



The thyroid-stimulating hormone (TSH) receptor (TSHR) is a major regulator of thyroid function and growth, and is the key antigen in several pathological conditions including hyperthyroidism, hypothyroidism, and thyroid tumors. Various effective treatment strategies are currently available for many of these clinical conditions such as antithyroid drugs or radioiodine therapy, but they are not devoid of side effects. In addition, treatment of complications of Graves’ disease such as Graves’ ophthalmopathy is often difficult and unsatisfactory using current methods. Recent advances in basic research on both in vitro and in vivo models have suggested that TSH analogs could be used for diagnosis and treatment of some of the thyroid diseases. The advent of high-throughput screening methods has resulted in a group of TSH analogs called small molecules, which have the potential to be developed as promising drugs. Small molecules are low molecular weight compounds with agonist, antagonist and, in some cases, inverse agonist activity on TSHR. This short review will focus on current advances in development of TSH analogs and their potential clinical applications. Rapid advances in this field may lead to the conduct of clinical trials of small molecules related to TSHR for the management of Graves’ disease, thyroid cancer, and thyroid-related osteoporosis in the coming years.
Tanto el crecimiento como la función de la glándula tiroides son regulados por la tirotropina (TSH), la cual ejerce su función a través de su receptor (TSHR). El TSHR es el antígeno clave en varias alteraciones tiroideas, entre las que se incluyen el hipertiroidismo y el hipotiroidismo, o en algunos tumores. Actualmente, se disponen de diversas estrategias terapéuticas para muchos de estos trastornos, como son los fármacos antitiroideos o la administración de radioyodo. Desgraciadamente, estas terapias no están exentas de efectos secundarios. Además, el tratamiento de las complicaciones derivadas de la enfermedad de Graves, como la oftalmopatía tiroidea, resulta muchas veces difícil e insatisfactorio. Los recientes avances en investigación básica, tanto en modelos in vitro como in vivo, sugieren que los análogos de la TSH podrían emplearse en el diagnóstico y en el tratamiento en algunas de las enfermedades tiroideas. La llegada de los métodos de cribado de alto rendimiento ha proporcionado un grupo de análogos de la TSH, también llamados pequeñas-moléculas, que son fármacos con características potencialmente prometedoras. Estas pequeñas-moléculas son sustancias con bajo peso molecular que pueden poseer actividad agonista, antagonista y, en algunos casos, agonista inversa sobre el TSHR. En esta revisión nos centraremos en los avances actuales en el desarrollo de los análogos de la TSH y su posible aplicación clínica. Los rápidos avances en este campo podrían propiciar que en los próximos años se desarrollen ensayos clínicos con pequeñas-moléculas relacionadas con el TSHR para pacientes con enfermedad de Graves, cáncer de tiroides y osteoporosis de origen tiroideo.
Thyrotropin (TSH) receptor (TSHR), a member of the glycoprotein hormone receptor family, is the major antigen for Graves’ disease. In addition to being an auto antigen, this heptahelical G protein couple receptor is also the key regulator of thyroid growth and function.1–3 Recent studies have shown that this receptor is not restricted to thyroid rather it is expressed in other extra thyroidal tissues such as heart, brain, bone adipocytes, immune cells etc., though the function of the receptor in these extrathyroidal sites is far from clear.4,5 In addition to its wide expression, TSHR has also an in-built constitutive activity, which is unique, to this receptor.2,6 Controlling this activity in thyroid cancers could be of therapeutic importance. Since current therapeutic approaches in controlling thyroid diseases are far from satisfactory and not without side effects there has been concerted effort in developing small-molecules (SM) as potential second line therapeutic agents.
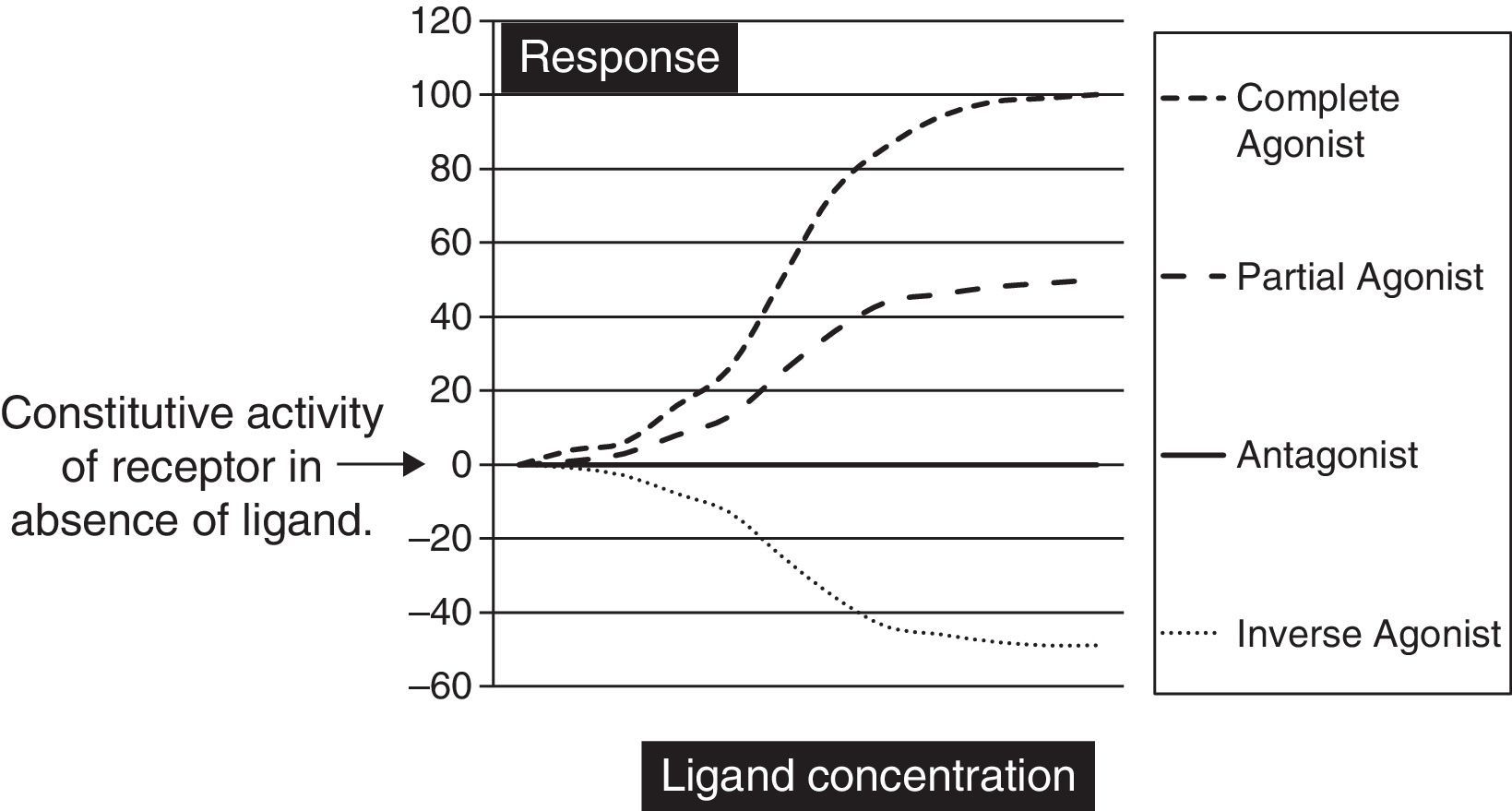
Biochemically SM are low molecular weight compounds (usually smaller than 800Da) that bind to cell surface receptors modifying their signaling responses. These responses could be activation of receptor (agonistic), or suppression of the natural activity (antagonistic) or even suppression of the constitutive activity (inverse agonistic). Fig. 1 gives a very simplistic view of their classification based on the responses they can generate. SM have become attractive candidates in this therapeutic panorama because of their (1) potential pharmacological activity; (2) ease of manufacturing in large amounts at low cost; (3) their ability to penetrate the cell membrane rapidly; and finally (4) ability to be administered orally. Though there is not yet a clear understanding on the pharmacodynamic or molecular aspects of these SM, but progressive advances in molecular medicine have clarified the possible pathways by which these molecules can exert their effects (Table 1).

