


Turner syndrome is characterized by a great variability of clinical manifestations caused by a total or partial loss of X-chromosome.
Patients and methodsA retrospective, descriptive study of the diagnosis, course, and current status of patients with Turner syndrome followed up at our section over the past 40 years, based on review of medical records supplemented with a telephone survey.
ResultsForty-five female patients with a current mean age of 22.95years (range 2–38) and a mean age at diagnosis of 4.71 were included. Sixty-three percent of them showed a mosaic karyotype. Short stature was the most common reason for consultation (54%), with increased prenatal diagnosis in most recent cases. Seventy-two percent have been treated with growth hormone, together with oxandrolone in 26%. Final stature was short in 69% of patients. Gonadal failure was found in 66%; most of whom received replacement therapy. Three patients achieved pregnancy by oocyte donation. The 31 adult patients are mainly monitored by the endocrinology (37.5%) and/or gynecology (34.4%) departments. As regards psychosocial aspects, 22% required support during school, and 80% completed middle to high level education. Two patients died, one due to dissecting aortic aneurysm and the other one, who had multiple pathological conditions, from respiratory failure.
ConclusionsShort stature is the main cause of diagnosis in patients with Turner syndrome; most cases show genetic mosaicism. The most common clinical manifestations include short stature and gonadal failure. Eighty percent of patients complete middle or high education. In adulthood, follow-up is irregular, sometimes scarce, and clearly improvable.
El síndrome de Turner es definido por un conjunto de rasgos fenotípicos característicos resultantes de la alteración completa o parcial del cromosoma X.
Pacientes y métodosEstudio descriptivo retrospectivo en el que se analiza el diagnóstico, la evolución y la situación actual de pacientes controladas en los últimos 40años de síndrome de Turner en un hospital terciario (Bizkaia) mediante revisión de historias clínicas y encuestas telefónicas.
ResultadosEstudiamos 45 mujeres, con edad media actual de 22,95años (rango 2–38) y edad media de diagnóstico de 4,71años. El 63% presentaron mosaicismo en su cariotipo. El motivo de consulta más frecuente fue talla baja (54%), objetivándose un incremento de diagnóstico prenatal en los casos más recientes. Han recibido tratamiento con hormona de crecimiento el 72%, y el 26% además recibieron oxandrolona. En el 69% de las pacientes la talla final alcanzada fue baja. Han presentado fallo gonadal el 66%, recibiendo la mayoría tratamiento hormonal sustitutivo. Tres pacientes han tenido descendencia con óvulo de donante. El seguimiento de las 31 pacientes adultas es llevado a cabo fundamentalmente por endocrinología (37,5%) y/o ginecología (34,4%). En el ámbito psicosocial han precisado ayuda durante la escolarización el 22%, alcanzando el 80% estudios de nivel medio-alto. Dos pacientes han fallecido, una por disección de aneurisma aórtico y la otra paciente, con múltiples patologías, por insuficiencia respiratoria.
ComentariosLa talla baja es el motivo más frecuente de consulta en pacientes con síndrome de Turner. En la mayoría de los casos el cariotipo es mosaico. Las manifestaciones clínicas más frecuentes son talla baja y fallo gonadal. El 80% consiguen realizar estudios de nivel medio-alto. En la edad adulta el seguimiento es irregular y en ocasiones escaso, siendo claramente mejorable.
