


Central nervous system (CNS) receives peripheral relevant information that are able to regulate individual's energy balance through metabolic, neural, and endocrine signals. Ingested nutrients come into contact with multiple sites in the gastrointestinal tract that have the potential to alter peptide and neural signaling. There is a strong relationship between CNS and those peripheral signals (as gastrointestinal hormones) in the control of food intake. The purpose of this review is to give updated information about the role of gut hormones as mediators of feeding behavior and of different nutrients in modulating gut hormones production. The role of gut hormones in the pathogenesis of emerging diseases as obesity and non-alcoholic fatty liver disease (NAFLD) is also discussed together with the possible role of these peripheral signals as targets of future therapeutic options.
El sistema nervioso central (SNC) recibe cierta información periférica capaz de regular el equilibrio energético del individuo a través de señales metabólicas, neuronales y endocrinas. Los nutrientes ingeridos entran en contacto con varios sitios del tracto gastrointestinal que son capaces de alterar las señales nerviosas y pépticas. Existe una fuerte relación entre el sistema nervioso central y estas señales periféricas (hormonas gastrointestinales) en el control de la ingesta de alimentos. El objetivo de esta revisión es aportar información actualizada sobre el papel de las hormonas intestinales como mediadoras de la conducta alimentaria, y de los diversos nutrientes que modulan la producción de hormonas intestinales. También se analiza el papel de las hormonas intestinales en la patogénesis de enfermedades incipientes tales como la obesidad y la esteatohepatitis no alcohólica (EHNA), así como la posibilidad de que estas señales periféricas sean dianas en futuras alternativas terapéuticas.
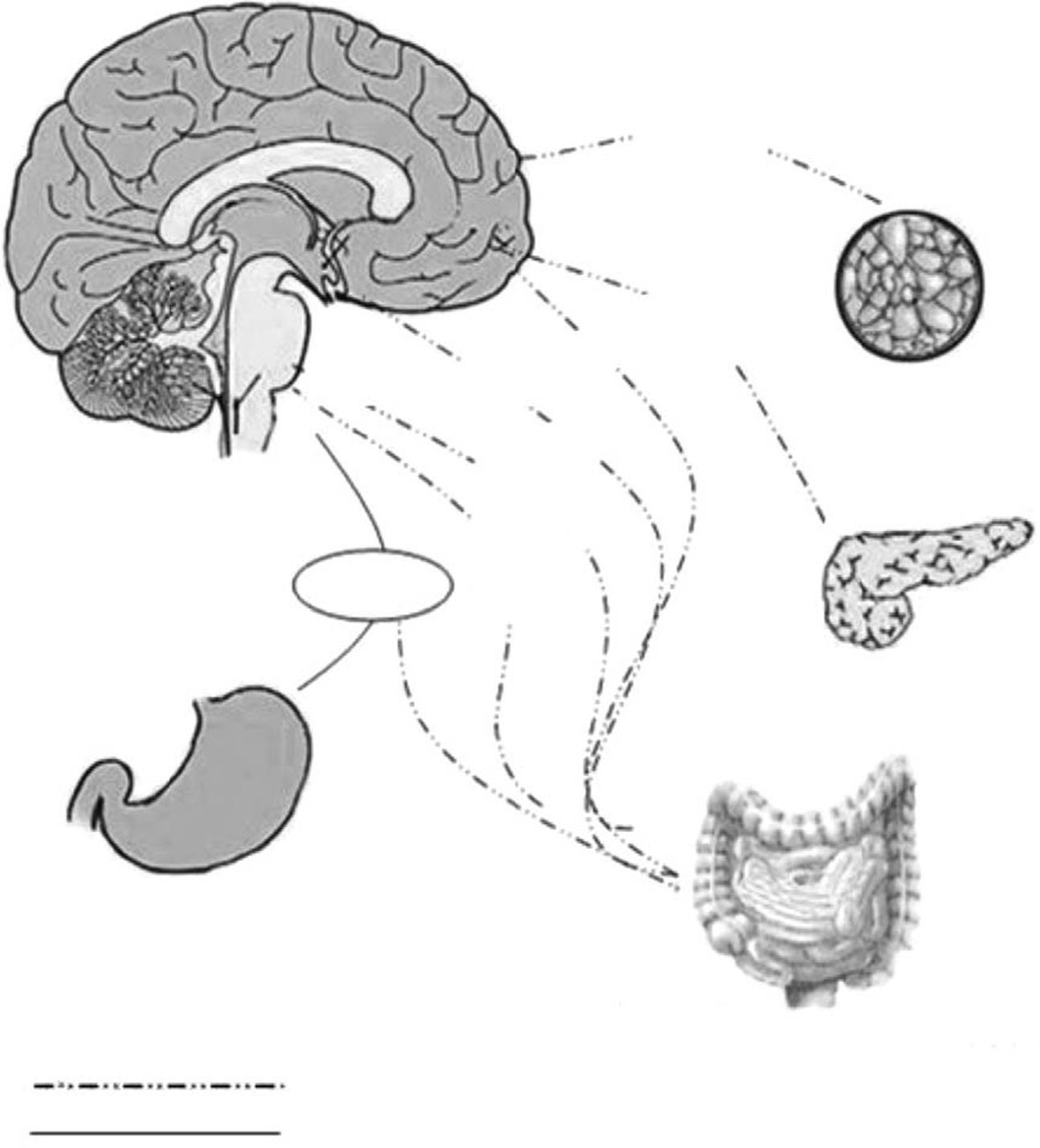
The energy balance is regulated to defend a “set point” in body weight, whereas the food intake has a wide range of variations within each individual. New regulative mechanisms regarding gut–brain relationship have been recently discovered, allowing a better comprehension about the way in which energy homeostasis is regulated. Long-term and short-term hormonal signals connect periphery to the central nervous system influencing feeding behavior. Hypothalamus, specifically the arcuate nucleus and the dorsal vagal complex in the brain stem, are the most relevant structures involved in this function.1 The role of the arcuate nucleus and the brain stem consists in an integration within signals coming from periphery and recognized in the gut hormones2 (Fig. 1). Distinct subsets of neurons are involved, acting both as stimulants and as inhibitors toward food intake. Stimulant neurons are represented by neuropeptide Y (NPY) and agouti-related peptide (AgRP), while inhibitors include alpha-melanocyte stimulating hormone (alpha-MSH) and cocaine- and amphetamine-regulated transcript (CART). Typically, when one of these subsets is activated, the other is inhibited. Neurons responsiveness to hunger and satiety signals (as ghrelin, cholecystokinin, and PYY) are also modulated by body energy stores (represented by leptin and insulin).3