The graph is a simple schematic representation of a Gsα coupled receptor activation by agonists and antagonists. Among the glycoprotein hormone receptors, TSH receptor has constitutive activity which leads to a higher basal activity as represented by the arrow on the Y axis. An inverse agonist would suppress this natural basal activity whereas an antagonist would suppress any ligand or antibody stimulated activity of the receptor. On the other hand agonists and partial agonists have the ability to stimulate receptor activity by binding to receptors and eliciting a positive conformational change.
Glossary of cellular receptor activity.
| Concept | Definition |
| Receptor | It is a protein embedded in either the plasma membrane or the cytoplasm of a cell that detects a stimulus (usually a specific signal molecule or a ligand) and thereupon initiates a response in the cell. The response is associated with gain of or loss of protein activity ordinarily leading to some sort of cellular responseReceptor agonists, antagonists and inverse agonists bind to the same receptor types. The agonist has a positive value, the inverse agonist has a negative value, and the antagonist for the receptor takes both the agonist and the inverse agonist back to a neutral state |
| Constitutive or basal activity | It is the receptor intrinsic activity without the action of a ligand upon them. Thus, cellular receptors display some signal action in absence of ligands. The constitutive activity could be promoted or inhibited |
| Ligand | It is a substance that binds (attaches) to a specific site on a biomolecule (receptor). It is a signal triggering substance. Ligand binding to a receptor (docking or association) is usually reversible (dissociation). Ligands could be inhibitors and activators. The tendency or strength of binding is called affinityThe ligand mediated receptor activation depends on the capacity of the ligand for saturating its receptor |
| Signaling | Sequence of linked protein reactions acting as a relay chain to transmit a signal within a cell. In cell biology signal transduction is the process by which a cell produces a response to an extracellular signal |
| Full agonist | It is a chemical molecule that binds to a cell receptor and cause a complete (∼100%) response. An agonist causes an action which is the same reaction or activity typically mimics the action of a naturally occurring ligand |
| Partial agonist | It is a chemical substance capable of combining with a cellular receptor and initiate similar reaction produced by the natural ligand, although the response is incomplete |
| Antagonist | It is a substance that does not trigger a biological response itself upon binding to a receptor, but blocks or constraint agonist- or inverse-agonist- mediated responses. In other words, antagonists have affinity but no efficacy for their cognate receptors. An antagonist blocks the action of the agonist and an inverse agonist causes an action opposite to that of the agonistSometimes are called Neutral agonist (or neutral antagonist) because those ligands do not modify the receptor's constitutive activity. Antagonist response inhibits agonist-stimulated signaling but does not inhibit agonist-independent signaling |
| Inverse agonist | It is a chemical compound that binds to the same receptor binding-site as an agonist for that receptor and reverses constitutive activity of receptors. Inverse agonists exert the opposite biological effect of a receptor agonist. The effect of an inverse agonist is measured as the negative value of the agonistWhereas agonists cause an action, antagonists block the action of the agonists and inverse agonists cause an action opposite to that of the agonists |
| Superagonist | It is a substance capable of producing a greater maximal response than the endogenous agonist for the target receptor, and therefore has an efficacy greater than 100% |
Thyroid diseases have not been an exception to this advance. Currently there are several groups investigating on SM that interact either with the TSHR or the thyroid hormone receptors. An example of the latter is the recent development of a number of thyroid hormone analogs such as eprotirome7 and DITPA.8 These two compounds have already been evaluated in phase II clinical trials showing benefits in circulating lipid profile and in heart failure, respectively. Likewise the potential role of analogs that interact with TSHR is an object of intense investigation.