Turner syndrome (TS) is a genetic disease caused by complete or partial absence of the second X chromosome, with or without mosaicism, affecting approximately 1/2500 newborns with female phenotype. Its most common clinical manifestations include short stature, gonadal dysfunction, cardiac and renal malformations, ear and ophthalmological problems, and certain phenotypic traits. The literature shows that treatment with growth hormone (GH) is essential to improve short stature, and patients with ovarian insufficiency require estrogen therapy.1 Delaying estrogen therapy until 15 years of age to optimize potential stature, as previously recommended, appears inadvisable, because it underestimates the psychosocial importance of normal pubertal maturation. Van Pareren et al.2 and Rosenfield et al.3 suggested that starting estrogen therapy at 12 years of age allows for a normal pubertal development without interfering with the positive effect of GH on final adult stature. There are different routes to administer estrogens, of which the oral route has been most common to date. However, it is currently thought that transdermal preparations or depot injections of estrogens could be more convenient because they are more physiological alternatives.4–6 It is essential to educate patients on the importance of continuing hormone replacement therapy (HRT) until the normal age of menopause to maintain feminization and prevent osteoporosis.7
Women with TS have greater morbidity and mortality, caused mainly by cardiovascular diseases. They have an increased risk of other conditions such as osteoporosis, hypothyroidism, diabetes, and dyslipidemia.1 Specialists in pediatrics, gynecology and obstetrics, endocrinology, cardiology, genetics, otorhinolaryngology (ORL), and ophthalmology, amongst others, should participate in management.8 Intellectual development is usually normal, except in patients with a ring X chromosome, who have a greater risk of showing a variable intellectual deficit. A specific neurocognitive profile with defects in different areas of development related to visual and spatial perception, non-verbal communication, motor coordination, perceptive abilities, and visual memory is usually found.9 Recent international guidelines for management of women with TS recommend annual laboratory tests including blood glucose, liver and lipid profiles, and thyroid hormones, as well as frequent blood pressure (BP) monitoring, audiometry every 2–3 years, and cardiac imaging tests every 5–10 years.7
The results of a retrospective study conducted to describe the endocrine and psychosocial characteristics of patients with TS diagnosed at the pediatric endocrinology section of Hospital Universitario de Cruces in the past 40 years and their adult status are discussed below.
Patients and methodsA retrospective, descriptive study was conducted on patients diagnosed with TS at Hospital Universitario de Cruces in the past 40 years (Table 1). Data were collected from two sources, retrospective review of clinical histories from the pediatric endocrinology outpatient clinic and collection of current variables through a telephone survey.
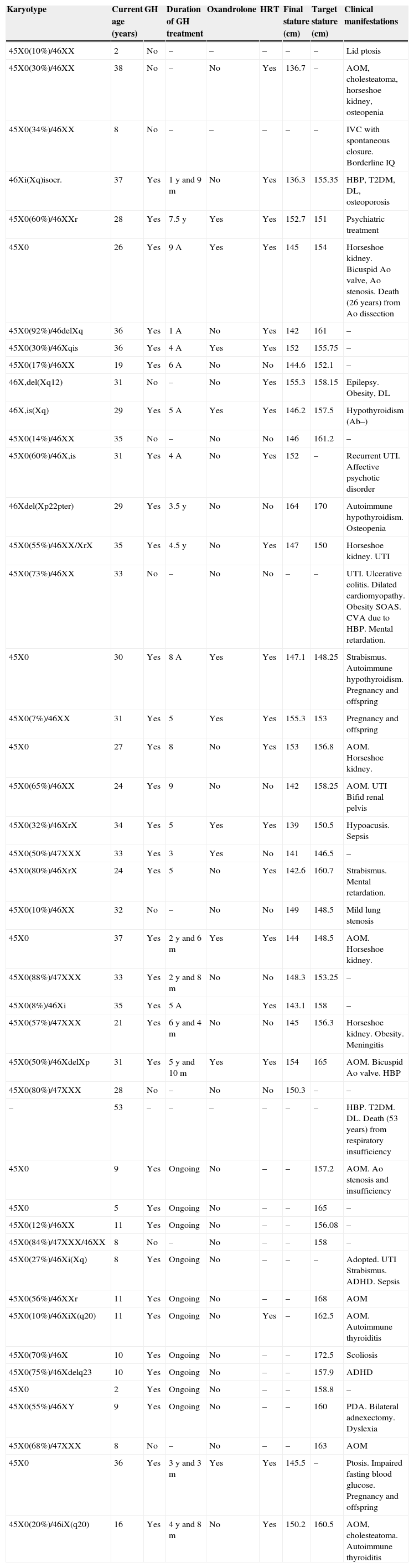
Summary of individual data from the 45 patients with Turner syndrome.
