


A pesar de la mejor optimización posible de los fármacos disponibles en la enfermedad inflamatoria intestinal (EII), un grupo importante de pacientes precisará de nuevas alternativas terapéuticas. Afortunadamente, existen ya incorporaciones recientes como el vedolizumab o próximas como el ustekinumab; sin embargo, son muchas más las que previsiblemente estarán disponibles en un futuro no muy lejano. En esta revisión repasamos las evidencias y posibles usos de las alternativas con más probabilidad de llegar a la práctica clínica. Entre ellas destacamos los fármacos que bloquean la adhesión leucocitaria, los inhibidores de las JAK quinasas, anticitocinas, con especial hincapié en el ustekinumab, y el mongersen.
In spite of the best possible optimisation of the available drugs in inflammatory bowel disease (IBD), an important group of patients will require new therapeutic alternatives. Fortunately, there are already new drugs such as vedolizumab or ustekinumab, but, there are many more that will be available in a near future. In this article, a review is made of the evidence and the possible uses of these alternatives. Some of them are drugs that inhibit white cell adhesion, the JAK-kinase inhibitors, the anti-cytokines (especially, ustekinumab), and mongersen.
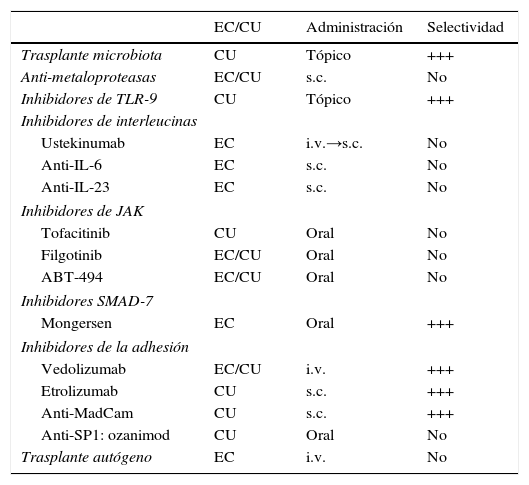
La introducción de los anti-TNF supuso un gran avance en el manejo de la enfermedad inflamatoria intestinal (EII) y a pesar de que su manejo esté cada vez más optimizado (determinación de niveles y anticuerpos, tratamiento combinado, etc.) existe un grupo amplio de pacientes en los que no se logra el control de la enfermedad. Así, hasta un 36-40% no responden inicialmente en los ensayos clínicos, siendo este porcentaje algo inferior en la práctica clínica (13-33%)1. Por otro lado, la pérdida de respuesta en la enfermedad de Crohn (EC) a lo largo del tiempo se da en un 10-21% de pacientes/año2,3 empleándose un segundo anti-TNF (en ausencia de disponibilidad de niveles de fármaco) cuya durabilidad es menor que el primero4. En el caso de la colitis ulcerosa (CU) estas cifras de pérdida de respuesta pueden ser aún mayores, oscilando entre el 46 y el 67%. Por todo ello, parece clara la necesidad de nuevos tratamientos preferentemente con mecanismo de acción diferente del bloqueo anti-TNF. En esta revisión nos detendremos en los fármacos con desarrollo clínico más avanzado (tabla 1) y en las novedades de los ya disponibles bajo la fórmula de fármaco fuera de indicación pero próximos a su comercialización (ustekinumab).
Nuevas dianas terapéuticas en la enfermedad inflamatoria intestinal
| EC/CU | Administración | Selectividad | |
|---|---|---|---|
| Trasplante microbiota | CU | Tópico | +++ |
| Anti-metaloproteasas | EC/CU | s.c. | No |
| Inhibidores de TLR-9 | CU | Tópico | +++ |
| Inhibidores de interleucinas | |||
| Ustekinumab | EC | i.v.→s.c. | No |
| Anti-IL-6 | EC | s.c. | No |
| Anti-IL-23 | EC | s.c. | No |
| Inhibidores de JAK | |||
| Tofacitinib | CU | Oral | No |
| Filgotinib | EC/CU | Oral | No |
| ABT-494 | EC/CU | Oral | No |
| Inhibidores SMAD-7 | |||
| Mongersen | EC | Oral | +++ |
| Inhibidores de la adhesión | |||
| Vedolizumab | EC/CU | i.v. | +++ |
| Etrolizumab | CU | s.c. | +++ |
| Anti-MadCam | CU | s.c. | +++ |
| Anti-SP1: ozanimod | CU | Oral | No |
| Trasplante autógeno | EC | i.v. | No |
Este fármaco es un anticuerpo monoclonal IgG1 humano frente a la proteína p40 que forma parte de las interleucinas (IL) 12 y 23, ambas implicadas en la etiopatogenia de la EC, bloqueando de esta forma a ambas. La IL-12 está implicada en la polarización de los linfocitos naive a polarizarse hacia una respuesta TH-1 que aparece en las fases iniciales de la EC5,6. Sin embargo, en fases tardías de la enfermedad predomina una respuesta TH-17 en la que participa la IL-23, de esta forma el ustekinumab bloquearía ambos tipos de respuesta.
Este fármaco tiene una vida media de 3 semanas aproximadamente (15-32 días)7, alcanzando la concentración sérica máxima después de una dosis de 90mg a los 8,5 días. Está comercializado para la psoriasis desde el año 2009 (posteriormente se ha añadido la indicación en pediatría y para la artritis psoriásica). La posología recomendada en estas indicaciones es de 45mg iniciales por vía subcutánea (s.c.) seguida de otra dosis a las 4 semanas y luego cada 12 semanas. En pacientes con un peso superior a 100kg la dosis se eleva a 90mg por administración. Dada la disponibilidad en el mercado desde hace años, la experiencia con este fármaco en España en la EC bajo la fórmula de tratamiento fuera de indicación es amplia y se ha comunicado recientemente8.