TSH/TSHR analogs or SM can be used in the management of patients with thyroid dysfunction either with Graves’ disease or TSHR mutations resulting in hyper- or hypothyroidism. In addition, SM would also be useful in the management of differentiated thyroid cancer, either to inhibit TSH signaling in those patients who need suppressive therapy or to stimulate sodium iodide symporter (NIS) expression to enhance radioactive iodine uptake and to have beneficial therapeutic effect, or even to stimulate a thyroglobulin (Tg) response during monitoring follow-up. Since the expression of TSHR has been described in a number of extra thyroidal tissues, TSHR analogs could be therapeutically active in other tissues besides the thyroid gland.
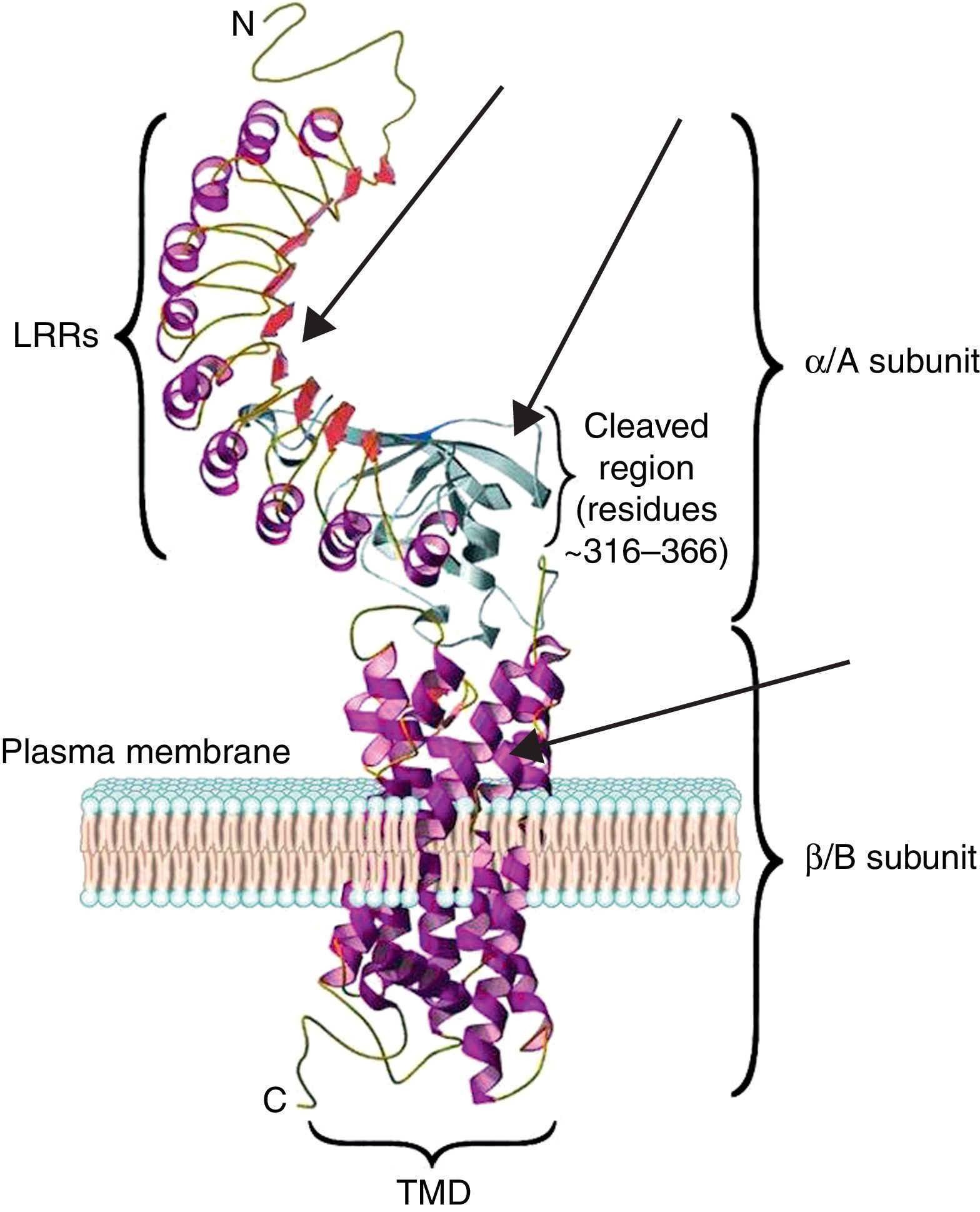
The TSH receptor structureTSHR is a G protein-coupled transmembrane receptor with two subunits: a large extracellular subunit known as the alpha subunit (α or A) and a membrane anchored signal transducing unit known as the beta subunit (β of B).9 The extracellular domain or ectodomain has a characteristic horseshoe-shaped structure and contains ten Leucine Rich Repeats (LRR) made up of α helices and β pleated sheets. The well-characterized leucine rich domain (LRD) starting from amino acids (AA) 22 to 260 constitutes the cavity where the ligand TSH binds, which is also known as the TSH-binding pocket. The LRD is followed by a large region of 130 AA known as hinge region which encompasses a 50 AA region that undergoes proteolysis resulting in different forms of receptor on the surface of the cell. Recent studies have shown that in addition to LRD region the hinge region plays a critical role in stabilization and even signaling of the TSH.10 The B subunit or the transmembrane domain (TMD) is composed of seven transmembrane domains and a large intracytoplasmic tail. The TMD is the main signaling unit of receptor with the seven helices that are connected to each other by extra and intra cellular loops. The crystallization of the TSHR with stimulating and blocking antibodies11,12 has further defined the exact binding residues that these pathogenic autoantibodies, thus opening avenues for targeting these region with small molecules or analogs to either stimulate or inhibit receptor responses. Fig. 2 is a model of the bipartite structure of TSH receptor and with predicted hot spots where TSHR analogs or small molecules could be targeted (Fig. 2).