| Karyotype | Current age (years) | GH | Duration of GH treatment | Oxandrolone | HRT | Final stature (cm) | Target stature (cm) | Clinical manifestations |
|---|---|---|---|---|---|---|---|---|
| 45X0(10%)/46XX | 2 | No | – | – | – | – | – | Lid ptosis |
| 45X0(30%)/46XX | 38 | No | – | No | Yes | 136.7 | – | AOM, cholesteatoma, horseshoe kidney, osteopenia |
| 45X0(34%)/46XX | 8 | No | – | – | – | – | – | IVC with spontaneous closure. Borderline IQ |
| 46Xi(Xq)isocr. | 37 | Yes | 1 y and 9 m | No | Yes | 136.3 | 155.35 | HBP, T2DM, DL, osteoporosis |
| 45X0(60%)/46XXr | 28 | Yes | 7.5 y | Yes | Yes | 152.7 | 151 | Psychiatric treatment |
| 45X0 | 26 | Yes | 9 A | Yes | Yes | 145 | 154 | Horseshoe kidney. Bicuspid Ao valve, Ao stenosis. Death (26 years) from Ao dissection |
| 45X0(92%)/46delXq | 36 | Yes | 1 A | No | Yes | 142 | 161 | – |
| 45X0(30%)/46Xqis | 36 | Yes | 4 A | Yes | Yes | 152 | 155.75 | – |
| 45X0(17%)/46XX | 19 | Yes | 6 A | No | No | 144.6 | 152.1 | – |
| 46X,del(Xq12) | 31 | No | – | No | Yes | 155.3 | 158.15 | Epilepsy. Obesity, DL |
| 46X,is(Xq) | 29 | Yes | 5 A | Yes | Yes | 146.2 | 157.5 | Hypothyroidism (Ab–) |
| 45X0(14%)/46XX | 35 | No | – | No | No | 146 | 161.2 | – |
| 45X0(60%)/46X,is | 31 | Yes | 4 A | No | Yes | 152 | – | Recurrent UTI. Affective psychotic disorder |
| 46Xdel(Xp22pter) | 29 | Yes | 3.5 y | No | No | 164 | 170 | Autoimmune hypothyroidism. Osteopenia |
| 45X0(55%)/46XX/XrX | 35 | Yes | 4.5 y | No | Yes | 147 | 150 | Horseshoe kidney. UTI |
| 45X0(73%)/46XX | 33 | No | – | No | No | – | – | UTI. Ulcerative colitis. Dilated cardiomyopathy. Obesity SOAS. CVA due to HBP. Mental retardation. |
| 45X0 | 30 | Yes | 8 A | Yes | Yes | 147.1 | 148.25 | Strabismus. Autoimmune hypothyroidism. Pregnancy and offspring |
| 45X0(7%)/46XX | 31 | Yes | 5 | Yes | Yes | 155.3 | 153 | Pregnancy and offspring |
| 45X0 | 27 | Yes | 8 | No | Yes | 153 | 156.8 | AOM. Horseshoe kidney. |
| 45X0(65%)/46XX | 24 | Yes | 9 | No | No | 142 | 158.25 | AOM. UTI Bifid renal pelvis |
| 45X0(32%)/46XrX | 34 | Yes | 5 | Yes | Yes | 139 | 150.5 | Hypoacusis. Sepsis |
| 45X0(50%)/47XXX | 33 | Yes | 3 | Yes | No | 141 | 146.5 | – |
| 45X0(80%)/46XrX | 24 | Yes | 5 | No | Yes | 142.6 | 160.7 | Strabismus. Mental retardation. |
| 45X0(10%)/46XX | 32 | No | – | No | No | 149 | 148.5 | Mild lung stenosis |
| 45X0 | 37 | Yes | 2 y and 6 m | Yes | Yes | 144 | 148.5 | AOM. Horseshoe kidney. |
| 45X0(88%)/47XXX | 33 | Yes | 2 y and 8 m | No | No | 148.3 | 153.25 | – |
| 45X0(8%)/46Xi | 35 | Yes | 5 A | Yes | 143.1 | 158 | – | |
| 45X0(57%)/47XXX | 21 | Yes | 6 y and 4 m | No | No | 145 | 156.3 | Horseshoe kidney. Obesity. Meningitis |
| 45X0(50%)/46XdelXp | 31 | Yes | 5 y and 10 m | Yes | Yes | 154 | 165 | AOM. Bicuspid Ao valve. HBP |
| 45X0(80%)/47XXX | 28 | No | – | No | No | 150.3 | – | – |
| – | 53 | – | – | – | – | – | – | HBP. T2DM. DL. Death (53 years) from respiratory insufficiency |
| 45X0 | 9 | Yes | Ongoing | No | – | – | 157.2 | AOM. Ao stenosis and insufficiency |
| 45X0 | 5 | Yes | Ongoing | No | – | – | 165 | – |
| 45X0(12%)/46XX | 11 | Yes | Ongoing | No | – | – | 156.08 | – |
| 45X0(84%)/47XXX/46XX | 8 | No | – | No | – | – | 158 | – |
| 45X0(27%)/46Xi(Xq) | 8 | Yes | Ongoing | No | – | – | – | Adopted. UTI Strabismus. ADHD. Sepsis |
