


La cardiopatía congénita cianótica (CCC) se refiere a un grupo de cardiopatías que aparece tras el nacimiento, afecta a 1/1.000 nacidos vivos y cursa con hipoxia sistémica. La incidencia de cardiopatías congénitas es de 12-14/1.000 nacidos vivos1. Los diferentes defectos cardiacos congénitos pueden causar un aumento de la resistencia vascular pulmonar e hipertensión pulmonar, de forma que alrededor del 8% del total de cardiopatías congénitas y el 11% de las que tienen cortocircuitos de izquierda a derecha desarrollan un síndrome de Eisenmenger, caracterizado por progresiva afectación vascular pulmonar y cianosis resultante de una comunicación sistémica-pulmonar que provocan la inversión del cortocircuito. El síndrome de Eisenmenger es la causa más frecuente de CCC en adultos2.
El feocromocitoma (FC) y paraganglioma (PG) son tumores neuroendocrinos originados en tejido cromafín, que se presentan con baja incidencia en la población general y con una prevalencia del 0,2-0,6% entre los pacientes con hipertensión arterial. Se ha descrito que en más del 30% de los casos su aparición está relacionada con alteraciones genéticas3. La coexistencia de ambas entidades ha sido descrita en algunos trabajos, y se postula una posible asociación patogénica.
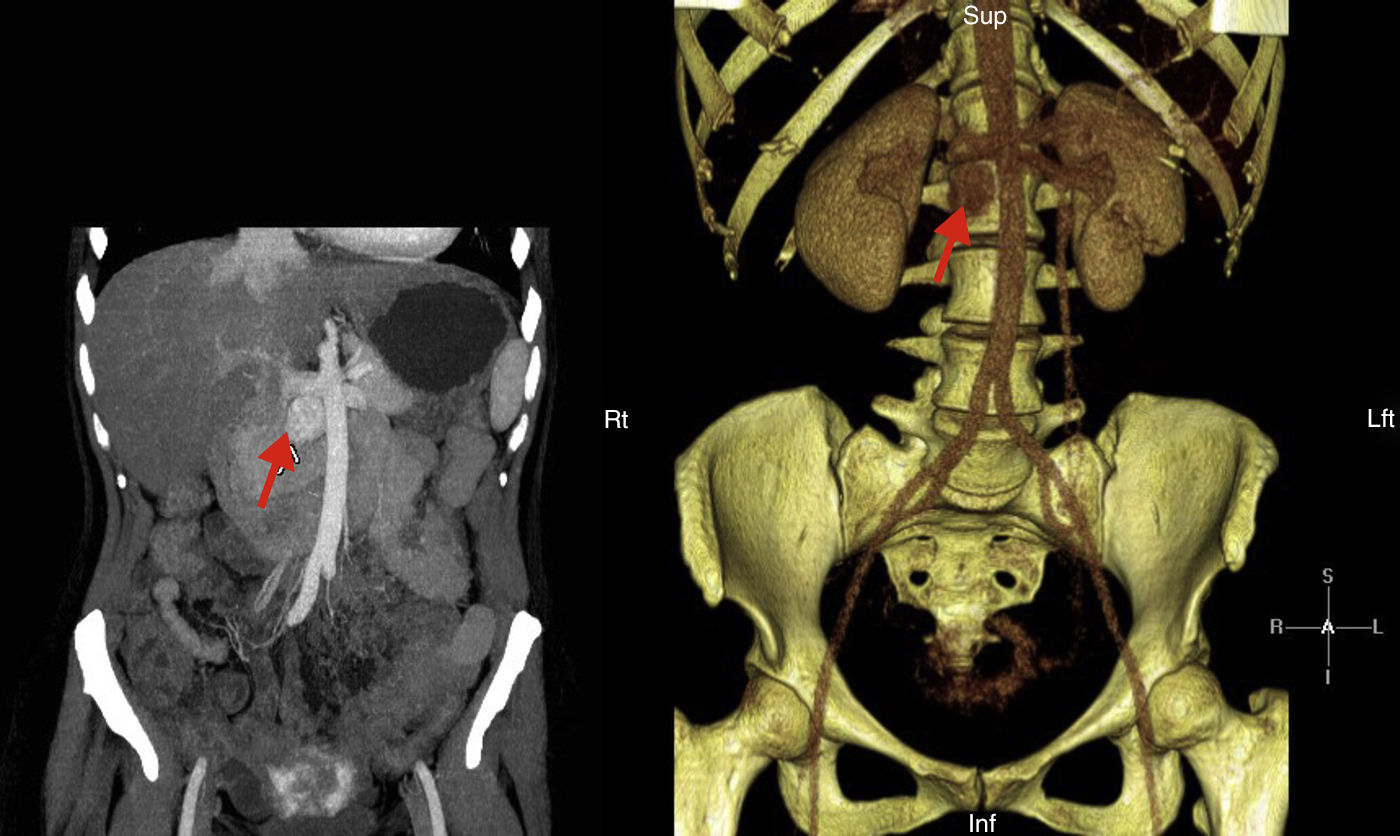
Presentamos el caso de una paciente de 41 años de edad diagnosticada de una cardiopatía congénita en la infancia consistente en: ventrículo único izquierdo de doble cámara con L-malposición de los grandes vasos e hipertensión pulmonar severa en situación Eisenmenger, con importante cianosis y eritrocitosis compensadora, en tratamiento con sildenafilo y bosentan. El diagnóstico se realizó a los 5 meses de edad y se desestimó la cirugía de reconstrucción cardiaca. Consultó por HTA, astenia, palpitaciones y malestar torácico, evidenciándose una descompensación de la insuficiencia cardiaca. Ante la sospecha de hipersecreción catecolamínica se realizó estudio hormonal que mostró metanefrina en orina de 24h: 215μg/24h (VN<341) y normetanefrina 2.491μg/24h (VN<444). El estudio de imagen realizado mediante TC abdominal mostró una masa retroperitoneal hipercaptante de 3×2cm en espacio interaorto-cava compatible con la existencia de un PG (fig. 1). El estudio gammagráfico de glándulas suprarrenales realizado con Iodo-123MIBG y fusión con imágenes de TC evidenció una zona de captación patológica del radiotrazador a nivel del espacio interaortocava coincidente con la lesión visualizada mediante TC y compatible con el diagnóstico de sospecha clínica, sin demostrarse otras lesiones a distancia. Se realizó estudio molecular mediante secuenciación de los exones