



El conocimiento de la patogenia de la enfermedad inflamatoria intestinal ha ido evolucionando con el tiempo. La hipótesis más aceptada en el momento actual es que la enfermedad inflamatoria intestinal se origina como consecuencia de una respuesta inmunológica aberrante en un huésped genéticamente susceptible. Las últimas 2 décadas han supuesto un enorme avance en este conocimiento a partir de investigaciones que complementan esta hipótesis desde varios frentes distintos. El objetivo de esta revisión es el de recorrer los trabajos que han supuesto un descubrimiento cualitativo en esta materia, abarcando desde la influencia de la microbiota hasta los avances en el estudio de la genética y, por último, el enfoque de la biología de sistemas.
Knowledge of the pathogenesis of inflammatory bowel disease has evolved over time. The most accepted hypothesis nowadays supports the fact that inflammatory bowel disease is the result of an aberrant immune response in a genetically susceptible host. The last 2 decades have signified a huge advance in this knowledge coming from different perspectives. The objective of this review is to explore studies that represent a qualitative breakthrough in this area, ranging from the influence of microbiota to advances in genetics and finally systems biology approach.
Tanto para la enfermedad de Crohn (EC) como para la colitis ulcerosa, las 2 formas más conocidas de enfermedad inflamatoria intestinal (EII), la hipótesis más aceptada en el momento actual es que la EII se origina como consecuencia de una respuesta inmunológica aberrante a bacterias no (necesariamente) patógenas en un huésped genéticamente susceptible. Anteriormente se pensaba que la EII se debía a inmunodeficiencia (defectos fundamentalmente en la inmunidad innata) o a infección micobacteriana crónica1–3. Todas estas hipótesis, incluso la más reciente, tienen un fondo común, comparten mecanismos patológicos y es probable que describan fenómenos que pueden coexistir en lo que fenotípicamente se conoce como EII y que probablemente represente a diversas entidades4.
El conocimiento de la patogenia de la EII ha ido evolucionando con el tiempo. Las últimas 2 décadas han supuesto un enorme avance en este conocimiento a partir de investigaciones que complementan esta hipótesis desde varios frentes distintos. El objetivo de esta revisión es el de recorrer los trabajos que han supuesto un descubrimiento cualitativo en esta materia.
Microbiota intestinalLa microbiota intestinal está conformada por los microorganismos que habitan en el intestino. Se estima que el intestino alberga alrededor de 1.800 géneros y más de 3.500 especies de bacterias5, algunas de ellas todavía desconocidas, constituyendo en conjunto un mayor número de células procariotas que el de las células somáticas y germinales de un ser humano6. La microbiota es esencial para el correcto desarrollo del sistema inmune intestinal, provee elementos nutricionales y también modula el metabolismo energético7. Los sujetos sanos presentan una microbiota intestinal de alta biodiversidad, rica en especies comensales y baja en concentración de patógenos intestinales. Cuando se altera la homeostasis en el epitelio intestinal, la misma flora comensal puede actuar como un patógeno, lo que perpetúa la respuesta inflamatoria. Varias especies comensales pueden inducir enfermedad de forma selectiva en huéspedes con diversos trasfondos genéticos y también pueden causar distintos fenotipos de enfermedad en un único huésped.
Son varios los argumentos que vinculan la flora entérica con la patogenia de la EII, entre ellos cabe destacar los efectos fisiológicos de la flora entérica en la estructura mucosa, en el recambio celular, la motilidad y el funcionamiento y desarrollo inmune8–10; la reactividad inmunológica hacia la flora en pacientes con EII (pérdida de tolerancia)11,12; la atenuación de inflamación en modelos animales de EII cuando se encuentran en condiciones exentas de bacterias13; el hecho de que las lesiones ocurran predominantemente en sitios con mayor exposición bacteriana (íleon y colon)14; la respuesta clínica a la derivación del contenido fecal, recidiva ante la restauración o exposición de materia fecal15, y por último, la eficacia de los antibióticos y probióticos en pouchitis y para la prevención de recurrencia posquirúrgica16,17.