The aim of this article is to review the role of gut hormones as mediators of feeding behavior and of the different nutrients in modulating gut hormones production. The action and the burden of gut hormones in pathogenesis of emerging diseases as obesity and NAFLD are also emphasized as well as their possible role as future therapeutic targets.
Materials and methodsA medline and Pubmed search was performed to identify relevant past and recent (2000–2010) literature using the following keywords: control of food-intake, gut hormones and food intake, glucagon like peptide-1 (GLP-1), cholecystokinin (CCK), ghrelin, peptide YY (PYY), non-alcoholic fatty liver disease (NAFLD), and obesity.
ResultsGut hormones and food intakeDuring and after meal, ingested nutrients alter the release of gut peptides that may potentially modulate the food intake and contribute to a “three-steps process”: meal initiation, within-meal satiety and across-meal satiety influences.4
Meal initiationGhrelinOriginally isolated as the endogenous ligand for the growth hormone secretagogue receptor (GHS-R) and for its capacity to stimulate growth hormone (GH) secretion, ghrelin is also the only orexigenic signal from the gastrointestinal tract and a long-term regulator of energy homeostasis.5 This 28 amino acid peptide is modified post-translationally with an eight-carbon fatty acid by the enzyme ghrelin-O-acyl-transferase (GOAT). The acylation of ghrelin is required to activate the GHS-R and to mediate its effects on GH secretion and food intake.6
Ghrelin is mainly synthesized in the endocrine stomach cells and has a series of different biologic actions, including effects on glucose homeostasis, gut motility, pancreatic exocrine secretion, cardiovascular function, immunity and inflammation.7 The physiological relevance of these actions remains unclear, and the major role of the ghrelin seems to be exerted in the regulation of energy balance. Ghrelin stimulates the synthesis of neuropeptide Y (NPY) and agouti-related protein (AgRP) in neurons of the arcuate nucleus of the hypothalamus, which in turn enhances food intake.8 Ghrelin also stimulates appetite and food intake when it is administered systemically in humans.9 Ghrelin also regulates the preprandial hunger. Plasma ghrelin peaks preprandially in human subjects, who have been deprived of time cues, initiating meals voluntarily.10 In animal models, during postprandial period, plasma ghrelin is suppressed in proportion to the calories ingested, when macronutrient content and volume are kept constant.11 Interestingly, fat appears to suppress ghrelin less potently per calorie than carbohydrates or proteins.12,13 This may, in part, explain the reduced satiety and higher weight gain associated with high-fat diets.