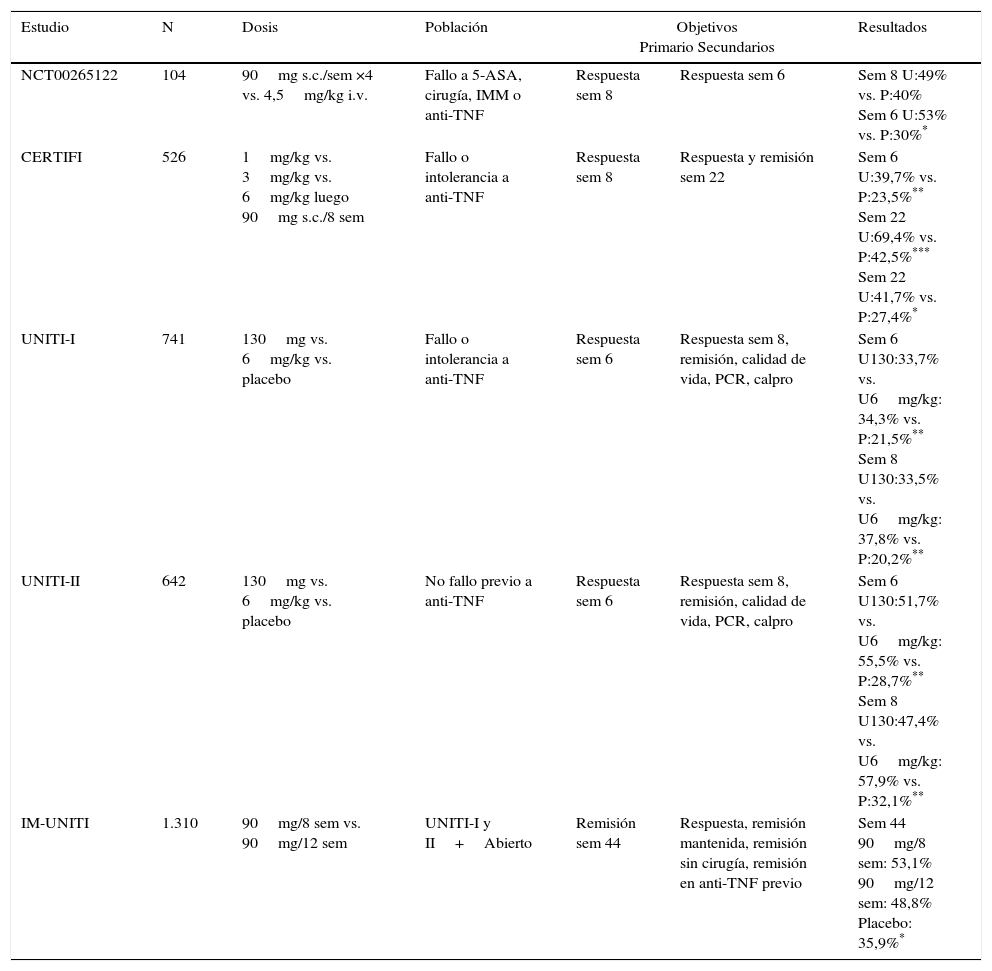
Los ensayos clínicos publicados se muestran en la tabla 2. El primero de ellos es un ensayo controlado fase IIa9 que incluye a 2 poblaciones diferentes con los mismos criterios de inclusión: CDAI entre 220 y 450 y fallo a tratamientos previos incluyendo anti-TNF pero no reciente (>16 semanas). En la primera población analizada los 104 pacientes incluidos se aleatorizaban a uno de los siguientes grupos de tratamiento: placebo s.c. en las semanas 0, 1, 2 y 3; ustekinumab s.c. 90mg en las mismas semanas; placebo intravenoso (i.v.) dosis única inicial y ustekinumab 4,5mg i.v. dosis única. El objetivo del estudio era valorar la respuesta en la semana 8, y en todos los grupos se administraba a partir de esa semana el tratamiento complementario, es decir, si habían recibido placebo s.c. en las primeras semanas luego se administraba el fármaco por la misma vía. No se encontraron diferencias significativas entre los grupos placebo y los de tratamiento valorados conjuntamente (40% vs. 49% respectivamente); sin embargo, en las semanas 4 y 6 sí existían diferencias a favor del grupo tratado con ustekinumab (53% vs. 30%; p=0,02). Lo más llamativo es que en los pacientes con exposición previa a anti-TNF las diferencias alcanzaban significación en todos los puntos analizados desde la semana 2 a la 8. En la segunda población de 27 pacientes se comparaba de forma abierta la administración i.v. con la s.c. sin encontrar diferencias. Un aspecto interesante es que un CDAI más elevado y un menor peso corporal se asociaban con una mejor respuesta; esto último podría estar en relación con una posible infradosificación del fármaco en el estudio.