Más específicamente, una de las bacterias más estudiadas en la patogenia de la EC es Escherichia coli (E. coli). Los pacientes con EC (sobre todo aquellos con afectación ileal) presentan mayor número de cepas enteroadhesivas/enteroinvasivas de E. coli en comparación de los controles sanos18. Estas cepas son además más propensas a sobrevivir y replicarse dentro de los macrófagos que generan altos niveles de TNFα en respuesta a la infección19. Se han descrito diversos factores de virulencia asociados a otras bacterias comensales y que también podrían inducir enfermedad (citotoxina en Bacteroides fragilis, citotoxina del Clostridium difficile, superoxidasa en el Enterococcus faecalis, etc.)20.
La EII, sobre todo la EC, se caracteriza por presentar un desequilibrio en la composición de la microbiota intestinal, lo que se conoce como disbiosis. Los individuos afectos de EC presentan una flora inestable que se caracteriza por una reducción de la diversidad en las familias más abundantes de la flora intestinal, sobre todo las asociadas a la mucosa, tales como Firmicutes (bacterias grampositivas que incluyen la familia de Clostridium y Bacillus) y Bacteroidetes5. Otras especies que se encuentran disminuidas son Dialister invisus (del grupo de Clostridium), Faecalibacterium prausnitzii y Bifidobacterium adolescentes. Asimismo, presentan un aumento de Enterobacteriaceae, Proteobacteria y Fusobacteria21. Se ha demostrado cómo miembros de la familia de los Clostridiales (Faecalibaterium y Roseburia) se encuentran claramente disminuidos en fases de intensa actividad en los pacientes con EC ileal. Estos grupos son potentes fuentes de ácidos grasos de cadena corta, como el butirato, el cual ha demostrado proteger la mucosa en los modelos murinos de colitis. Por el contrario, Ruminococcus gnavus22 está aumentada. Estas bacterias tienen capacidad para degradar las mucinas intestinales por lo que esto, añadido a la baja producción de butirato, podría ser una de las causas por la que la disbiosis podría alterar la barrera epitelial intestinal22. Inicialmente se pensaba que la disbiosis era resultado de la inflamación, por ejemplo por las diferencias en las condiciones ecológicas del medio intestinal debidas a la inflamación, con cambios en el pH, el potencial redox y en la disponibilidad de sustratos. Actualmente se sabe que existe disbiosis en los pacientes con EC incluso en ausencia de inflamación21. Por otra parte, parece que la disbiosis podría ser uno de los vínculos entre genética y microbiota, ya que se ha visto que familiares sanos de pacientes con EC tienen también cierto grado de disbiosis (menor cantidad de Collinsella aerofaciens y de un miembro no identificado de la familia E. coli-Shigella, así como aumento de Ruminococcus torques) que los diferencia de sujetos sanos sin antecedentes familiares de EC22.
A pesar de los avances en el estudio de la influencia de la microbiota en la patogenia de la EII (tabla 1) quedan aún asuntos importantes por abordar en profundidad que confirmen una relación causa-efecto entre las alteraciones de la microbiota y la EII: identificar el rol de la microbiota intestinal en el inicio de la enfermedad, determinar si la composición microbiana puede predecir el riesgo subsecuente de brotes de actividad, o examinar si la flora luminal puede predecir la respuesta al tratamiento. En resumen, conocer en profundidad la relación de la microbiota intestinal con el huésped afecto de EII.
Cambios en el microbioma asociados a la EII
| Composición microbiana | Disminución en la α-diversidad (o riqueza de especies en términos de ecología) Disminución en Bacteroides, Clostridium, Ruminococcaceae, Bifidobacterium, Lactobacillus, Fecalibacterium prausnitzii y Firmicutes Aumento en Bacteroides, Pectinatus, Sutterella, Fusobacterium, Verrumicrobium, Clostridia, Mycobacterium paratuberculosis, Mycobacterium paramyxovirus, Listeria monocitogenes, Escherichia coli Presencia de Escherichia coli enteroadhesiva-enteroinvasiva Presencia de Fusobacterium |
| Función microbiana | Disminución en butirato y otros ácidos grasos de cadena corta Disminución del metabolismo del butirato y el propionato Disminución de la biosíntesis de aminoácidos Aumento de la auxotrofía (incapacidad de proliferar de un microorganismo sin el soporte de sustancias externas) Aumento del transporte de aminoácidos Aumento del transporte de sulfatos Aumento del estrés oxidativo Aumento de la secreción de toxinas |
Modificada de Kostic et al.48.