Taken together, these data strongly suggest a role for ghrelin as a meal initiator. Whether ghrelin is the only hunger signal is yet undetermined. In addition, ghrelin appears to participate in long-term energy balance. Chronic administration of ghrelin in rodents results in fact in prolonged hyperphagia and weight gain.14,15
Within-meal satiety signalingAfter a meal, nutrients pass into the stomach and intestine, and a number of gastrointestinal signals are released. Peptide signals also act to optimize the digestive process, and as short-term satiety signals. Three examples are: cholecystokinin (CCK), pancreatic glucagons and amylin.
CCKCCK is widely distributed within the gastrointestinal tract, but the majority is synthesized in the duodenum and jejunum. It is rapidly released into the surrounding tissues and circulation in response to nutrients, in particular, fat and protein-rich meals in the gut, with levels rising approximately 5-fold postprandially.
Its main actions include delaying gastric emptying, stimulating pancreatic enzyme secretion and stimulating gall bladder contraction. Together with these actions, it also promotes the effective digestion of fat and protein in the small intestine by matching the delivery of nutrient with the capacity to digest it.16 In addition it inhibits food intake. Administration of CCK, in humans and animals, inhibits food intake by reducing meal size and its duration.17,18
Recent work suggests that CCK-A receptors expressed by vagal afferent neurons are an important target for CCK in producing sensations of satiety, as well as inhibiting gastric emptying and stimulating pancreatic digestive enzyme secretion.19 In animals it is thought that the reduction in food intake may be mediated by a paracrine/neurocrine effect since high concentrations of CCK occur only locally to the site of release.20 Thus, locally released CCK may increase vagal tone, without a significant increase in plasma CCK level.
CCK-A receptor antagonists increase calorie intake and reduce satiety, suggesting a possible role of endogenous CCK in appetite regulation.21 CCK may be a very short-term modulator of appetite; it has a half-life of only 1–2min and is not effective in reducing meal size if the peptide is administered more than 15min before a meal.
Pancreatic peptides and amylinMeal termination may be attributed to the action of endogenous amylin and glucagon since receptorial antagonists or specific antibodies increase food intake.
The feeding-inhibitory action of glucagon is peripherally mediated and dependent on intact hepatic vagal signaling; nevertheless the actions of amylin are mediated by the area postrema, a circumventricular organ with a porous blood–brain barrier.22,23
Across-meal satiety signalingOther gut peptides show patterns of effects that act after the meal that have stimulated their release. Both peptide YY (PYY) and glucagon-like peptide 1 (GLP-1) are synthesized and released from L-cells located in the distal intestine. In contrast to the pattern of release of within-meal satiety peptides, elevations in plasma PYY and GLP-1 occur more slowly, not peaking until meal termination and remaining high for several hours after a meal.