| 45X0(56%)/46XXr | 11 | Yes | Ongoing | No | – | – | 168 | AOM |
| 45X0(10%)/46XiX(q20) | 11 | Yes | Ongoing | No | Yes | – | 162.5 | AOM. Autoimmune thyroiditis |
| 45X0(70%)/46X | 10 | Yes | Ongoing | No | – | – | 172.5 | Scoliosis |
| 45X0(75%)/46Xdelq23 | 10 | Yes | Ongoing | No | – | – | 157.9 | ADHD |
| 45X0 | 2 | Yes | Ongoing | No | – | – | 158.8 | – |
| 45X0(55%)/46XY | 9 | Yes | Ongoing | No | – | – | 160 | PDA. Bilateral adnexectomy. Dyslexia |
| 45X0(68%)/47XXX | 8 | No | – | No | – | – | 163 | AOM |
| 45X0 | 36 | Yes | 3 y and 3 m | Yes | Yes | 145.5 | – | Ptosis. Impaired fasting blood glucose. Pregnancy and offspring |
| 45X0(20%)/46iX(q20) | 16 | Yes | 4 y and 8 m | No | Yes | 150.2 | 160.5 | AOM, cholesteatoma. Autoimmune thyroiditis |
Ab, antibodies; CVA, cerebrovascular accident; Ao, aortic; IQ, intelligence quotient; IVC, interventricular communication; DL, dyslipidemia; DM, diabetes mellitus; HBP, high blood pressure; UTI, urinary tract infection; PDA, patent ductus arteriosus; ADHD, attention deficit hyperactivity disorder.
Data collected referring to the pediatric stage included: age at diagnosis, reason for consultation, karyotype result, anthropometric data (baseline and over time), schooling (normal or with support), treatment with GH (with or without associated oxandrolone), adverse effects of this therapy, presence or absence of gonadal failure and whether treated or not with HRT, presence of immune, ophthalmological, ORL, renal, and cardiac disease.
Data collection was completed with a telephone survey to adult patients (aged 16 years or older) to collect the following information: adult stature; presence or absence of a planned transition from pediatric to adult specialists; current or absent medical monitoring; type of specialist caring for them; knowledge that they had been done some supplemental examinations (densitometry, lipid and carbohydrate profile, BP recording) since they were discharged from pediatric endocrinology; presence or absence of HRT; presence or absence of offspring; educational level and social position reached; social and couple relationships; and potential death.
The overall patient sample was divided into subgroups based on current age (older than 30 years, 16–30 years old, younger than 16 years) to compare some of the study variables, find out the different therapeutic trends over time, and see their potential impact on final stature.
Statistical methodsA statistical Chi-square test or a Fisher's exact test was used to compare two categorical variables. A Student's t tests or an ANOVA test was used to compare means. SPSS vs 21.0 statistical software was used to perform these calculations.
ResultsForty-five patients with TS were studied (Table 2). Their mean age at the time of the study was 22.95±11.7 years (range, 2–38 years), and their mean age at diagnosis was 4.71±4.33 years. Sixty-one percent of patients were older than 16 years n=31). The most common reason for consultation was short stature (54%), followed by a peculiar phenotype (17%), both manifestations together (11%), and prenatal diagnosis (13%). It should be noted that when the pediatric and adult groups were compared, prenatal diagnosis by amniocentesis was more common among younger patients (36% vs 3%; p=0.008).