Ensayos clínicos con ustekinumab en la enfermedad de Crohn
| Estudio | N | Dosis | Población | Objetivos Primario Secundarios | Resultados | |
|---|---|---|---|---|---|---|
| NCT00265122 | 104 | 90mg s.c./sem ×4 vs. 4,5mg/kg i.v. | Fallo a 5-ASA, cirugía, IMM o anti-TNF | Respuesta sem 8 | Respuesta sem 6 | Sem 8 U:49% vs. P:40% Sem 6 U:53% vs. P:30%* |
| CERTIFI | 526 | 1mg/kg vs. 3mg/kg vs. 6mg/kg luego 90mg s.c./8 sem | Fallo o intolerancia a anti-TNF | Respuesta sem 8 | Respuesta y remisión sem 22 | Sem 6 U:39,7% vs. P:23,5%** Sem 22 U:69,4% vs. P:42,5%*** Sem 22 U:41,7% vs. P:27,4%* |
| UNITI-I | 741 | 130mg vs. 6mg/kg vs. placebo | Fallo o intolerancia a anti-TNF | Respuesta sem 6 | Respuesta sem 8, remisión, calidad de vida, PCR, calpro | Sem 6 U130:33,7% vs. U6mg/kg: 34,3% vs. P:21,5%** Sem 8 U130:33,5% vs. U6mg/kg: 37,8% vs. P:20,2%** |
| UNITI-II | 642 | 130mg vs. 6mg/kg vs. placebo | No fallo previo a anti-TNF | Respuesta sem 6 | Respuesta sem 8, remisión, calidad de vida, PCR, calpro | Sem 6 U130:51,7% vs. U6mg/kg: 55,5% vs. P:28,7%** Sem 8 U130:47,4% vs. U6mg/kg: 57,9% vs. P:32,1%** |
| IM-UNITI | 1.310 | 90mg/8 sem vs. 90mg/12 sem | UNITI-I y II+Abierto | Remisión sem 44 | Respuesta, remisión mantenida, remisión sin cirugía, remisión en anti-TNF previo | Sem 44 90mg/8 sem: 53,1% 90mg/12 sem: 48,8% Placebo: 35,9%* |
El objetivo del segundo ensayo clínico fase IIb (estudio CERTIFI) es analizar la utilidad del mantenimiento y corroborar la eficacia tras la inducción en los pacientes con fracaso previo a anti-TNF10. Se incluyeron a 526 pacientes que se asignaban aleatoriamente a 4 dosis únicas de inducción por vía i.v. (placebo, 1, 3 y 6mg/kg) valorando la respuesta (disminución de 100 puntos en el CDAI) en la semana 6. Se encontraron diferencias significativas entre los grupos de 1 y 6mg/kg (36,6%, p=0,02; 39,7%, p=0,005) y el grupo tratado con placebo (23,5%). No se encontraron diferencias en los porcentajes de remisión clínica. En una segunda fase, los pacientes que habían respondido inicialmente se aleatorizaban a recibir 90mg s.c. en las semanas 8 y 16 o placebo, alcanzándose diferencias significativas en respuesta (69,4% vs. 42,5%; p<0,001), remisión (41,7% vs. 27,4%; p=0,03) y remisión sin corticoides evaluadas en la semana 22.
Recientemente se han comunicado los resultados de los ensayos fase III previos a su comercialización tanto en inducción (UNITI-I y UNITI-II)11 como en mantenimiento de la respuesta (IM-UNITI)12. El diseño de los 2 primeros es similar aleatorizando a 741 y 628 pacientes, respectivamente, a recibir 130mg de ustekinumab en dosis única frente a 6mg/kg o placebo, evaluando la respuesta (▿CDAI 100) en la semana 6. En el UNITI-I se incluyeron a pacientes con EC moderada con fallo previo o intolerancia al menos a un anti-TNF (51% más de uno), mientras que en el UNITI-II se permitía el uso previo de anti-TNF mientras no hubiera habido fallo al tratamiento. Los resultados se muestran en la tabla 2. En ambos estudios el fármaco fue superior a placebo tanto en respuesta como en remisión, sin diferencias entre las 2 formas de dosificación. Dos aspectos son interesantes: en primer lugar es que ya en la semana 3 se detectan diferencias significativas con respecto a placebo (30% vs. 17,8% UNITI-I y 39% vs. 21,5%; ambas p<0,05), es decir, que tiene un inicio de acción rápido, y en segundo lugar, de nuevo la eficacia en pacientes con fracaso previo a anti-TNF es menor que en pacientes sin exposición o fallo previo a estos fármacos.
En el estudio de mantenimiento (IM-UNITI) se aleatorizaron a 388 pacientes con respuesta en la semana 8 en los estudios de inducción a recibir placebo o 90mg de ustekinumab cada 8 o cada 12 semanas. La valoración de los resultados se hace en la semana 44, encontrando diferencias significativas entre las 2 dosis y el placebo en todas las variables analizadas: respuesta (objetivo primario), remisión, remisión sin corticoides, remisión en pacientes con fracaso previo a anti-TNF, remisión mantenida, etc. Si bien es cierto que aunque las diferencias no alcanzan la significación, los resultados son consistentemente mejores con la dosificación cada 8 semanas12. Con estos datos se espera la próxima comercialización en nuestro país dado que ya ha sido autorizado por la European Medicines Agency (EMA) con fecha 15 de septiembre de 201613.