PYYPYY is released into the circulation following meals and it is suppressed by fasting. PYY has been known for long to exert numerous effects on the gastrointestinal tract. Administration of PYY increases the absorption of fluids and electrolytes from the ileum and inhibits pancreatic and gastric secretions, gallbladder contraction, and gastric emptying.24 Peripheral administration of PYY, like ghrelin, also exerts effects on numerous other body systems. For example, in rats it reduces cardiac output, causes vasoconstriction and reduction in glomerular filtration rate, plasma renin and aldosterone activity. The physiologic significance of these numerous actions has not been established.25
The pattern of PYY secretion in response to a meal raises the hypothesis that it may play a role as physiologic satiety signal, acting in terminating the meal and stimulating coordinated gastrointestinal responses in order to aid digestion and absorption. PYY levels rise to a plateau at 1–2h postprandially, with peak levels influenced both by number of calories and by composition of the macronutrient.26
Long-term augmentation of dietary proteins in mice increased plasma levels of PYY, decreased food intake and reduced “adiposity”. A study showed that PYY null mice developed marked obesity that may be reversed by exogenous PYY treatment, suggesting that this hormone plays a role not only in postprandial early satiety but also in long-term, by regulating energy balance through unknown mechanism.27
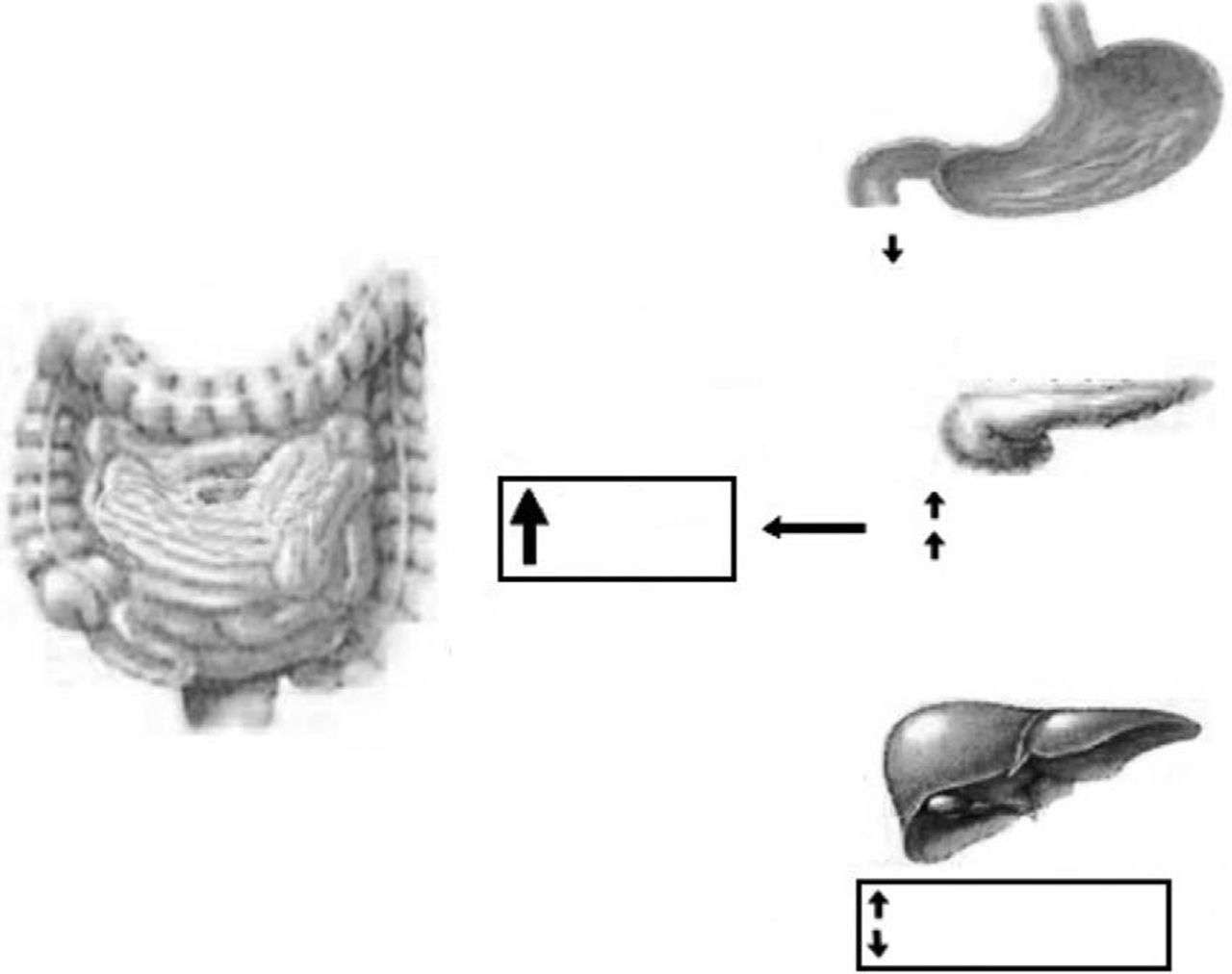
Glucagon like peptide-1 (GLP-1) and oxyntomodulin (OXM)GLP-1 and OXM are products of the preproglucagon gene, which is expressed in the CNS, in the L-cells of the small intestine, and in pancreas. Secreted preproglucagon is cleaved by prohormone convertases 1 (PC1), 2 (PC2) and 3 (PC3) into different products depending on the tissue. In pancreas, the predominant proglucagon posttranslational processing products are glicentin-related polypeptide, glucagon, intervening peptide-1 and the major proglucagon fragment; this process is mediated by PC2. Differently, posttranslation processes in enteroendocine L-cells and CNS liberate glicentin, oxyntomodulin (OXM), GLP-1, intervening peptide-2 and GLP-2; these processes are mediated by PC1 and PC3.28–30
OXM inhibits gastrointestinal secretion and motility (so probably promoting satiety) and stimulates pancreatic enzyme secretion and intestinal glucose uptake.31 Among others (cardiovascular and central effects), GLP-1 exerts a number of gastrointestinal actions: stimulation of glucose-induced insulin secretion by upregulating insulin gene expression and potentiating all steps of insulin biosynthesis; improvement of glucose sensitivity of resistant pancreatic beta-cells; reduction in gastric emptying32 (Fig. 2). An intravenous infusion of GLP-1 is capable to completely normalized blood glucose levels in patients with long-standing type 2 diabetes that cannot be controlled with sulphonylurea therapy.33

As well as OXM, GLP-1 is released rapidly into the circulation after oral nutrient ingestion (from 5 to 30min) and in proportion to meal calorie intake, inhibiting both gastric acid secretion and motility.