. The 7 transmembrane domains (spirals) are embedded within the plasma membrane followed with short cytoplasmic tail. This part of receptor is known as the B or β subunit. The ectodomain of the receptor is made 10 LRR followed by cleaved region and uncleaved regions of receptor. The region from AA 260 onwards to AA 410 is also known as hinge region of the receptor. The entire region of the receptor outside the plasma membrane is known as the A or α subunit of receptor. The thick gray arrows represent the possible locations on the receptors where these small molecules or receptor analogs can be directed to elicit a positive or a negative response.")
A modified version of the computer model of the TSH receptor (adapted from Davies et al.9). The 7 transmembrane domains (spirals) are embedded within the plasma membrane followed with short cytoplasmic tail. This part of receptor is known as the B or β subunit. The ectodomain of the receptor is made 10 LRR followed by cleaved region and uncleaved regions of receptor. The region from AA 260 onwards to AA 410 is also known as hinge region of the receptor. The entire region of the receptor outside the plasma membrane is known as the A or α subunit of receptor. The thick gray arrows represent the possible locations on the receptors where these small molecules or receptor analogs can be directed to elicit a positive or a negative response.
The characteristics of the TSH-binding pocket are critical for the correct ligand-receptor binding. The AA 22–44, AA 246–260, AA 277–296 and AA 317–385 are especially important in this union. All of them are located in the THS binding region either within or outside of the LRR.4,9 These specific areas are the epitopes with potential antigenic capacity. Therefore, SM that are targeted to the extracellular region or the transmembrane portion of the receptor may potentially alter the signaling characteristic of the receptor.
TSH receptor signalingThe transmembrane domain of the TSHR mainly activates the classical G-protein-coupled effectors such as Gs, Gq, different subtypes of Gi and Go, as well as G12 and G13.13 Stimulation of the receptor leads to dissociation of trimeric G proteins into Gα and Gβγ subunits which in turn trigger a complex signaling network.14
Most of the activities of the TSHR are mediated by Gs protein which activates the adenylate cyclase (AC)/cAMP cascade leading to the activation of either the PKA-dependent cascade or the PKA-independent signaling pathway in order to regulate thyroid function.14 Both these pathways include the Extracellular Regulated Kinase (ERK), which is a downstream component of a well-conserved signaling module.15 An alternate TSHR effector pathway, via Gq, mediates the activation of the PLC-β and the Gγ subunit.16 Once activated, PLC hydrolyzes phosphatidylinositol bisphosphate (PIP2) to inositol 3,4,5-trisphosphate (IP3) and diacylglycerol (DAG).17 AKT is activated as a result of PI3-kinase activity and activation of AKT is critical in several thyroid cancers. Recent studies have shown that the proliferative response to chronic TSHR stimulation by TSH relies heavily on the activation of the mTOR pathway.18
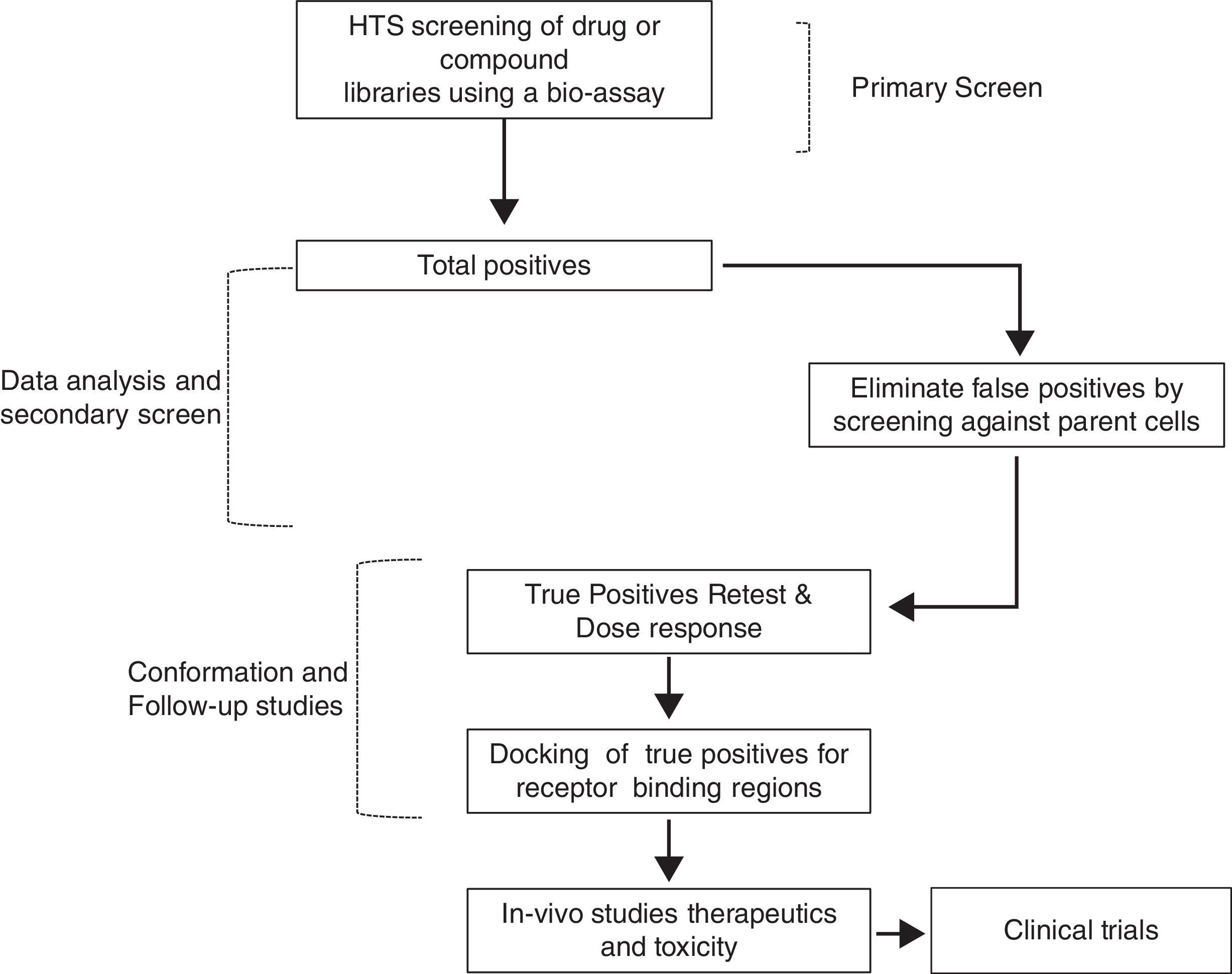