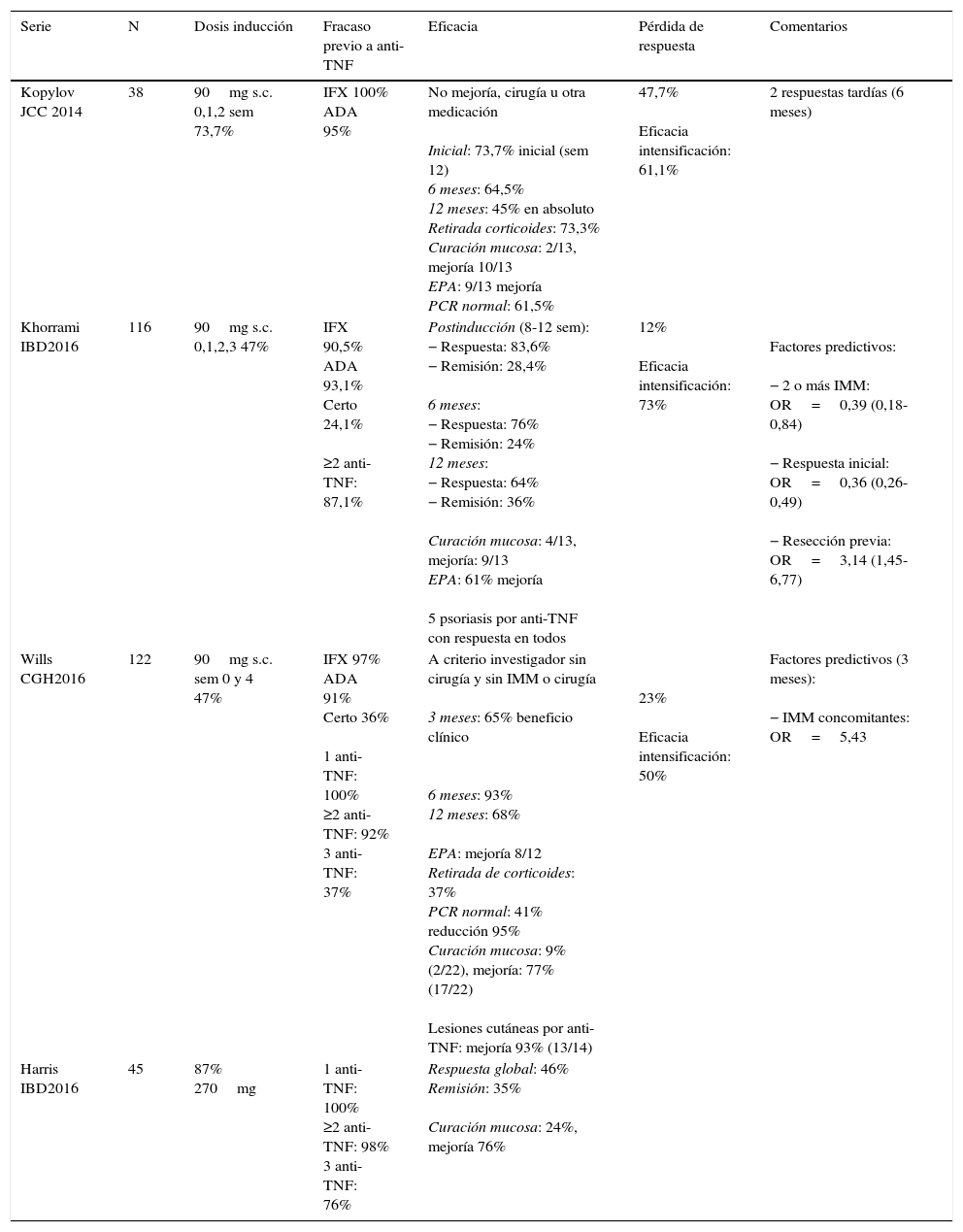
Las series abiertas publicadas (tabla 3) reproducen estos resultados con una respuesta inicial entre el 74 y el 83%8,14 y al año del 45 al 68%, y esto teniendo en cuenta que todos los pacientes habían fracasado previamente a un anti-TNF y la mayoría a 2 (87-98%)8,14-16. Las dosis de inducción utilizadas son muy heterogéneas dado que la formulación i.v. aún no está disponible, aunque lo más habitual es administrar 3-4 dosis s.c. de fármaco el primer mes8,14. Los datos sobre cicatrización mucosa son muy escasos, consiguiéndose de forma completa entre el 15 y el 24% y parcial en un 69-77%. Los pacientes tratados con enfermedad perianal (EPA) también mejoran en esta localización en el 61-69%8,14, aunque de nuevo los números son pequeños. La pérdida de respuesta en el seguimiento es muy variable oscilando entre el 12 y el 47,7%, siendo lo habitual en esta situación acortar el intervalo de administración con una eficacia del 50-73%8,14,15. La respuesta inicial al fármaco y el empleo de inmunosupresores (IMM) se asocian a una mayor probabilidad de respuesta (OR=5,43; IC 95%:1,14-25,7), mientras que una resección previa disminuye las probabilidades de éxito8,15.
Series abiertas con ustekinumab en la enfermedad de Crohna
| Serie | N | Dosis inducción | Fracaso previo a anti-TNF | Eficacia | Pérdida de respuesta | Comentarios |
|---|---|---|---|---|---|---|
| Kopylov JCC 2014 | 38 | 90mg s.c. 0,1,2 sem 73,7% | IFX 100% ADA 95% | No mejoría, cirugía u otra medicación Inicial: 73,7% inicial (sem 12) 6 meses: 64,5% 12 meses: 45% en absoluto Retirada corticoides: 73,3% Curación mucosa: 2/13, mejoría 10/13 EPA: 9/13 mejoría PCR normal: 61,5% | 47,7% Eficacia intensificación: 61,1% | 2 respuestas tardías (6 meses) |
| Khorrami IBD2016 | 116 | 90mg s.c. 0,1,2,3 47% | IFX 90,5% ADA 93,1% Certo 24,1% ≥2 anti-TNF: 87,1% | Postinducción (8-12 sem): − Respuesta: 83,6% − Remisión: 28,4% 6 meses: − Respuesta: 76% − Remisión: 24% 12 meses: − Respuesta: 64% − Remisión: 36% Curación mucosa: 4/13, mejoría: 9/13 EPA: 61% mejoría 5 psoriasis por anti-TNF con respuesta en todos | 12% Eficacia intensificación: 73% | Factores predictivos: − 2 o más IMM: OR=0,39 (0,18-0,84) − Respuesta inicial: OR=0,36 (0,26-0,49) − Resección previa: OR=3,14 (1,45-6,77) |
| Wills CGH2016 | 122 | 90mg s.c. sem 0 y 4 47% | IFX 97% ADA 91% Certo 36% 1 anti-TNF: 100% ≥2 anti-TNF: 92% 3 anti-TNF: 37% | A criterio investigador sin cirugía y sin IMM o cirugía 3 meses: 65% beneficio clínico 6 meses: 93% 12 meses: 68% EPA: mejoría 8/12 Retirada de corticoides: 37% PCR normal: 41% reducción 95% Curación mucosa: 9% (2/22), mejoría: 77% (17/22) Lesiones cutáneas por anti-TNF: mejoría 93% (13/14) | 23% Eficacia intensificación: 50% | Factores predictivos (3 meses): − IMM concomitantes: OR=5,43 |
| Harris IBD2016 | 45 | 87% 270mg | 1 anti-TNF: 100% ≥2 anti-TNF: 98% 3 anti-TNF: 76% | Respuesta global: 46% Remisión: 35% Curación mucosa: 24%, mejoría 76% |
La determinación de los niveles del fármaco puede ser de utilidad, ya que se han correlacionado con la eficacia17. Se ha visto que unos niveles superiores a 4,5μg/ml en la fase de mantenimiento se asocian a una mayor respuesta endoscópica (S=72%, E=83%, AUC=0,782), aunque en este estudio el 78% de los pacientes recibían el fármaco cada 4 semanas18. En este sentido, los niveles que se detectan son proporcionales a las dosis administradas, de forma que la pauta de 90mg cada 8 semanas consigue niveles 3 veces superiores a los conseguidos con la administración cada 12 semanas (1,97-2,24 vs. 0,61-0,76μg/ml)17, pero aun así claramente inferiores al nivel propuesto en la serie comentada. En todo caso, los datos son muy escasos para extraer conclusiones. La inmunogenicidad observada en los estudios UNITI después de un año de seguimiento es bastante contenida cifrándose en un 2,3%17, similar a la encontrada en los estudios en psoriasis (3,8-5,4%)19.