GLP-1 has been shown to decrease calorie intake in animal models by intracerebroventricular and hypothalamic paraventricular nucleus (PVN) administration.34 An infusion of OXM to normal-weight human subjects produced a reduction in calorie intake of 19.3%, whereas a recent meta-analysis which studied the effect of GLP-1 infusion demonstrated an average reduction in calorie intake of 11.7%. Reduction in calorie intake is dose-dependant and does not differ between obese and lean subjects.35 The feeling of satiety produced by OXM and GLP-1 is likely due to their effects on the CNS as well as on gastric emptying.
Effect of protein, fat, carbohydrate and fiber on gastrointestinal peptide release in humansShort-term regulation of food intake controls what, when and how much we eat within a single day or a meal.
Nutrients and ghrelin releaseIngestion of simple and complex carbohydrates has been described to exert the major suppressing role on postprandial ghrelin concentration,36,37 as well as intravenous or oral administration of glucose.38
About the effect of dietary fiber on postprandial ghrelin (including those with different physical and chemical properties), a limited number of studies have been conducted. However a high content of fiber in the meal has been shown both to decrease postprandial ghrelin concentration and unexpectedly to inhibit this event.39
The effects of protein on postprandial ghrelin have given contradictory results. After ingestion of protein-rich meal, postprandial ghrelin concentrations do not change40 or do not increase41; in contrast, a high protein test breakfast (enriched with milk-based proteins) or liquid preloads of whey, casein, soy and gluten caused a prolonged suppression of ghrelin.42 The ability of ingested proteins and carbohydrates to cause ghrelin suppression is similar, even if they act with a different kinetic.43
Ghrelin concentrations may both decrease or increase after ingestion of a high-fat meal, with a slower return to baseline than after an ingestion of high-carbohydrate meal.44,45 The effect of fatty acids on ghrelin secretion may be dependent on their chain length.45 In a study, long-chain fatty acids (sodium oleate, 18 carbon atoms) showed to inhibit ghrelin, whereas medium-chain fatty acids (sodium caprylate, 8 carbon atoms) were ineffective.46
Nutrients and CCK releaseIngested fats and proteins cause a greater, faster and longer postprandial release of CCK than carbohydrates.47 In fact CCK increase after carbohydrates is short-lived and returns to baseline within 1h.48
Fiber content of the meal also affects postprandial CCK release. Different dietary fibers, including hydrolyzed guar gum,49 beta-glucan in barley pasta50 or fiber in bean flakes, oatmeal and in oat bran,51 have been shown to induce high levels of postprandial CCK.
Proteins are also involved in stimulating CCK release via inhibition of trypsin-mediated digestion of intestinal CCK releasing peptides.52 This effect has been shown to be significant especially after ingestion of liquid whey, but also after casein, soy and gluten or after a whey protein meal. Whey demonstrated to be more satiating than casein.53 Higher CCK responses after proteins, however, did not affect food intake.