Therefore, TSH analogs or SM that targets these pathways either in an orthogonic or allosteric manner would be critical in the treatment of thyroid diseases and differentiated thyroid cancers. However, developing these SM that are specific to the target is a long and laborious process that follows a general scheme as shown in Fig. 3. They appear to be like low hanging fruits but it takes a lot of stretching to get the right one.
Small-molecules and Graves’ disease treatmentLimitations of current Graves’ disease therapy
Graves’ disease has been reasonably treated since more than a century.19 However, there has been little (if any) progress in Graves’ disease therapeutics over the last decades. The three main therapeutic approaches (drugs, radioactive iodine or surgery) have been practically unchanged since several years ago. At the same time it is also well-reckoned that there exist a number of limitations or drawbacks in current management of Graves’ disease.20
1. Medical therapy. Patients medically treated normally receive a long-term treatment with drugs that are not free of side effects risks. Antithyroid drugs can cause severe or even occasionally life threatening complications.21 On top of that, in some series the number of relapses after pharmacological therapy increases up to 80%.22 Unfortunately, reliable markers for predict individual patient's response to antithyroid drugs are lacking and similarly there are no consistent clues that foretell which patient will relapse. Therefore it becomes difficult to assess the response to medical therapy in a given patient. An additional antithyroid drug drawback is the difficulty to titrate the adequate dose in some subjects, especially in those in whom triiodothyronine (T3) secretion is the predominant thyroid hormone. Finally, it should also be considered that antithyroid drug effect is not directly related to the specific pathophysiological mechanism that causes Graves’ disease. Medical therapy does not repair the specific autoimmune alteration.
2. Ablative therapy (surgery or radioactive iodine). Permanent residual hypothyroidism is the most characteristic sequelae of ablative therapies. Albeit unusual in experienced centers, complications related to thyroid surgery are permanent hypoparathyroidism and vocal cord palsy. In addition, surgery and radioactive iodine have elevated costs, normally associated to admissions to hospital. The administration of radioactive iodine is contraindicated during pregnancy and can exacerbate Graves’ orbitopathy, especially in severe cases. It is also worth to considerer that surgery removes healthy thyroid tissue that contains not only follicular cells but also parafollicular cells. Parafollicuar cells are not involved in the development of hyperthyroidism. Although calcitonin is never replaced after total thyroidectomy without apparent disadvantages, there is no doubt that this hormone contributes to the correct bone homeostasis.
All these factors seem very good reasons to look for new therapeutic options for patients with Graves’ disease. In accordance with the limitations of current therapy, innovative therapies should meet the following requirements:
- (1)
Reduce or eliminate side effects;
- (2)
Preserve normal thyroid function;
- (3)
Be safe during pregnancy;
- (4)
Target the pathophysiologic mechanism;
- (5)
Be swifter, cheaper and straightforward than current options.