codificantes y las regiones de unión exón-intrón de los genes: SDHD, SDHC, SDHB, VHL, SDHAF2, MAX y TMEM127, no encontrándose alteraciones. Tras el adecuado bloqueo alfa con doxazosina, y bajo estrecha vigilancia cardiológica fue sometida a tratamiento quirúrgico con resultado histológico de PG. No se identificó invasión capsular, ni linfovascular. La inmunohistoquímica reveló expresión intensa y difusa para cromogranina, sinaptofisina y tinción S100 en las zonas sustentaculares; índice de proliferación Ki-67: 1%. Tras la intervención se objetivó una concentración urinaria de normetanefrina dentro del rango de referencia, normalización de la TA, así como una mejoría clínica evidente.
.")
En los últimos años se ha descrito la asociación entre la hipoxia y los síndromes genéticos relacionados con la existencia de FC y PG (SDHx, von Hippel-Lindau, HIF2A). La mayoría de estos síndromes condicionan una activación aberrante de vías de señalización que activan la síntesis de factores inducidos por la hipoxia (HIF), responsable de la patogénesis de FC y PG4. Se ha sugerido que la exposición a la hipoxia crónica en pacientes con CCC puede aumentar el riesgo de desarrollo de FC y PG5. Esta paciente fue diagnosticada en la infancia, por lo que la evolución de la enfermedad conlleva una situación prolongada de cianosis. Por otra parte, en cuanto al fenotipo bioquímico se refiere, nos encontramos con producción exclusiva de noradrenalina. En los últimos años, en los pacientes con FC/PG, se ha descrito la utilidad del fenotipo bioquímico como guía para la realización del estudio genético, y así se han diferenciado 2 grupos (cluster 1 y 2) que difieren en la vía de señalización alterada. En el grupo 1, asociada con errores en activación anormal de HIF, se observa aumento en la expresión de factores angiogénicos que conducen a la aparición de los tumores. Este grupo se caracteriza por presentar un fenotipo noradrenérgico con secreción de adrenalina normal6. El grupo 2 comprende un grupo de tumores causados por mutaciones del Rearranged during transfection (RET), el gen de la neurofibromatosis tipo 1 (NF 1) y el gen TMEM127 con un fenotipo adrenérgico y secreción predominante de adrenalina. El resultado del estudio genético realizado a nuestra paciente descarta la predisposición genética. El diagnóstico de FC/PG en estos pacientes puede ser difícil de sospechar debido al solapamiento de los síntomas, sin embargo la hipersecreción catecolamínica puede empeorar el cuadro clínico, por lo que consideramos importante tener en consideración que en pacientes con CCC que presenten un empeoramiento de la función cardiaca se debe descartar la presencia de FC/PG.