En cuanto a su eficacia en la EPA, los datos aún son muy escasos. A la espera de los subanálisis de los estudios UNITI, en las series abiertas aproximadamente 2/3 de los pacientes mejoran8,14, por lo que podría representar una alternativa a las opciones actuales. Una de las situaciones en que más eficaz se ha mostrado es en la psoriasis inducida por anti-TNF, donde la respuesta se consigue en la mayor parte de los pacientes, al menos en las lesiones cutáneas8,15.
En cuanto a la seguridad del fármaco, no se han detectado diferencias con el grupo placebo en los principales ensayos clínicos10,12,20 con respecto a los efectos adversos graves, aunque se han descrito casos aislados de infecciones oportunistas como histoplasmosis9 o listeria20. En las series abiertas los hallazgos son similares, aunque en la serie francesa en 2 pacientes el fármaco tuvo que ser retirado por mialgias y hubo un caso de neumonía. En las revisiones sistemáticas en pacientes con psoriasis los resultados son similares21,22 y solamente en una de ellas se detecta un mayor número de infecciones con ustekinumab que con el grupo placebo23. En los registros prospectivos dermatológicos las tasas de infecciones graves se sitúan entre el 0,18 y el 1,19% paciente/año19,24,25 y sin diferencias con los fármacos no biológicos y no metotrexato25. Los efectos adversos más frecuentemente descritos son la nasofaringitis (20%), las infecciones respiratorias altas y la cefalea19. No obstante, los resultados de los estudios en psoriasis en cuanto a seguridad no necesariamente son extrapolables a la EC, puesto que lo habitual en esta enfermedad es el empleo en monoterapia. Por último, aunque de forma excepcional, se han descrito en la literatura reacciones paradójicas de forma similar a lo encontrado con los anti-TNF. Entre estas reacciones se han descrito sarcoidosis26, dermatitis atópica27, artritis psoriásica28, psoriasis pustular29 y enfermedad desmielinizante30, entre otras. En cuanto a la gestación no hay datos suficientes, aunque no se han encontrado malformaciones en los animales de experimentación7 y se ha descrito algún caso de exposición fetal sin complicaciones en el recién nacido31; no obstante, de momento se aconseja su no utilización en la ficha técnica salvo que los beneficios superen a los riesgos. Se desconoce si se excreta en la leche materna, por lo que de momento se aconseja evitar la lactancia materna.
Perfil del paciente y posicionamiento en el algoritmo de tratamientoCon los datos disponibles es difícil establecer un perfil definido de paciente para este fármaco. Quizás la situación más clara sea la psoriasis inducida por anti-TNF que obligue a la suspensión del fármaco donde consigue la desaparición de las lesiones cutáneas en la mayoría de los casos8,15. Ante un fallo o intolerancia a los anti-TNF con artropatía asociada, el ustekinumab podría tener un papel más destacado que el vedolizumab dado que es eficaz en la artropatía psoriásica, aunque los datos aún son escasos. La posición en el algoritmo terapéutico dependerá de su coste aunque con la introducción de los biosimilares probablemente se situará después de los anti-TNF ya sea por fallo, intolerancia o contraindicación a los mismos. Hay muchos aspectos que aclarar con el tiempo, tanto en segu- ridad (gestación y lactancia) como en eficacia (prevención de recurrencia, EPA, empleo con IMM o no, etc.).
Otras anticitocinasExisten múltiples estudios que bloquean otras tantas citocinas implicadas en la etiopatogenia de la EII con resultados negativos32-44 a pesar de que se haya demostrado su utilidad en entidades con aspectos comunes como la psoriasis34 o la espondilitis32,33. De todos los fármacos anticitocinas en desarrollo destacan el bloqueo de la IL-23 y el de la IL-6.
Anti-IL-23: risankizumabEste fármaco es un anticuerpo IgG1 humanizado frente a la subunidad p19 de la IL-23 que se ha demostrado eficaz en la psoriasis y en la artritis psoriásica, con unos resultados significativamente mejores que ustekinumab45. En la EC se ha presentado un ensayo clínico fase II que incluye a 121 pacientes con lesiones endoscópicas (CDEIS ≥7, ≥4 en íleon) y CDAI ≥220. La mayoría tienen exposición previa a anti-TNF (94%) y son aleatorizados a recibir 2 dosis de risankizumab (200 y 600mg) frente a placebo en las semanas 0, 4 y 8 por vía i.v. En la semana 12, el grupo de 600mg conseguía unos porcentajes de remisión clínica (36,6% vs. 15,4%; p=0,025), respuesta y remisión endoscópica significativamente superiores al grupo placebo con unos efectos adversos similares, por lo que resulta un fármaco muy prometedor.