Lipids significantly stimulate CCK release.54 To do this, however, triglycerides must be hydrolyzed to fatty acids. CCK release55 is influenced by the length of the fatty acid carbon chain. Several studies showed that carbon chain length N10C are the most potent stimulants for CCK release. Furthermore, C12 as compared with C10 suppressed appetite and energy intake.56 High fat intake seems to have long-lasting effect on CCK: after a high-fat evening meal, CCK concentrations remained elevated until the following morning.57 Cholecystokinin initiates satiety by acting at cholecystokinin type 1 receptor (CCK1R) located on vagal afferent nerve terminals. It plays an important role in regulating meal patterns, so that its absence impairs the ability to terminate meals. In CCK1R null mice the alteration of meal patterns is more marked when ingesting a high fat diet: fat seems to be a potent inhibitor of food intake via CCK release.58
Nutrients and PYY releaseNutrients stimulate PYY release within 30min from ingestion of a meal, usually reaching a maximum within 60min59 and proportionally to calorie intake. Meal composition is also able to influence postprandial PYY release, whereas gastric distension is not implicated in influencing plasma PYY concentration.60
Dietary fat, carbohydrates and protein all stimulate PYY release, but to different degrees and time-courses.61,62 About the role played by carbohydrates, Essah et al.26 found that low-carbohydrate, high-fat (LCHF) diet stimulates PYY secretion more than low-fat, high-carbohydrate (LFHC) diet. Higher PYY release after lipids than after carbohydrates has also been reported by others.63,64 In contrast, a study reported opposite results about the role of fat in increasing PYY concentration, which has been shown to rise slightly after fat ingestion. Proteins also stimulate PYY secretion, but moderately, whereas glucose solution caused only a transient and minor release.65
Fat hydrolysis and fatty acids chain length may differently influence PYY release. Intraduodenal administration of dodecanoic (“C12”), markedly increased PYY compared with decanoic (“C10”), while C10 had no effect.66 Serrano et al.67 showed that plasmatic PYY levels were consistently higher in group following a diet containing olive oil than those on a diet containing sunflower oil.
Solid meals enriched with psyllium fiber strongly modified postprandial signals arising from the GI tract, in particular decreasing PYY responses.68
Moreover, it has been observed that oat- and wheat-fiber consumptions result in different postprandial responses of PYY and ghrelin, but interestingly do not differ in satiety effects.69
About the effect of protein on PYY secretion, the response of plasmatic PYY to feeding with cow milk protein solutions in humans are independent of the degree of protein fractionation and are not altered by small differences in the amino acid composition or protein solubility.70
Nutrients and GLP-1 releaseCarbohydrates are strong stimuli of GLP-1 release consistent with the role of GLP-1 as incretin.71 A study showed no differences in GLP-1 response between different carbohydrates by intraduodenal infusion of glucose and fructose.72 Otherwise, another study showed that oral glucose exerted a bigger effect on GLP-1 release than fructose.73
The type or amount of dietary fiber may elevate, inhibit or not influence GLP-1 responses. Weickert et al.74 found that ingestion of insoluble fibers significantly accelerated early insulin response, whereas GLP-1 postprandial levels were not influenced. In addition, postprandial GLP-1 levels 24h after the intake of highly fermentable fiber compared with non-fermentable fiber were unchanged. On the other hand a study observed that in mice fed with a diet containing dietary-fiber, an elevation of GLP-1 concentration occurred. GLP-1 levels slightly decreased in high fat diet and cellulose diet groups whereas significantly increased in psyllum and sugar cane fiber groups, likely due to a different degree of fiber solubility.75
Proteins stimulate GLP-1 release, even more than carbohydrates.43 When compared to meals rich in fat, carbohydrate or alcohol, in protein-rich meals, GLP-1 responses were the highest one.76 Veldhorst et al.77 showed that whey triggered the strongest responses in concentrations of active GLP-1 compared with casein and/or soy, whereas there were no differences in energy intake.
Regarding the effect of fat on GLP-1 concentration, diets enriched in monounsaturated fatty acids (MUFA) induce higher GLP-1 concentrations with a positive effect on glycemic control.78 However, previous study did not find the same effect.79 The postprandial GLP-1 response to intraluminal fats may also depend on the chain length of fatty acids. C12 stimulate GLP-1 secretion, whereas C10 do not.56 Regarding the relationship between GLP-1 and macronutrients in controlling food intake, a study showed that maintenance on high-fat diet in rats impairs GLP-1s ability to reduce food intake, contributing to overconsumption of high-fat foods.80
Gut hormones and obesityThe epidemic “obesity” is fastly becoming one of the leading causes of mortality and morbidity worldwide. Over the past 30years, gastrointestinal hormones have been increasingly understood to have an important role as regulators of appetite and energy balance in obese individuals. Among others, the levels of these hormones are also modulated by bariatric surgery and the comprehension of the underlying reciprocal mechanism is fundamental. In others words, it is now partially clear that gut hormones play a role in the regulation of body weight, representing potential therapeutic targets for treatment of obesity.