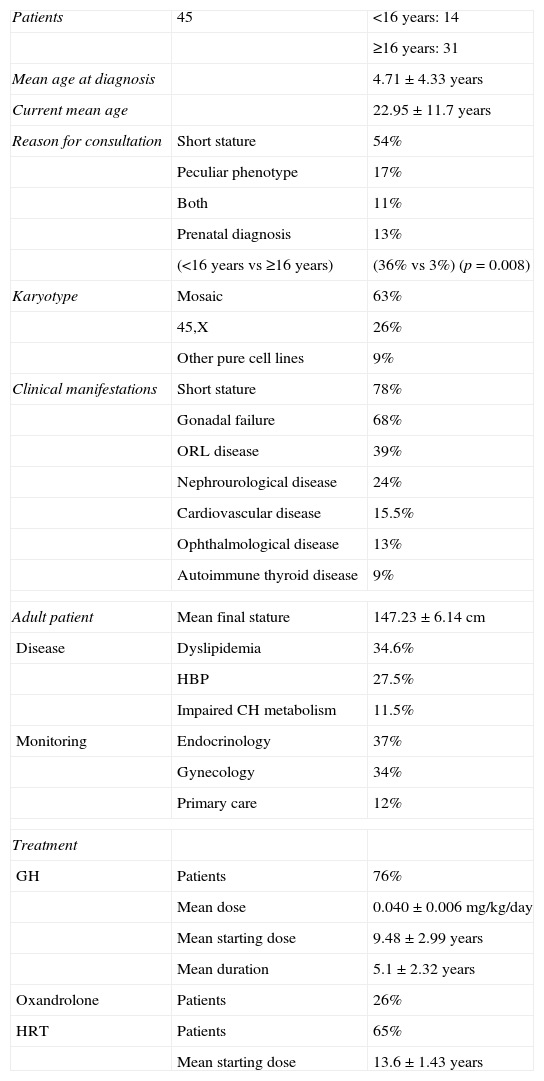
Initial and subsequent characteristics of patients with Turner syndrome.
| Patients | 45 | <16 years: 14 |
| ≥16 years: 31 | ||
| Mean age at diagnosis | 4.71±4.33 years | |
| Current mean age | 22.95±11.7 years | |
| Reason for consultation | Short stature | 54% |
| Peculiar phenotype | 17% | |
| Both | 11% | |
| Prenatal diagnosis | 13% | |
| (<16 years vs ≥16 years) | (36% vs 3%) (p=0.008) | |
| Karyotype | Mosaic | 63% |
| 45,X | 26% | |
| Other pure cell lines | 9% | |
| Clinical manifestations | Short stature | 78% |
| Gonadal failure | 68% | |
| ORL disease | 39% | |
| Nephrourological disease | 24% | |
| Cardiovascular disease | 15.5% | |
| Ophthalmological disease | 13% | |
| Autoimmune thyroid disease | 9% | |
| Adult patient | Mean final stature | 147.23±6.14cm |
| Disease | Dyslipidemia | 34.6% |
| HBP | 27.5% | |
| Impaired CH metabolism | 11.5% | |
| Monitoring | Endocrinology | 37% |
| Gynecology | 34% | |
| Primary care | 12% | |
| Treatment | ||
| GH | Patients | 76% |
| Mean dose | 0.040±0.006mg/kg/day | |
| Mean starting dose | 9.48±2.99 years | |
| Mean duration | 5.1±2.32 years | |
| Oxandrolone | Patients | 26% |
| HRT | Patients | 65% |
| Mean starting dose | 13.6±1.43 years | |
Karyotype was determined in peripheral blood lymphocytes, after culture for 72h and various treatments, through microscopic study of at least 20 metaphases, magnified 50 times in the event of mosaicism. Mosaicism was found in 63% of patients (in one case, 45,X/46,XY). It should be noted that five out of the six patients with prenatal diagnosis had a mosaic karyotype. Of the remaining cases, 26% were 45,X and 9% other pure cell lines: 46,Xi(Xq), 46,Xdel(Xq), or 46Xdel(Xp).
The most common clinical manifestation was short stature, found in 78%. All patients but one were treated with GH. Doses administered ranged from 0.030 to 0.054mg/kg/day, with a mean of 0.040±0.006mg/kg/day. Mean age at treatment start was 9.5±2.99 years. Treatment duration was highly variable (range, 1–9 years), with a mean of 5.1±2.32 years. No side effects of this therapy were recorded.
Oxandrolone was also administered to 26% of patients. A higher proportion of patients received oxandrolone in the older subgroup (<16 years, 0%; 16–30 years, 27.3%; ≥30 years, 47.4%; p=0.005). Oxandrolone was discontinued in three patients due to severe adverse effects: mild hirsutism in one patient and advanced bone age in the other two.
Mean final stature was 147.23±6.14cm (range, 136.3–164cm), with 69% of patients below percentile 3. Mean deviation from target stature was −8.22±6.45cm. There were no differences in the final stature reached over time (women younger or older than 30 years of age: 148.7±6.4 vs 146.3±6.0cm). No differences were also found between those with or without concomitant oxandrolone therapy (147.1±5.3 vs 147.3±6.8cm). There was no statistically significant difference between patients with 45X karyotype and all other karyotypes (147.65±3.68 vs 147.16±6.50cm).