Anti-IL-6Esta citoquina tiene un papel determinante en la EII y en la EC en particular, siendo el principal estímulo para la producción de la proteína C reactiva (PCR). El primer fármaco ensayado fue el tocilizumab46 que bloquea el receptor de la IL-6 en un pequeño ensayo con 36 pacientes, demostrando una mayor respuesta que el placebo pero sin diferencias en cuanto a remisión clínica, por lo que abandonó su desarrollo en esta indicación. Recientemente se han presentado los resultados en EC de un anticuerpo monoclonal humano frente a la IL-6 (PF-04236921) denominado estudio Andante47, que incluye a 247 pacientes con PCR ≥5, úlceras en la colonoscopia y fallo o intolerancia previa a anti-TNF. Los pacientes son aleatorizados en 4 grupos (placebo, 10, 50 y 200mg), aunque el grupo de la dosis más elevada se termina de forma precoz por cuestiones de seguridad. La administración es s.c. en los días 1 y 28, analizando los resultados en las semanas 8 y 12. El grupo de 50mg consigue mejores resultados que placebo tanto en respuesta como en remisión (47,4 y 27,4% vs. 28,6 y 10,9% respectivamente; p<0,05). Sin embargo, el problema potencial de este fármaco que habrá que confirmar en estudios futuros es su seguridad, ya que en este estudio se recogen 2 perforaciones intestinales y 7 abscesos. No obstante, podría ser aceptable en pacientes refractarios a múltiples tratamientos.
Inhibidores de la adhesiónEstos fármacos se han consolidado como una opción de tratamiento en la EII: primero el natalizumab demostró eficacia tanto en inducción como sobre todo en el mantenimiento de la EC48,49, aunque lamentablemente la aparición de casos de leucoencefalopatía multifocal progresiva impidió su implantación; posteriormente, el vedolizumab se ha consolidado como una opción más de tratamiento, no solo tras fallo de los anti-TNF, sino que también está posicionado como primer biológico por algunas guías clínicas50 en la CU. No es objetivo de esta revisión comentar en detalle este fármaco, ya que no se puede considerar nuevo al estar autorizado desde septiembre de 2015 y existir una amplia experiencia clínica51-53 y revisiones excelentes sobre el tema54,55. Nos detendremos en aquellos fármacos en desarrollo que tienen más posibilidades de llegar a estar disponibles en la clínica.
EtrolizumabLa selectividad intestinal del vedolizumab deriva de la inhibición de la integrina α4β7 ligando de la molécula del endotelio Mad-CAM específica de la mucosa intestinal (también se expresa en menor medida en páncreas, bazo, hígado, pulmón y vejiga). El etrolizumab mantiene esta selectividad pero inhibiendo el componente β7 de esta integrina, pero además tiene capacidad de bloquear la interacción entre la integrina αEβ7 y la E-Cadherina en el compartimento epitelial y responsable de la permanencia de los linfocitos y células dendríticas a ese nivel56. Esto permitiría una capacidad mayor que la del vedolizumab en reducir el acúmulo de linfocitos CD8+ y CD4+ en el intestino57, aunque aún falta conocer si esto se traduce en una mayor eficacia clínica.
En el desarrollo clínico de este fármaco solo contamos en la actualidad con un estudio fase I58 que explora su eficacia y otro en fase II59 que comentaremos, aunque hay al menos 8 ensayos clínicos en marcha tanto en CU (inducción y mantenimiento: NCT02165215, fracaso o intolerancia a anti-TNF previo: NCT02100696, anti-TNF naive; NCT02136069, NCT02165215 y estudios de extensión correspondientes: NCT02118584) como en EC (NCT02394028), y lo que es más interesante: estudios comparativos con adalimumab (NCT02163759) e infliximab (NCT02136069) en pacientes sin anti-TNF previo.
El estudio fase II59 incluye a 124 pacientes con CU moderada-severa (índice de Mayo ≥6) sin respuesta al tratamiento convencional (anti-TNF: 64%) que se aleatorizan a recibir placebo o 2 dosificaciones de etrolizumab: 100mg en las semanas 0, 4 y 8 con placebo en la semana 2 o bien 420mg i.v. iniciales seguidos de 300mg s.c. en las semanas 2, 4 y 8. Ningún paciente del grupo placebo alcanza la remisión clínica en la semana 10 frente al 21% (p=0,004) y 10% (p=0,048) en los grupos de 100 y 300mg, respectivamente. La respuesta clínica no alcanza significación estadística en ninguno de los puntos analizados, al igual que la remisión en la semana 6. En el subanálisis, los pacientes con corticoides, sin IMM y sin anti-TNF obtienen mejores resultados. Algo interesante que podría seleccionar a los pacientes candidatos a este tratamiento es el hecho de que una mayor expresión en las biopsias antes del tratamiento de αE se asocia a unos mejores resultados59,60. En cuanto a la seguridad, no hubo más efectos adversos que con placebo excepto en síntomas pseudogripales, artralgias y rash. La inmunogenicidad está contenida apareciendo anticuerpos frente al fármaco en un 5% de los pacientes y sin efecto sobre su farmacocinética. Aún es pronto para situar este fármaco en el algoritmo de tratamiento, pero de confirmarse estos resultados ocuparía un lugar similar al vedolizumab, pendiente de confirmar esa teórica mayor eficacia.
Anti-MadCAM: PF-00547659Este fármaco es un anticuerpo monoclonal tipo IgG2 dirigido frente a la Mad-CAM, el ligando endotelial de la α4β7, por lo que conserva la especificidad intestinal de vedolizumab y etrolizumab. En la CU, el estudio fase II Turandot61 que incluye a 357 pacientes compara 4 dosis diferentes (7,5, 22,5, 75 y 225mg) frente a placebo administradas en las semanas 0, 4 y 8. Se alcanzan todos los objetivos del estudio al encontrar diferencias con placebo en la semana 12 con las dosis intermedias, aunque los porcentajes alcanzados son discretos (13 y 12% en los grupos de 22,5 y 75mg respectivamente). Los resultados son mejores en aquellos pacientes no expuestos previamente a anti-TNF, como ocurre con otros inhibidores de la adhesión. El estudio Opera62 en EC, sin embargo, no encuentra eficacia del fármaco más que en el subgrupo con PCR elevada. A la espera de los ensayos en fase III, el papel potencial de este fármaco en el manejo de los pacientes con CU parece similar al del vedolizumab, pero aún los datos son escasos.