A role for ghrelin in the etiology of human obesity has been proposed. Ghrelin has an inverse relationship with body mass index and it is significantly lower in obese individuals compared with lean ones.81 Ghrelin is the “hormone of hunger,” and this picture would fit with the notion of homeostatic control of body weight; high circulating ghrelin in thin individuals would favor increased food intake and positive energy balance. Weight loss in obese people results in an elevation in ghrelin level,82 which may contribute to the difficulty in maintaining ideal weight after weight loss.
Individuals with Prader–Willi syndrome have elevated levels of ghrelin. This hormone presents a reduced ability in inducing GH secretion and a preserved ability to stimulate food intake (patients with Prader–Willi syndrome show hyperphagia). This aspect could reflect an altered action of ghrelin on its specific receptors implicated in feedback mechanisms.83
Regarding the role of CCK in inhibiting food intake, CCK-A receptor knockout rats are hyperphagic and obese.84 In animals, as expected, repeated preprandial administration of CCK reduces food intake but it also increases meal frequency, with no resulting effect on body weight.85
Circulating levels of PYY are not elevated in obese individuals; in fact obese subjects retain full sensitivity to anorectic actions of PYY3–36. It has been also reported that obese subjects have lower fasting and postprandial circulating PYY than lean subjects. To produce an equivalent stimulation of PYY and equivalent satiety, obese individuals needed to consume a much greater caloric load than their lean counterparts.86–88 However, not all studies have detected a difference in fasting PYY concentrations between lean and obese groups.89
Current data suggest that impaired postprandial PYY release may impair satiety and help to maintain obesity, if not act as a primary driver of initial development of obesity.90
The anorectic effects of GLP-1 are preserved in obesity. A reduced secretion of GLP-1 could therefore contribute to the pathogenesis of obesity. GLP-1 in physiological concentrations powerfully reduces the rate of entry of nutrients into the circulation by a reduction of gastric emptying rate in obese subjects. So, the effect of GLP-1 on appetite and food intake may be beneficial in weight reduction.91
Furthermore, a 6-wk subcutaneous infusion of GLP-1 to type 2 diabetics normalizes glycosylated fructosamine and reduces HbA1c by 1.3%. The infusion of GLP-1 was also found to reduce body weight by 2kg.92 This evidence makes GLP-1 an excellent drug not only for diabetes mellitus, but also for appetite control and the treatment of obesity. However, the therapeutic potential of this gut hormone is limited by its rapid breakdown. GLP-1 is deactivated by dipeptidyl peptidase IV (DPP-IV), which cleaves off the two NH2-terminal amino acid residues, transforming the peptide into an antagonist of the GLP-1 receptor. However, recent trials have shown that inhibition of DPP-IV may be an effective treatment for type II diabetes mellitus. Various resistant analogs, such as exendin 4 (exenatide, Amylin), and albumin-based forms, such as liraglutide (NovoNordisk), may improve glycemic control and reduce body weight.93
Finally, among bariatric surgery techniques, intestinal bypass surgery is successful in reducing food intake and in causing weight loss because of enhanced secretion of satiety signals as GLP-1 and PYY.94
Gut hormones and liver diseaseNAFLD (non-alcoholic fatty liver disease) represents a disease spectrum ranging from simple steatosis to steato-hepatitis and fibrosis.95 The prevalence of NAFLD is about 17–33% in the Western world countries so that it shows to be the most prevalent liver disease, more than chronic hepatitis C virus or alcoholic liver disease. Non-alcoholic fatty liver disease (NAFLD) is closely linked with obesity and with insulin resistance; therefore improvement of insulin resistance, or insulin sensitivity, may have therapeutic potentiality in preventing the progression of NAFLD.96 Insulin sensitizers have been tried.97 Exendin-4 is a 39 amino acid agonist of the glucagon-like peptide 1 (GLP-1) receptor. It has a significantly longer half-life than GLP-1 and could be significant in the treatment of type 2 diabetes mellitus and NAFLD. Exendin-4 appears to effectively reverse hepatic steatosis in ob/ob mice by improving insulin sensitivity. Exendin-4 treated hepatocytes resulted in a reduction in mRNA expression of stearoyl-CoA desaturase 1 (SCD-1), a gene associated with fatty acid synthesis. This aspect may be critical in order to limit triglyceride accumulation in hepatocytes.98
Liver cirrhosis is often associated with alteration in carbohydrate metabolism similar to type 2 diabetes. The incretin hormone, glucagon-like peptide-1 (GLP-1), has therapeutic potential for the treatment of type 2 diabetes in cirrhotic patients. Hyperinsulinemia observed in liver cirrhosis is not due to an increase of insulin secretion from islets, but could be explained by decreased hepatic degradation of insulin.99 The mechanism through which GLP-1 may ameliorate diabetes in patients with liver cirrhosis needs to be investigated.