Sixty-eight percent of patients had gonadal failure. All patients with gonadal failure but one (because of family refusal) received HRT starting with oral ethinyl estradiol 50ng/kg/day, followed by dose titration every 4–6 months up to 20μg/day at 18–24 months. Mean age at treatment start was 13.6±1.43 years (range, 11 and 16 years). A comparison of age of start of treatment between the patient subgroups showed that the therapy is being prescribed at increasingly younger ages in recent years (subgroup <16 years, 11.65±0.91 years; subgroup 16–30 years, 12.55±0.42 years; subgroup ≥30 years, 14.33±1.43 years; p=0.004). Three patients had offspring using donor oocytes.
The next most common associated manifestations include ORL conditions, prevalent in 39% of patients (recurrent otitis media, mild to moderate hearing loss, and cholesteatoma), nephrourological conditions, occurring in 24% of patients (horseshoe kidney or urinary tract infections), and ophthalmological conditions in 13% of cases. As regards autoimmune diseases, two patients each developed autoimmune thyroiditis and autoimmune hypothyroidism.
Associated cardiovascular conditions included congenital cardiac problems in six patients: three aortic abnormalities (bicuspid aortic valve and mild aortic stenosis and insufficiency), one interventricular communication, one mild pulmonary stenosis, and patent ductus arteriosus in one patient. A seventh patient was diagnosed with dilated cardiomyopathy in adulthood. Two of the 45 patients have died, one at 26 years of age due to aortic aneurysm and dissection, and the other at 53 years from acute respiratory insufficiency in the setting of a massive pleural effusion.
In the subgroup of 31 adult patients, transition from pediatric to adult endocrinological care occurred after the final stature was reached and pubertal development was completed, although transition was not planned. These patients are currently monitored at different departments: 37% by endocrinologists, 34% by gynecologists, and 12% by primary care physicians.
Complete medical data are available for 26 patients. Of these, 34.6% have an impaired lipid profile and 27.5% high blood pressure (HBP), and carbohydrate metabolism is altered in three patients. Six patients have undergone echocardiography in the past five years, but magnetic resonance imaging has been performed in one patient only. Sixty-two percent of patients continue on HRT. Osteopenia and osteoporosis were found each in one of the six patients performed densitometry.
Twenty-two percent of patients required cognitive assistance during schooling, and two had a significant mental retardation. One of these had 45,X/46,XX mosaicism. Socioemotional level was assessed in 24 women. Most of them (80%) had a middle-high educational level. As regards ability to relate, 6% reported that they had little social relations, and 41% of patients had had a couple at any time.
DiscussionIn TS, both the correlation between genotype and phenotype and age at diagnosis are highly variable. When diagnosis is made by amniocentesis,9 fetal phenotype is difficult to predict. Women with the pure 45X cell line have been shown to have higher risk of fetal death or greater clinical expression of TS.10 This study found that more cases had been diagnosed by amniocentesis in girls as compared to adult women, which reflects the higher proportion of amniocenteses currently performed. The most common karyotype in this study was a mosaicism, which contrasts with previous reports by other authors of a greater prevalence of the 45X karyotype. A possible explanation for this difference could be that pregnancies with 45X are voluntarily terminated, a fact that we cannot prove.
In the series reported by Savendahl and Davenport,11 15% of postnatal diagnoses were done in the neonatal stage and 38% at adult age. Thus, the mean age at diagnosis of 4.7 years is acceptable. As the most common reason for consultation was short stature, we emphasize the importance of including the karyotype when girls with short stature are studied, in accordance with recommendations in the clinical practice guidelines of the TS study published by Bondy7 in 2007.
Treatment with GH has been established to be effective for increasing final stature12 in these patients, but controversy exists about the dose and best time of start of therapy.8,13 Mean stature in the series is similar to that reported in the Canadian study, the only randomized and controlled study showing a mean benefit of 6.5cm with GH treatment in these patients.12 Mean dose in our study was within the recommended range.7,9
Mean stature in our series was shorter than reported in other studies, such as the multicenter US study conducted by Rosenfeld et al.,14 where patients treated with GH alone achieved a mean final height of 150.4±5.5cm, while those given GH and oxandrolone reached 152.1±5.9cm with early start and long duration of treatment.