OzanimodEste fármaco representa un mecanismo de acción diferente a los anteriores al modular los receptores de S1P (esfingomielina 1 fosfato). Esta sustancia tiene múltiples funciones biológicas diferentes en función de la unión a uno u otro de los 5 tipos de receptores diferentes (S1P1-5) que existen. En concreto, el S1P1 está implicado en la circulación por el ganglio linfático de los linfocitos. En el ganglio linfático existe un gradiente de S1P que es detectado por los recptores S1P1 del linfocito permitiendo su migración. El ozanimod es un agonista de este receptor que ocasiona la internalización y degradación del mismo, de forma que el linfocito no detecta el gradiente y permanece en el ganglio linfático; es por tanto un «secuestrador» de linfocitos. De hecho, la administración del fármaco conlleva una reducción reversible de hasta el 70-80% de estas células en sangre periférica63.
Hasta el momento hay un estudio fase II publicado en CU que compara la dosis de 0,5 y 1mg con placebo en administración única diaria por vía oral durante 8 semanas, con una segunda fase de mantenimiento hasta la semana 32 en los respondedores. Únicamente es eficaz la dosis de 1mg en la inducción mejorando los resultados del grupo placebo tanto en remisión (16% vs. 6%; p=0,048) como en respuesta (57% vs. 37%; p=0,02) y cicatrización mucosa (34% vs. 12%; p=0,03), pero no así en remisión histológica. Al final del seguimiento ambas dosis eran superiores al placebo, por lo que la de 0,5mg podría ser útil como mantenimiento. La exposición previa a anti-TNF era el único factor que condicionaba una peor respuesta, aunque el porcentaje de pacientes expuestos con anterioridad al estudio con estos fármacos era bajo (18%).
En cuanto a la seguridad, fármacos similares como el fingolimod utilizado en la esclerosis múltiple tiene efectos adversos importantes como bloqueos auriculoventriculares, edema de mácula o hipertransaminasemia consecuencia de un bloqueo no selectivo del receptor S1P1, ya que modula también otros como el S1P3, S1P4 y S1P5. Con el ozanimod, dada su mayor selectividad por los receptores S1P1 y S1P5, es esperable que estos efectos adversos sean menores. En este estudio, un paciente tuvo un bloqueo auriculoventricular y otro elevación de las transaminasas x3VN, ambos reversibles tras la suspensión del fármaco. La linfopenia es muy frecuente, de forma que en la dosis de 1mg más de la mitad de los pacientes presentaban recuentos inferiores a la normalidad, aunque solo un 15% estaban por debajo de 500cel/mm3. No obstante, la tasa de infecciones fue baja probablemente porque el secuestro en los ganglios linfáticos es selectivo para linfocitos naive y de memoria centrales y no para los linfocitos de memoria efectores. Los ensayos en marcha en CU y EC aclararán esta cuestión.
Es pronto para establecer su lugar en la práctica clínica, pero su administración oral y la ausencia de inmunogenicidad son ventajas con respecto a los otros inhibidores de la adhesión, si bien la eficacia es aparentemente discreta y está por definir la seguridad del fármaco.
MongersenEl TGF-β1 tiene un papel regulador de la respuesta inmune determinante en la EC ya que inhibe la proliferación celular, reduce la activación de macrófagos y la maduración de células dendríticas, teniendo de esta forma importante efecto antiinflamatorio. En la EC su señalización intracelular está bloqueada por la proteína SMAD7 que se encuentra elevada en cualquier momento de la evolución de la enfermedad64.
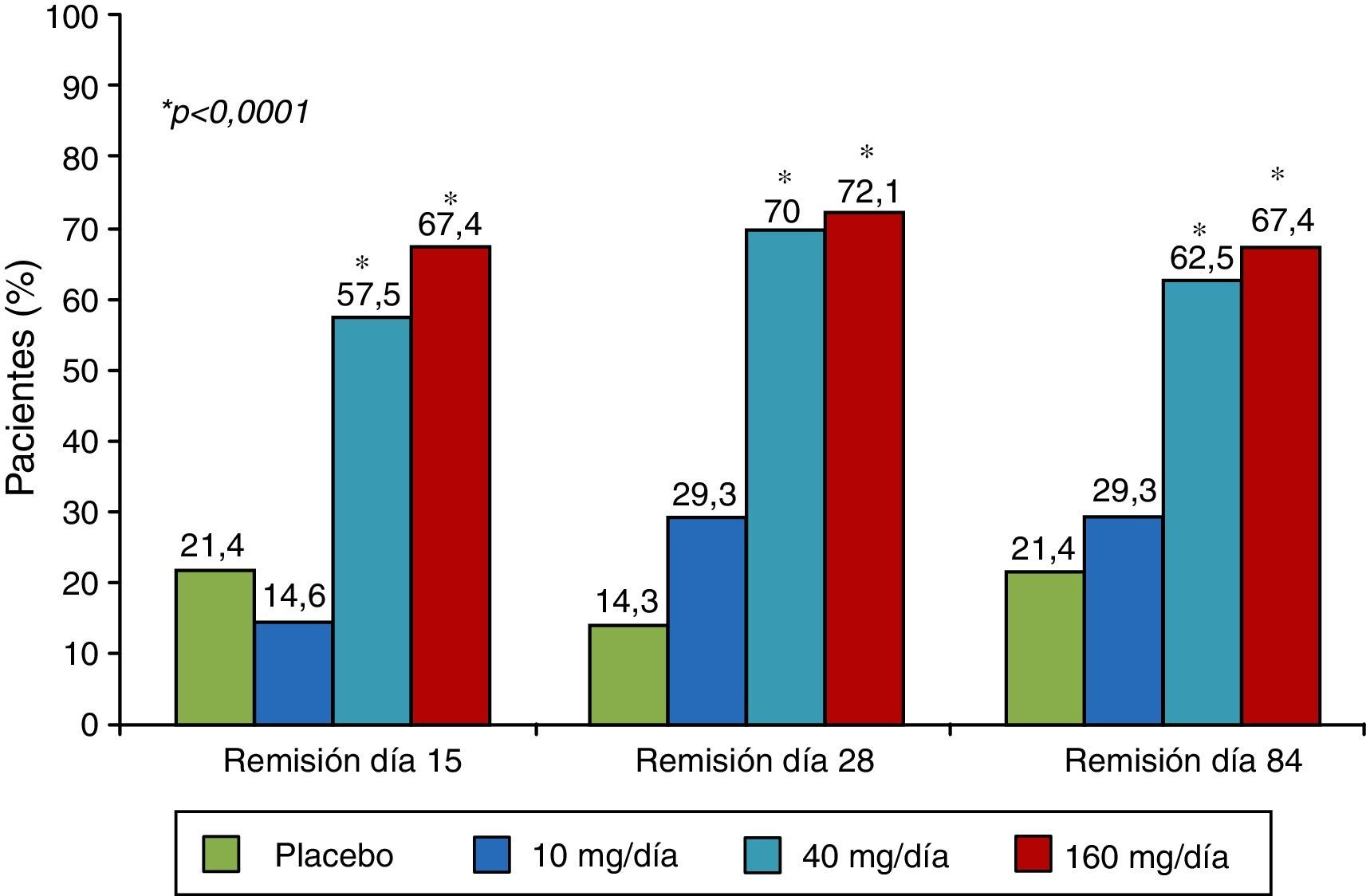