About anorexia in cirrhosis, we could affirm that its pathogenesis is complex and the hormone ghrelin may be involved. Normal, increased and decreased levels of ghrelin have been found in patients with hepatic failure in several studies.
Acylated ghrelin is the active form of ghrelin that is able to exert its biological effect influencing insulin sensitivity and body composition. El-Shehaby et al. found that in cirrhotic patients, plasma levels of both acylated and total ghrelin were significantly higher than in controls and the same finding was observed in patients with Child C cirrhosis when compared to Child A and B. These data suggest that ghrelin (as hunger hormone) may help cirrhotic patients to weight gain or to restore energy homeostasis.100 The same evidences were obtained by Tacke et al.101 Other studies did not find differences in plasma levels of ghrelin in normal and cirrhotic patients.102,103 Finally, Diz-Lois et al. studied fasting and post-OGTT (oral glucose tolerance test) ghrelin concentrations in liver failure patients before and after liver transplantation. They observed a reduction both in fasting and in post-OGTT ghrelin concentrations in liver failure patients; ghrelin levels normalized after liver transplantation. This study suggests a possible role of ghrelin in pathogenesis of anorexia occurring in advanced liver failure patients.104,105
DiscussionAs firstly described, food intake and energy expenditure are regulated by hormones released from the gut and adipose tissue, which act on CNS creating a link and feedback mechanisms that allows a maintenance of a steady body weight despite daily variation of energy expenditure and nutrient intake. Recently, this issue has become a great area of interest, so that a considerable amount of articles about this aspect has been published. It has been clearly demonstrated that composition of diet modifies secretion of gut hormones; however, the amount of existing literature is still limited. As prevalence of obesity increases, the importance to find innovative therapeutic options advanced, together with bariatric surgery and nutritional interventions. Gut hormones seem to satisfy this request by representing a potential and physiological drug targets for the treatment of obesity and its comorbidities (NAFLD, NASH, diabetes mellitus). For example, they demonstrated to improve insulin-sensitivity and to reduce, or at least to limit, hepatic injury due to accumulation of triglycerides in liver. However, the exact mechanism through which they may act as a beneficial and therapeutic role is still unclear. More information exist about GLP-1 analogs (as exendine-4) and their efficacy in controlling the evolution of NAFLD. Hence, we would like to emphasize that GLP-1 has not only an indirect incretin effect which improves key parameters associated with NAFLD, but it also shows a direct insulin-like role, repressing regulatory genes associated with obesity, NASH and hepatic triglyceride accumulation. Thus, synthetic GLP-1 like proteins would be of biological and therapeutic benefit in NAFLD. Obese individuals seem to lose their ability to respond to satiety signals; whether the use of CCK, OXM and ghrelin as a potential novel obesity therapy is in some doubt, the retain of PYY sensitivity in obese individuals makes it an attractive therapeutic target. Many of the current drug formulations available require injection, negatively influencing patients’ compliance to therapy, and present important side effects due to the ubiquitous distribution of the targeting receptors. So the attention is now focused on the necessity to develop orally administering, safe and more effective drugs. There still remains several scientific, regulatory and economic barriers to the development and mainstream clinical use of antiobesity pharmacotherapies.
ConclusionA better understanding of the pathophysiology of these hormones is of a great interest in order to clarify their burden in emerging diseases (such as obesity) not only for their treatment, but also in order to prevent their appearance and progression.
Conflict of interestThe authors declare no conflicts of interest.
Please cite this article as: Del Prete A, Iadevaia M, Loguercio C. The role of gut hormones in controlling the food intake. What is their role in emerging diseases? Endocrinol Nutr. 2012;59(3):197–206.