A pesar de que el estudio de la influencia de la genética en la fisiopatología de la EC empieza formalmente a mediados de los 90, con los primeros estudios de ligamiento, es en 2001 cuando se describe el primer locus de susceptibilidad significativa en una región del cromosoma 16 (conocido como IBD1), que puede albergar las mutaciones específicas R702W, G908R y L1007Fs, todas parte del gen NOD2 (conocido en ese momento también como CARD15)23,24. Con este descubrimiento, uno de los mayores logros en el estudio de la patogenia de la EC, se ha demostrado la importancia de los defectos de inmunidad innata. Estas variantes suponen un riesgo relativo de padecer la enfermedad de 2-4 en heterocigotos y de 20-40 en homocigotos, estando por lo menos una de estas variantes en hasta un 40% de los pacientes con EC en comparación con un 6-7% del resto de población europea sin la enfermedad25. El hallazgo de las mutaciones en CARD15/NOD2 pone en evidencia la importancia de las células de Paneth (y su producción de defensinas) en el mantenimiento de la homeostasis intestinal, sobre todo en el íleon terminal19, localización más frecuente de la EC. Esta mutación se asocia, por tanto, a la localización ileal de la EC, pero también al patrón estenosante, al comienzo en edades tempranas, a mayor translocación bacteriana y a mayor riesgo de resecciones intestinales26.
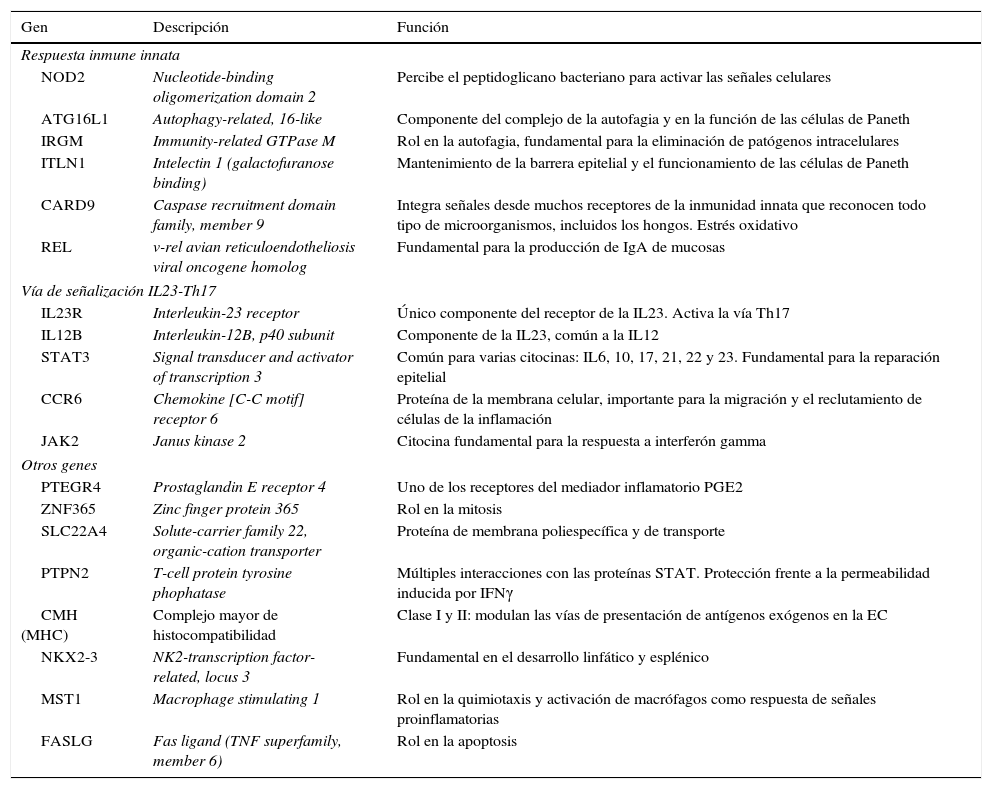
Otra de las grandes aportaciones del estudio de la genética en la EII radica en la exploración del genoma humano y el estudio de los polimorfismos de nucleótido único (SNP, del inglés single nucleotide polimorphism) de susceptibilidad de la enfermedad. Los avances tecnológicos en materia de microarrays han permitido que sea posible realizar el genotipado de cientos de miles de SNP dispersos por todo el genoma. Estos eventos han dado paso al inicio de los estudios de asociación a través del genoma (GWAS, del inglés genome-wide association studies) para poder identificar los loci asociados a rasgos complejos o riesgo de padecer determinada enfermedad. El primer estudio de GWAS en la EII data de 200527 y a partir de este momento se han estudiado entre 100.000 y 600.000 SNP. A partir de estos hallazgos se han descrito genes y vías de señalización que han permitido inferir los procesos biológicos y los tipos celulares involucrados en la patogenia de la EII, así como el fenotipo final de la enfermedad. Así, estos nuevos descubrimientos resaltan la importancia de la función-barrera intestinal, las respuestas microbianas específicas (autofagia, estrés del retículo endoplasmático, fagocitosis) y la complejidad de las respuestas de inmunidad innata (proteínas nucleotide oligomerization domain [NOD], toll-like receptor [TRL]) y adaptativa (vía de las Th17)28. Otra ventaja de los estudio de GWAS ha sido demostrar la superposición entre la EC y otras enfermedades relacionadas con la autoinmunidad. Hasta un 30% de las variantes de asociación se comparten con la colitis ulcerosa, mientras hasta