Several years ago in a patent application, Organon has described a range of bicyclic heteroaromatic compounds including thienopyrimidines as LH/hCG receptor agonists.23 Following the discovery of thienopyrimidine (org41841) in 2002 – the first SM with agonist activity over LH/hCG receptor – the group of Marvin C. Gershengorn, from the National Institutes of Health (NIH) predicted that org41841 might activate TSHR because of the homology of both receptors. Four years later the NIH group published their results and Org41841 was found to be a partial agonist for TSHR.24 Further in 2009 this group described the first breakthrough in TSHR agonist, NCG00161870, which exhibited high affinity, potency and efficacy in in vitro model cell systems.25 Moreover, NCG00161870 was described to be highly selective and did not activate the two closely related glycoprotein hormone receptors, the LH/hCG receptor and the FSH receptor. NCG00161870 binds to the helices of the transmembrane domain and was therefore named as an allosteric ligand. In view of the fact that there was the competence of synthesizing agonists, it was speculated about the opportunity of produce SM with antagonist action by modifying the basic scaffold of these molecules or by searching large libraries of existing SM using assays developed for high throughput screening. If there is a partial agonist, there could be an antagonist and as well as an inverse agonist. This important finding heralded a new era to the pharmacological modulation of TSHR activity. Thus, low-molecular-weight antagonist for TSHR may have therapeutic potential as orally active drugs to block TSHR stimulatory antibodies (TSI) in subjects with Graves’ hyperthyroidism.
Potential therapy for Graves’ diseaseFrom a number of chemical modifications of org41841, the investigators eventually developed, in 2008, an antagonist (NIDDK/CEB-52) of TSHR.26 NIDDK/CEB-52 displayed no agonist activity. The compound was tested in vitro which selectively blocked both TSH and TSI activation of the TSHR. NIDDK/CEB-52 was shown to work not only in cultured cell lines over-expressing TSHR but also in primary cultures of human thyrocytes expressing endogenous TSHR too. The compound inhibited up-regulation of RNA transcripts for thyroperoxidase (TPO) before and after treatment with TSH and, even after TSI, which suggested as proof of principle, its potential clinical use in subjects with Graves’ disease.
The success in the development of a compound with an antagonist activity boosted further research. NIDDK/CEB-52 could be employed as a lead molecule for the development of more potent and efficacious analogs that could be used in animal studies and for the development of future drugs for hyperthyroid patients, including those subjects with Graves’ disease.
Three years later, in 2011, the same NIH group developed a new SM based on modifications of NIDDK/CEB-52 structure. The new SM, which was denominated NGC00229600, was able to inhibit basal (constitutive) and TSH-stimulated TSHR activity, as shown by its ability to reduce cAMP production in cell cultures.27 The NGC00229600 inhibition of TSH signaling was competitive even though it did not compete for TSH binding. In primary cultures of human thyrocytes, NGC00229600 inhibited TSHR-mediated basal and Graves’ disease sera mediated up-regulation of TPO mRNA levels by about 65%, demonstrating its potent inverse agonist activity. The authors concluded that NGC00229600 had a dual activity: it was an inverse antagonist of TSHR while it was a general antagonist of TSHR activation by sera of patients with Graves’ disease. Further in 2012, Org274179-0 developed by an Australian group inhibited TSH (and TSI)-mediated TSHR activation making it as the most potent allosteric antagonist, which exerts it action in nanomolar concentrations.28 This compound also inhibited TSH, TSH receptor antibodies from patients and a human stimulating monoclonal antibody M22 activation of the receptor. In addition, it also stimulated cAMP in orbital fibroblasts derived from patients with Graves’ ophthalmopathy.
Potential therapy for Graves’ ophthalmopathyThere are several elegant studies claiming the important role of TSHR in orbital fibroblast and also its role in driving adipogenesis behind the retro orbital tissue.29–31 In recognition of this important role of the TSHR in extra thyroidal tissue, a recent paper has shown the potential therapeutic effect of NCGC00229600 in Graves’ orbitopathy.32 The antagonist inhibits the activation of TSHR endogenously expressed in fibroblast from patients with Graves’ ophthalmopathy. Future studies on the real efficacy of SM in controlling this disease awaits additional investigations, which is further compounded due to the lack of animal models for Graves’ eye disease.
Management of thyroid cancerIn the management of many patients with differentiated thyroid cancer it is crucial to maintain undetectable blood levels of circulating TSH during the follow-up. This approach, called suppressive therapy, involves treatment with supra-physiological doses of levothyroxine rendering patients in a subclinical hyperthyroid status. This approach is not entirely harmless, since subclinical hyperthyroidism is associated with some unwanted side effects like osteoporosis and heart issues.33
In addition, individuals with thyroid cancer are required to raise serum TSH levels periodically for improving the ablative (or therapeutic) radioactive iodine lethal effects and/or evaluate the presence of circulating Tg as tumor marker.34 This is achieved by withdrawal of levothyroxine replacement therapy or by the administration of a dose of recombinant human TSH (rhTSH). The availability of SM with potent agonist activity can be a future replacement for rhTSH therapy in the management of thyroid cancers.
Taking advantage from the inverse agonist effect on TSHRBesides their above-mentioned possible application in Graves’ hyperthyroid patients, TSHR inverse agonists can be used as probes to study the role of the basal (constitutive) receptor signaling. Physiologically, TSHR expresses high basal activity, which stimulates follicular cell proliferation. This activity could promote follicular thyroid cancer cells to growth. Thus, TSHR inverse agonists may play a therapeutically potential role by inhibiting TSHR basal signaling in individuals with differentiated thyroid carcinoma. This approach could be equivalent to suppressive therapy.
By chemical modifications of scaffold of existing antagonist Neumann et al., in 2010, have described a SM ligand that exhibits inverse agonist properties at TSHR.35 The new molecule 1;2-(3-((2,6-dimethylphenoxy)methyl)-4-methoxyphenyl)-3-(furan-2-ylmethyl)-2,3-dihydroquinazolin-4(1H)-one, was referred to simply as “1” by the authors in their publication. In primary cultures of human thyrocytes “1” inhibited basal and TSH-stimulated signaling as it was demonstrated by the decreased basal levels of mRNA transcripts for Tg, TPO and NIS. The authors suggested that “1” could be a lead for the development of higher-potency inverse agonists that may be tested as probes of TSHR biology with therapeutic potential, especially in patients with differentiated thyroid cancer. The compound inhibited both basal and TSH-stimulated cAMP production by TSHR up to 58% and 86% respectively. The authors also showed that this SM acts as a competitive antagonist of TSH signaling. All these characteristics support that “1” could be used therapeutically to inhibit TSHR signaling in patients with advanced or progressive differentiated thyroid cancer who are under suppressive therapy.
