) in exon 3 of MEN1 gene in 2008. Previously, two MEN1 related disorders had been identified: a prolactin secreting pituitary microadenoma responsive to cabergoline had been diagnosed in 1996, and primary hyperparathyroidism in 2002, with resection of a right lower parathyroid adenoma. Hyperparathyroidism recurred in 2008. After the molecular diagnosis of MEN1 was established, abdominal computed tomography and magnetic nuclear resonance imaging detected 2 small, 1cm-diameter pancreatic lesions located in body and tail regions of the gland; a cytologic specimen obtained by echoendoscopy confirmed a low grade neuroendocrine neoplasia. There was no evidence of liver metastases.
Neck ultrasound performed prior to surgery for hyperparathyroidism recurrence identified multiple nodules in the left thyroid lobe. Fine needle aspiration biopsy of a 20mm poorly delimited hypoechoic nodule and of a 16mm left cervical lymph node led to a cytologic diagnosis of a neuroendocrine carcinoma, with positive chromogranin staining. Both lesions had increased octreoscan uptake. Blood calcitonin was normal, while progastrin-releasing peptide was elevated. The patient underwent total thyroidectomy, subtotal parathyroidectomy and left lateral neck node dissection. She signed informed consent for genetic analysis.
The histological study of the left thyroid lobe showed a tumor composed of sheets, nests and trabecula of polygonal cells, separated by thin fibrovascular stroma, with moderate cytoplasm and clear large nuclei with prominent nucleoli. Immunohistochemical staining of tumor cells was positive for chromogranin A, synaptophysin and CD56, confirming its neuroendocrine origin; it was also focally positive for calcitonin. TTF1 and thyroglobulin were negative. Ki67 was 15%. Staining for Congo red did not identify stromal deposits of amyloid. On the right thyroid lobe, two microscopic papillary thyroid carcinoma foci were identified. Metastatic neuroendocrine carcinoma was identified in one out of seven lymph nodes. The parathyroidectomy specimen was consistent with an adenoma.
MEN1 gene mutation had been previously identified as described, and MEN2 gene was now investigated. Genomic DNA was extracted from peripheral leukocytes by standard procedures QIAmp DNA. Protooncogen RET was studied by standardized PCR protocol in which coding regions of exons 8, 10, 11, 13, 14, 15 and 16 were amplified. PCR products were sequenced by BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and purification with Millipore system (96 well plates Multiscreen PCRu96 & Montage seq96), followed by analysis with ABI Prism Genetic Analyzer 3130.xl (Applied Biosystems). No germline mutations were identified.