un 50% de los loci se han encontrado también asociados a diabetes tipo 1, celiaquía o artritis reumatoide29. El estudio más reciente utilizando esta herramienta demuestra la presencia de 140 loci de susceptibilidad para la EC30. Estos, junto a los 23 loci relacionados específicamente con la colitis ulcerosa, componen los 163 loci asociados a la EII, uno de los conjuntos de loci más extensos en el estudio de cualquier enfermedad compleja hasta la fecha31. En la tabla 2 se resaltan los loci mejor estudiados en la EII.
Asociaciones genéticas en la EII más conocidas
| Gen | Descripción | Función |
|---|---|---|
| Respuesta inmune innata | ||
| NOD2 | Nucleotide-binding oligomerization domain 2 | Percibe el peptidoglicano bacteriano para activar las señales celulares |
| ATG16L1 | Autophagy-related, 16-like | Componente del complejo de la autofagia y en la función de las células de Paneth |
| IRGM | Immunity-related GTPase M | Rol en la autofagia, fundamental para la eliminación de patógenos intracelulares |
| ITLN1 | Intelectin 1 (galactofuranose binding) | Mantenimiento de la barrera epitelial y el funcionamiento de las células de Paneth |
| CARD9 | Caspase recruitment domain family, member 9 | Integra señales desde muchos receptores de la inmunidad innata que reconocen todo tipo de microorganismos, incluidos los hongos. Estrés oxidativo |
| REL | v-rel avian reticuloendotheliosis viral oncogene homolog | Fundamental para la producción de IgA de mucosas |
| Vía de señalización IL23-Th17 | ||
| IL23R | Interleukin-23 receptor | Único componente del receptor de la IL23. Activa la vía Th17 |
| IL12B | Interleukin-12B, p40 subunit | Componente de la IL23, común a la IL12 |
| STAT3 | Signal transducer and activator of transcription 3 | Común para varias citocinas: IL6, 10, 17, 21, 22 y 23. Fundamental para la reparación epitelial |
| CCR6 | Chemokine [C-C motif] receptor 6 | Proteína de la membrana celular, importante para la migración y el reclutamiento de células de la inflamación |
| JAK2 | Janus kinase 2 | Citocina fundamental para la respuesta a interferón gamma |
| Otros genes | ||
| PTEGR4 | Prostaglandin E receptor 4 | Uno de los receptores del mediador inflamatorio PGE2 |
| ZNF365 | Zinc finger protein 365 | Rol en la mitosis |
| SLC22A4 | Solute-carrier family 22, organic-cation transporter | Proteína de membrana poliespecífica y de transporte |
| PTPN2 | T-cell protein tyrosine phophatase | Múltiples interacciones con las proteínas STAT. Protección frente a la permeabilidad inducida por IFNγ |
| CMH (MHC) | Complejo mayor de histocompatibilidad | Clase I y II: modulan las vías de presentación de antígenos exógenos en la EC |
| NKX2-3 | NK2-transcription factor-related, locus 3 | Fundamental en el desarrollo linfático y esplénico |
| MST1 | Macrophage stimulating 1 | Rol en la quimiotaxis y activación de macrófagos como respuesta de señales proinflamatorias |
| FASLG | Fas ligand (TNF superfamily, member 6) | Rol en la apoptosis |
Modificada de Abraham y Cho49.