Taking advantage from the agonist effect on TSHREarlier experiments aimed to discover TSHR agonist displayed some SM with low affinity for TSHR. These molecules were shown to activate the receptor only in a cell model system in which human TSHR was ectopically expressed. The compounds that demonstrated more potent activity were NCGC00057417, NCGC00038940, NCGC00054245, and NCGC00026086. Moving forward in the process of developing more effective TSHR agonists, the investigators have also identified a large number of TSHR agonists by quantitative high-throughput screening.36 After a selection process, 49 SM were confirmed as true TSHR agonist compounds and served as a starting point for further studies.36 Eventually the investigators selected the eighth-most potent molecules and re-tested them for efficacy and selectivity.25 They denominated Compound 1 as the most selective agonist for TSHR. This SM was highly selective, since it displayed no detectable agonist activity at the closely related LH/hCG or FSH receptors while working as a full agonist at the TSHR when compared to the endogenous TSHR activity. Using human thyrocytes in primary cultures, Neumann et al. have shown how some of these agonist compounds increased Tg mRNA as effectively as TSH and increased mRNAs for TPO, NIS, and type 2 deiodinase although somewhat less effectively than TSH.25 Further chemical modification of Compound 1 led to the identification of a new SM, which was denominated as Compound 2. This newer SM was a TSH analog with 16-fold improved potency than wild-type endogenous TSH. Interestingly, the authors tested its activity in vivo, administering Compound 2 orally to mice. As expected, the SM stimulated thyroid gland function (increased serum T4 secretion and thyroidal radioiodine uptake) in the animals. These encouraging results suggested that TSHR agonist could be as helpful as rhTSH in the control of thyroid cancer patients. Currently rhTSH is recommended for the management of individuals with differentiated thyroid cancer in several of the circumstances. The main indications of rhTSH in postoperative thyroid cancer patient are two: (1) to stimulate the radioactive iodide uptake and (2) to release of Tg from any residual thyroid cancer cells. This approach increases the sensitivity of measurements of radioiodine uptake or serum Tg levels in these patients avoiding the drawbacks of hypothyroidism.
Hence, the potential use of TSHR agonists in the management of differentiated thyroid cancer could be similar to those of rhTSH such as: (1) in initial therapy, to improve the diagnostic/therapeutic effect of radioactive iodine remnant ablation after surgery. (2) Diagnostic, to assess whether the initial therapy has been successful during the follow-up. Any TSHR agonist stimulus on potential latent follicular cells (if they remain or re-growth after surgery) will render an increase in circulating Tg levels. This is currently the most sensitive test for metastasis and recurrence in the surveillance of patients with a past history of differentiated thyroid cancer. (3) Prognostic, since the presence of high blood Tg levels or radioactive iodine uptake after a TSHR stimulus indicates presence of residual disease. (4) Therapeutic, especially in presence of remaining structural systemic disease, as TSHR agonists could enhance radioactive iodine lethal activity on thyroid cancer cells. Therefore, the development of SM agonists potentially could offer certain advantages over rhTSH such as a less expensive production and their convenient oral administration.
Small-molecules and non-autoimmune hyperthyroidismTSHR germline activating mutations are the cause of non-autoimmune hyperthyroidism.37 This rare disorder may be sporadic or familial and, like Graves’ disease, is associated with a diffuse enlargement of the thyroid gland. Mutated TSHR from these patients exhibit constitutive signaling between 15-fold and 27-fold higher than wild-type TSHR. It was demonstrated that the TSH inverse-agonist analog “1” decreased the constitutive over-activity from mutated TSHR in an in vitro cell model by 72%.35
These observations indicate that inverse agonists could be used therapeutically in subjects with non-autoimmune hyperthyroidism to suppress TSH-independent signaling, since these compounds target the specific mechanism of the disease. This option might be specially considered in infants with inherited forms of this disease in which ablative treatment is less attractive.
The agonist effect on TSHR and hypothyroidismCertain unusual types of subclinical hypothyroidism are caused by mutations in the TSHR that confer TSH resistance.38 In spite of the mutation, this resistance could be theoretically circumvented by TSHR agonists, as it has been shown experimentally. It has been published that “C2”, a TSHR agonist, activates the TSH unresponsive TSHR ectodomain mutants C41S and L252P. However, the compound had no effect on the serpentine mutant L467P.39 Although limited, these results indicate that the small-molecule agonist “C2” (or other TSHR agonist developed in the future) may be a useful pharmacological tool for the study of TSHR mutants.