The Bondy7 clinical guideline recommends assessment of higher GH doses and concomitant use of a non-aromatizing anabolic steroid such as oxandrolone in patients over 9 years and/or with extremely short stature. In our study, the patient subgroup given concurrent oxandrolone did not achieve a higher final stature. Because of this, oxandrolone was discontinued 15 years ago.
In a recent study, Menke et al.15 showed that treatment with GH combined with low dose oxandrolone (0.03mg/kg/day) moderately increased the final stature achieved with an acceptable safety profile, excluding a small deceleration in breast development.
Only 20–30% of women with TS experience spontaneous puberty.9 According to this, 69% of patients in our series had primary hypogonadism and received estrogen therapy. Decreased estradiol secretion may cause lower bone mineral density (BMD) in both girls and young adults. HRT is a crucial treatment to prevent fast BMD decrease, to induce maximum bone mass peak in young women with TS without spontaneous pubertal development,7 and for subsequent maintenance of adequate bone mass.8 Densitometry was only performed in six of the patients, in whom osteopenia and osteoporosis were detected in one patient each. Three patients had offspring using donor oocytes. In women with TS, pregnancy represents a risk due to increased cardiovascular morbidity, and they should therefore be adequately informed.16,17
Cardiovascular abnormalities are among the most significant consequences of X chromosome haploinsufficiency. Incidence of cardiovascular lesions ranges from 23% to 45% depending on the series18,19 and on the type of supplemental examination used. Angio-MRI is the test that diagnoses more lesions.20 Bicuspid aortic valve (13–34%) and coarctation of the aorta (4–14%) are the most common abnormalities. A lower prevalence was found in this study (13%). Matura et al. showed that young women with TS are at high risk of aortic dissection and require close monitoring with measurements of indexed aortic size.17,21 Prevalence of HBP in our study was 27.5%, which agrees with data reported in the literature.22,23
Prevalence of ORL, renal, and ophthalmological changes in this group of patients agrees with reports in other series.7–9 It should be noted that the prevalence of autoimmune disease found in our study was lower than reported by other authors, despite annual screening in all girls.24
Medical control of adult patients with TS varies widely and is often inadequate, considering their high morbidity. Patient referral to units specialized in TS is recommended, and special attention should be paid to women with low educational and socioeconomic levels.1 Devernay et al.1 reported inadequate monitoring of their patients during the transition phase. In our study, patient referral to adult specialists was not homogeneous, partly because of the long time period covered.
The most recent clinical practice guidelines in TS recommend regular, multidisciplinary monitoring and follow-up of these patients including the following: clinical examination (general, cardiac, and ORL), BP measurement, echocardiography and/or cardiac angio-MRI, audiometry, densitometry, HRT monitoring, and psychosocial evaluation.7 Adult endocrinologists have been suggested to be most adequate for this monitoring,1 and were in charge of it in 37% of patients in our series. Since a majority of adult women were not performed the examinations recommended by experts,25 the transition phase of these patients and their monitoring in adulthood need to be improved.
Epidemiological studies show a three times higher mortality rate in patients with TS as compared to the general population based on the prevalence of HBP and cardiovascular diseases.21,26,27 In agreement with previous reports, two women from this series died at an early age.
According to some authors,9,28,29 women with TS have psychosocial and cognitive difficulties such as increased risk of social isolation, disorders related to anxiety and difficulty for couple relationships, with delayed exit from parental home and late start of sexual life. However, most women with TS get a good education and find paid jobs, as occurred in our series.
This study was intended to emphasize the need for improving both the transition phase and monitoring in adult patients, because monitoring (especially cardiovascular) is inadequate in many of them, and magnetic resonance imaging is required in all patients. We also think that the high rate of patients with middle to high educational levels may be an optimistic data when informing patients and their families.
Study limitations are mainly due to its retrospective nature, and in the temporal and spatial dispersion of patients, who were monitored at different healthcare centers over 40 years, which makes data collection difficult and impairs sample homogeneity.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Ríos Orbañanos I, Vela Desojo A, Martinez-Indart L, Grau Bolado G, Rodriguez Estevez A, Rica Echevarria I. Síndrome de Turner: del nacimiento a la edad adulta. Endocrinol Nutr. 2015;62:499–506.