Además de los SNP en el gen NOD2, el conjunto de las demás mutaciones descritas hasta el momento relacionadas con el riesgo a padecer la EII se han asociado con desequilibrios en funciones biológicas claves de la inmunidad innata como la autofagia y la señalización generada a través del receptor de la interleucina 23 (IL23R).
La autofagia puede disminuir su capacidad defensiva en los pacientes con EII por la existencia de SNP en genes como ATG16L1 (autophagy-related, 16-like), IRGM (immunity-related GTPase M), ULK1 (unc-51 like autophagy activating kinase) y NCF4 (neutrophil cytosolic factor 4), y cuyo buen funcionamiento comporta la generación de las vesículas autofágicas perfectamente funcionales. La autofagia es un proceso catabólico en el que la célula es capaz de reciclar sus organelas dañadas mediante la maquinaria lisosomal, permitiendo rehacer la célula después de una agresión. Por otra parte, la autofagia también desempeña un papel importante en la defensa contra bacterias intracelulares y la presentación antigénica realizada por las células fagocíticas. Defectos en el gen ATG16L1 se han asociado con la autofagia ineficaz frente a Salmonella typhymurium32, mientras que las células deficientes de IRGM tienen una respuesta inadecuada frente a organismos intracelulares como Toxoplasma gondii, Listeria monocytogenes y Mycobacterium tuberculosis33. También se ha demostrado que tanto ATG16L1 como IRGM son importantes para el control de la E. coli enteroadhesiva-enteroinvasiva34. Recientemente se ha descrito que IRGM no solo controla la autofagia sino que también tiene capacidad de inducir la apoptosis (o muerte celular programada)35.
La vía del IL23R se ha vinculado a la EC desde hace casi una década. La IL23 es esencial en la polarización de la respuesta inmune hacía Th17, muy relevante en la patogenia de la EC. En resumen, la IL23 actúa amplificando de forma potente la cascada inflamatoria a través de la vía de respuesta inmune adaptativa conocida como Th17. La producción de IL23 parece circunscrita a las células del sistema inmune innato, particularmente las células dendríticas y macrófagos. El IL23R también se expresa en las células dendríticas maduras así como las natural killer. Es por eso que los polimorfismos que resalten la producción de IL23 podrían ser considerados como factores genéticos de riesgo de padecer EC36. A la luz de estos hallazgos se ha priorizado esta vía como diana terapéutica: la administración de anticuerpos anti-p40, que bloquean la IL23, ha demostrado ser efectiva en el tratamiento de inducción y mantenimiento de la EC37.
Biología de sistemas en enfermedad inflamatoria intestinal: estudios -ómicosUno de los mayores retos actuales al que se enfrenta la investigación en la EII es la dificultad de integrar el gran volumen de datos procedentes de distintas fuentes de información generada mediante tecnología «ómica». Métodos de computación y bioanálisis avanzados permiten actualmente integrar toda la información «ómica» para generar nuevas hipótesis más reduccionistas, capaces de ser demostradas experimentalmente. De las fuentes más estudiadas, y que integran la biología de sistemas, destacan la genómica (con los estudios de GWAS que ya se han comentado previamente), la transcriptómica y la epigenética, sin olvidar el microbioma (plural de microbiota y que incluye la totalidad de los microbios del organismos, la genética aportada por los mismos, así como su interacción medioambiental) y el metaboloma (a través del estudio de los productos del metabolismo).
La transcriptómica es el estudio del conjunto de transcritos o perfiles de expresión de ARN producidos en una estado de desarrollo específico o condición fisiológica. Junto con los estudios de traducción y los de interacción proteína-proteína, la transcriptómica compone lo que se conoce como genética funcional, que es un campo de la biología molecular donde se abarcan los aspectos dinámicos de los genes (en oposición a los aspectos estáticos de la información genómica como la secuencia de ADN o su estructura). Se ha demostrado que existe un perfil diferencial en el transcriptoma del mismo paciente con EC y la zona estudiada (sea colon y sus diferentes segmentos o íleon) lo que hace pensar en que podría realizarse la caracterización anatómica exclusiva en base a su transcriptoma38. De todas formas, las diferencias más estudiadas entre zonas intestinales afectadas e indemnes se han focalizado en estudiar las vías que estudian la inflamación39, por lo que queda un amplio camino por explorar en el estudio transcriptómico de la EII.