A potential additional advantage of treating hypothyroidism with TSHR agonist is that these compounds would stimulate the thyroid gland to produce both T4 and T3, which constitutes a clear benefit over the present therapy with levothyroxine alone. It is known that there are about 10% of distressed subjects among hypothyroid patients receiving levothyroxine replacement therapy. Given the long half-life of T4 of one week, it is improbable that minor failures to adjust the replacement dose could give a satisfactory justification for these complaints. Nevertheless circulating free T3 has, in contrast to free T4, a circadian rhythm with an acrophase in early morning hours around 03:00hours. The presence of polymorphisms in deiodinase 1 has been associated with differences in serum T3 and T4 ratio. Thus, carriers of these polymorphisms may explain the complaints in a subset of patients treated with levothyroxine monotherapy.40
Helping bones with TSHR analogsAs it has been mentioned above, TSHR is known to be present in many extra-thyroidal tissues, although the role of TSHR in many of these organs is not apparent. The consequences of TSH action in bones are not fully understood. However, the role of TSH as a single molecular switch in the independent role of both bone formation and resorption was defined nearly a decade ago.41 Preliminary data have revealed that the TSHR was found on osteoblast and osteoclast precursors, and that the reduction of TSHR expression caused intense osteoporosis. It was also demonstrated that TSH inhibited osteoclast formation and survival. On the other hand, TSH also inhibited osteoblast differentiation. As a whole these results marked that TSH is a negative regulator of skeletal remodeling. Studies in mice and humans have evidenced that thyroid hormones and TSH have the opposite effects on bones. However, both basic and clinical studies in recent years have established the potent role of TSH in bone remodeling.42–45
A recently published paper from Davies et al.46 has compared the skeletal phenotypes of wild-type and TSHR knockout mice in which hyperthyroidism were artificially induced. TSHR knockout animals had greater bone loss and resorption than hyperthyroid wild-type counterparts. These results indicate that the absence of TSH signaling contributes to bone loss and therefore suggest that therapeutic suppression of TSH to very low levels may contribute to bone loss in people. The authors have highlighted that they have identified a splice variant of TSHβ chain, a TSH-like factor, which confers osteo-protection. This TSHβ chain variant, produced locally in bone marrow, was able to extract a bone-protective effect. Evidently, this effect should be absent when TSHR expression is down-regulated. Clearly the discovery opens up new possibilities in the use of TSH analogs as therapeutic agents for osteoporosis. In addition to this the TSHR specific small molecule agonist if given in a therapeutically effective dose should modulate osteoclast and osteoblast activity within bone like TSH. Like all drugs in the market the therapeutic dose of these analogs have to be established from phase II and phase III clinical trials. NCG00161870 and several of the discovered analogs are being further evaluated preclinically prior to their approval by the Food and Drug Administration to allow for testing in human subjects.47
Summary of potential advantages of small-molecules on TSHRC.S. Lewis in his masterpiece The Lion, the Witch, and the Wardrobe, transports us to a magical world where impossible things in real world became easily possible. In this marvelous novel the Pensive siblings’ dreams came true. Thanks to continuous advances in medicine with the collaborative effort between those who are at the bench and those who are at bedside is made feasible which not long time ago seemed unattainable. One of the most promising progresses in translational thyroidology is the eventuality of treating any thyroid disease by modulating TSHR using TSH analogs. Thus, paraphrasing C.S. Lewis, the SM, the TSH receptor and the thyroid diseases will introduce us to a promising field that not long ago could be considered as magical.
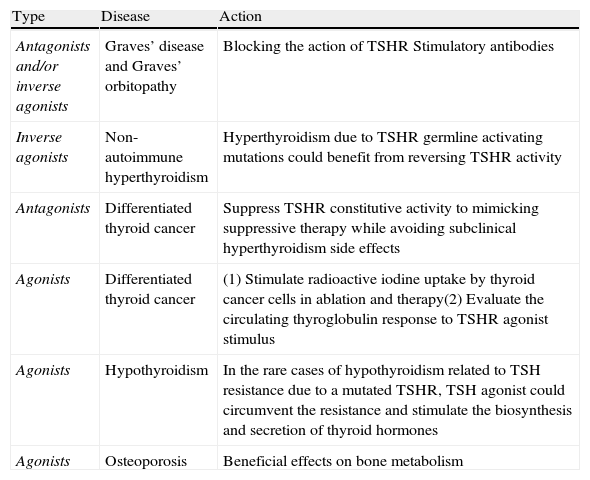
All the information regarding the biologic activity of TSH analogs summarized in this review arise from experimental and preclinical studies. All these molecules necessitate further development before they could be tested as therapeutic agents in clinical trials. There is almost no information about these compounds pharmacokinetics or pharmacodynamics. Data from in vivo models are scarce, thus the administration of TSH analogs could possibly elicit the development of unwanted side effects because of defects in the specificity. Nevertheless the outlook is highly promising especially for patients with differentiated thyroid cancer and Graves’ disease. A summary of potential applications on SM in thyroid diseases is presented in Table 2.
Summary of potential clinical applications of TSHR ligand analogs.
| Type | Disease | Action |
| Antagonists and/or inverse agonists | Graves’ disease and Graves’ orbitopathy | Blocking the action of TSHR Stimulatory antibodies |
| Inverse agonists | Non-autoimmune hyperthyroidism | Hyperthyroidism due to TSHR germline activating mutations could benefit from reversing TSHR activity |
| Antagonists | Differentiated thyroid cancer | Suppress TSHR constitutive activity to mimicking suppressive therapy while avoiding subclinical hyperthyroidism side effects |
| Agonists | Differentiated thyroid cancer | (1) Stimulate radioactive iodine uptake by thyroid cancer cells in ablation and therapy(2) Evaluate the circulating thyroglobulin response to TSHR agonist stimulus |
| Agonists | Hypothyroidism | In the rare cases of hypothyroidism related to TSH resistance due to a mutated TSHR, TSH agonist could circumvent the resistance and stimulate the biosynthesis and secretion of thyroid hormones |
| Agonists | Osteoporosis | Beneficial effects on bone metabolism |
The authors declare that they have no conflicts of interest.