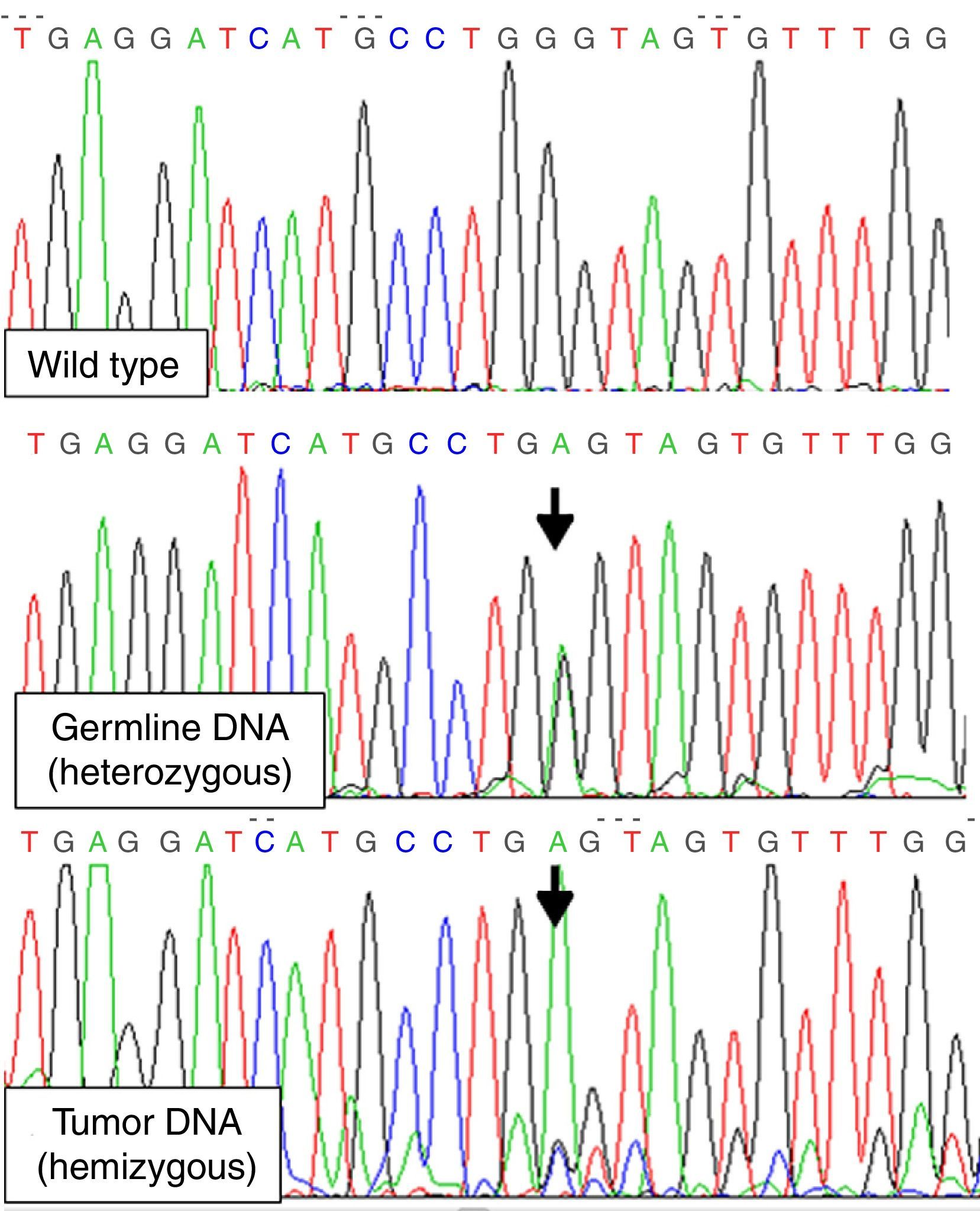
To detect LOH for MEN1 gene we amplified exon 3, where the patient's mutation was located, from tumor DNA isolated from paraffin-embedded thyroid-tumor tissue. A couple of exon 3 internal primers 5′GTGTGGCCTTTGCTGTGGTTG3′ and 5′ACTGTCTGGCCCCTGCGGTCC3′ were used in order to improve the efficiency of the PCR (short amplification). LOH involving chromosome 11q13 was demonstrated (mutation was found in hemizygosis) (Fig. 1). Exons 10, 11 and 16 of RET gene were also studied on DNA from tumor tissue, with no evidence of somatic RET mutations.

We report the concurrence of a MEN1 syndrome and a neuroendocrine thyroid neoplasm compatible with medullary thyroid carcinoma (MTC); to our knowledge, this association has been previously described just once.3
This association poses various possible explanations: the thyroid tumor could be a metastasis of her neuroendocrine pancreatic tumor, a MTC of either sporadic nature or due to an inherited RET gene mutation, or finally a thyroid neoplasia related with her MEN1 mutation. The first option is unlikely, as she presented neither local growth nor liver metastasis, as is expected in the progression of pancreatic neuroendocrine tumors. Although MEN1 and MEN2 coexistence is highly improbable, we excluded MEN2, as in any young patient with MTC. Sporadic MTC cannot be ruled out, though it presented atypical characteristics such as normal serum calcitonin and scarce calcitonin tumor staining. The last option, an atypical medullary/neuroendocrine carcinoma of the thyroid as a manifestation of her MEN1 status, seems the most feasible in view of the genetic findings, shared by all the neoplasia developed in the MEN1 context. More than 90% of tumors from MEN1 patients exhibit LOH on 11q13, an evidence of MEN1 gene activity as a tumor-suppressor gene, consistent with Knudson's two-hit hypothesis. This second hit may occur by LOH or other mechanisms including intragenic deletions and point mutations. On the other hand, LOH in the MEN1 locus has also been observed in 5–50% of sporadic endocrine tumors.4
In conclusion, we have identified a rare association of MEN1 and MTC, with LOH in the thyroid tumor involving chromosome 11q13 that had not been reported previously. As the complete inactivation of the MEN1 gene and subsequent absence of normal menin expression characterize MEN1 associated tumors, this finding relates her thyroid neoplasia to the MEN1 scope.
Conflict of interestThe authors have nothing to disclose.