Elementos de regulación postranscripcional, los microARN (miRNA) son un tipo de ARN de cadena simple, no codificantes, endógenos y de pequeño tamaño (18-24 nucleótidos). Regulan la expresión genética mediante el control de la estabilidad y translación de los mRNA que codifican proteínas. Regulan también diversos procesos biológicos como la diferenciación celular, proliferación, apoptosis y el control del ciclo celular. Por tanto, su desregulación se ha asociado a diversas enfermedades, entre ellas la EII; como ejemplo está el miR-196 que es un miRNA que se encuentra sobreexpresado en los pacientes con EC y que ha demostrado regular la expresión de IRGM40. El rol de los miRNA en la EII supone, al igual que la epigenética, como se verá más adelante, una nueva manera de entender esta enfermedad y permite su utilización como estrategia potencial de tratamiento (diana terapéutica). Estudiando sangre periférica se han identificado miRNA con capacidad de distinguir subtipos de EII, por lo que plantean la posibilidad de ser utilizados también como biomarcadores de la enfermedad41. Los miRNA asociados a EII también pueden servir de delineadores de la expresión génica, ya que controlan de forma estrecha los niveles de mRNA específicos. De esa forma, pequeñas alteraciones en los niveles de mRNA producen cambios importantes en la síntesis de proteínas42, como por ejemplo se ha demostrado que la disminución entre la unión del miRNA Let-73 y el Let-7f al gen IL23R da como resultado un aumento en los niveles de mRNA y su producción proteica, lo que a su vez conduce a una desregulación de la vía de señalización IL23R en la EII43.
La epigenética podría ser el vínculo de unión entre el genoma humano, el medio ambiente y el desarrollo fenotípico de la EII. Un gran número de factores ambientales ha demostrado inducir cambios epigenéticos incluyendo tabaco, polución ambiental, asbestos, metales, sílice y bencenos. De forma clásica, la epigenética engloba los cambios del fenotipo heredados e independientes a las secuencias de ADN. Representa los eventos asociados a la cromatina que regulan un amplio espectro de procesos que dependen del ADN, incluyendo la transcripción genética. Los avances recientes en el conocimiento de la epigenética se deben en gran medida al conocimiento cristalográfico de la estructura del nucleosoma, que es la unidad básica en empaquetamiento de la cromatina, pero también por las acciones de metilación y/o acetilación. La mayoría de los SNP relacionados con la EII están localizados en regiones no codificantes del genoma lo que muestra que pueden tener un rol en la regulación de la expresión génica44. Los primeros estudios de factores epigenéticos en la EII mostraron un perfil diferencial de miRNA (que también actúan como reguladores postranscripcionales de la expresión génica) en la mucosa cólica de pacientes con EC en comparación de controles sin EC45. Datos recientes han demostrado que las modificaciones de cromatina están relacionadas con la activación transcripcional de los genes del colágeno de tipo i en la transición intestinal endotelio-mesenquimal, lo que muestra que los cambios epigenéticos son capaces de regular los genes profibrogénicos, y por tanto la fibrogénesis intestinal46. Asimismo, se ha demostrado que las bacterias pueden regular la expresión génica epitelial y la respuesta inmune intestinal influyendo mediante mecanismos epigenéticos47.
ConclusiónEl conocimiento de la EII se está construyendo mediante el estudio de un gran número de procesos biológicos que determinan la patogenia de la enfermedad. La integración del exoma con los datos de secuenciación de los estudios de GWAS, el conocimiento de la regulación de la transcripción de genes clave, la realización de experimentos específicos según tipos celulares y los datos de la ecología compleja que representa la microbiota intestinal conforman el mayor reto actualmente. Es por esto que el futuro del estudio de la EII se centrará no solo en determinar genes causales, sino además en la demostración de su función patológica en la enfermedad, ensamblando estos genes en vías moleculares y redes celulares para la mejora del manejo de la enfermedad mediante la medicina personalizada (por ejemplo qué pacientes evolucionarán a fenotipos complicados, quiénes presentarán una progresión lenta de la EC, cuál será la frecuencia de los brotes de actividad, cómo predecir el fracaso terapéutico o quiénes presentarán recurrencia posquirúrgica precoz).
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLa autora declara no tener ningún conflicto de intereses.