El mongersen es un oligonucleótido antisentido frente al ARN mensajero de la proteína SMAD7 de forma que conseguiría revertir el bloqueo de TGF-β1. El fármaco se administra por vía oral y con una galénica que permite su liberación en la región ileocecal de forma que su efecto es tópico, detectándose niveles en sangre de forma excepcional65. Tras 2 estudios fase I para evaluar la toxicidad y dosificación66,67, se publicó el estudio de fase II denominado IGON168 que incluye a 160 pacientes con EC corticodependiente o corticorresistente y CDAI ≥220, a los que se aleatorizaba a recibir una de las 3 dosis evaluadas (10, 40 o 160mg) o placebo por vía oral durante 2 semanas. En el día 15 los grupos de 40 y 160mg alcanzan el objetivo de remisión clínica en un 55 y 65%, respectivamente, claramente superior a la conseguida con placebo (10%; p<0,001). Los porcentajes de respuesta son también muy llamativos, con un 72,4 y 81,4% en los grupos de las 2 dosis más altas frente a un 31% en el grupo placebo (p=0,0005 y p<0,0001 respectivamente).
Estos resultados, en cuanto al porcentaje de remisión alcanzada, son mucho mejores que los encontrados en cualquier otro ensayo clínico con biológicos en la EC, ya sea con infliximab (33% en el estudio de Targan o 32,5% en el estudio SONIC en la semana 6)69-71, adalimumab (36% en el Classic II)70 o vedolizumab (14,5% en la semana 6 y 39% en la semana 54, estudio GEMINI 2)72. Pero lo realmente llamativo es que, a pesar de que el fármaco solo se administra durante 14 días, estos resultados se mantienen hasta el día 84 (fig. 1), lo que daría pie a su utilización intermitente. De hecho, en el estudio fase III en marcha, 2 de las ramas incluyen esta estrategia de tratamiento (clinicaltrial: NCT02596893).
. El fármaco se administraba los primeros 14 días.")
Este estudio también tiene críticas. En primer lugar, no se tenía en cuenta para la inclusión la presencia de lesiones endoscópicas ni era un criterio de eficacia a pesar de que está recomendado por las principales agencias reguladoras. Por otro lado, los pacientes incluidos tenían una enfermedad leve-moderada con un CDAI medio de 240-260 y hasta en un 39% la PCR era normal. Además, claramente la actividad influía en los resultados, de forma que en un subanálisis publicado con posterioridad73 la dosis de 40mg no era diferente del placebo en el grupo con un CDAI ≥260, aunque la de 160mg conservaba la eficacia. Otro aspecto destacable es que solo se conseguía la normalización de la PCR en un 18% de los pacientes tratados con las dosis más altas, cifra no significativamente diferente de la conseguida con placebo (4%). Probablemente este hecho sea consecuencia de su selectividad intestinal72, ya que también se ha observado con el vedolizumab. Finalmente, el número de pacientes tratados hasta la fecha no permite extraer conclusiones en cuanto a la seguridad del fármaco. Una de las complicaciones teóricamente posible es la inducción de estenosis, ya que el TGF-β1 es un potente inductor de fibrosis. No se encontró esta complicación en un pequeño ensayo preliminar con 15 pacientes en ninguno de ellos, aunque en el estudio IGON1 un paciente del grupo de 40mg tuvo una obstrucción intestinal. Quedan, por tanto, muchos aspectos por aclarar, tanto de seguridad como de eficacia: cicatrización mucosa, combinación o no con IMM, forma de administración y eficacia a largo plazo e incluso el efecto ahorrador de corticoides, ya que solo un 17-32% de los pacientes del IGON1 tenían corticoides en la aleatorización74,75.
El perfil del paciente que se podría beneficiar de este fármaco es difícil de establecer en la medida en que faltan los estudios fase III en marcha, pero probablemente correspondería a un paciente con EC exclusivamente ileocecal (al menos en monoterapia), con actividad leve-moderada, sin manifestaciones extraintestinales o EPA y sin anti-TNF previo, ya que no está representado este grupo de pacientes en los estudios publicados. En el algoritmo de tratamiento se posicionaría antes de los fármacos anti-TNF tanto por eficacia como por seguridad por su aparente baja tasa de efectos adversos, aunque siempre el coste es determinante en este sentido.
En cuanto a la CU, está en marcha un ensayo fase II con mongersen en esta indicación que ya ha terminado su reclutamiento y está pendiente del seguimiento, por lo que pronto conoceremos los resultados (clinicaltrials: NCT02601300)
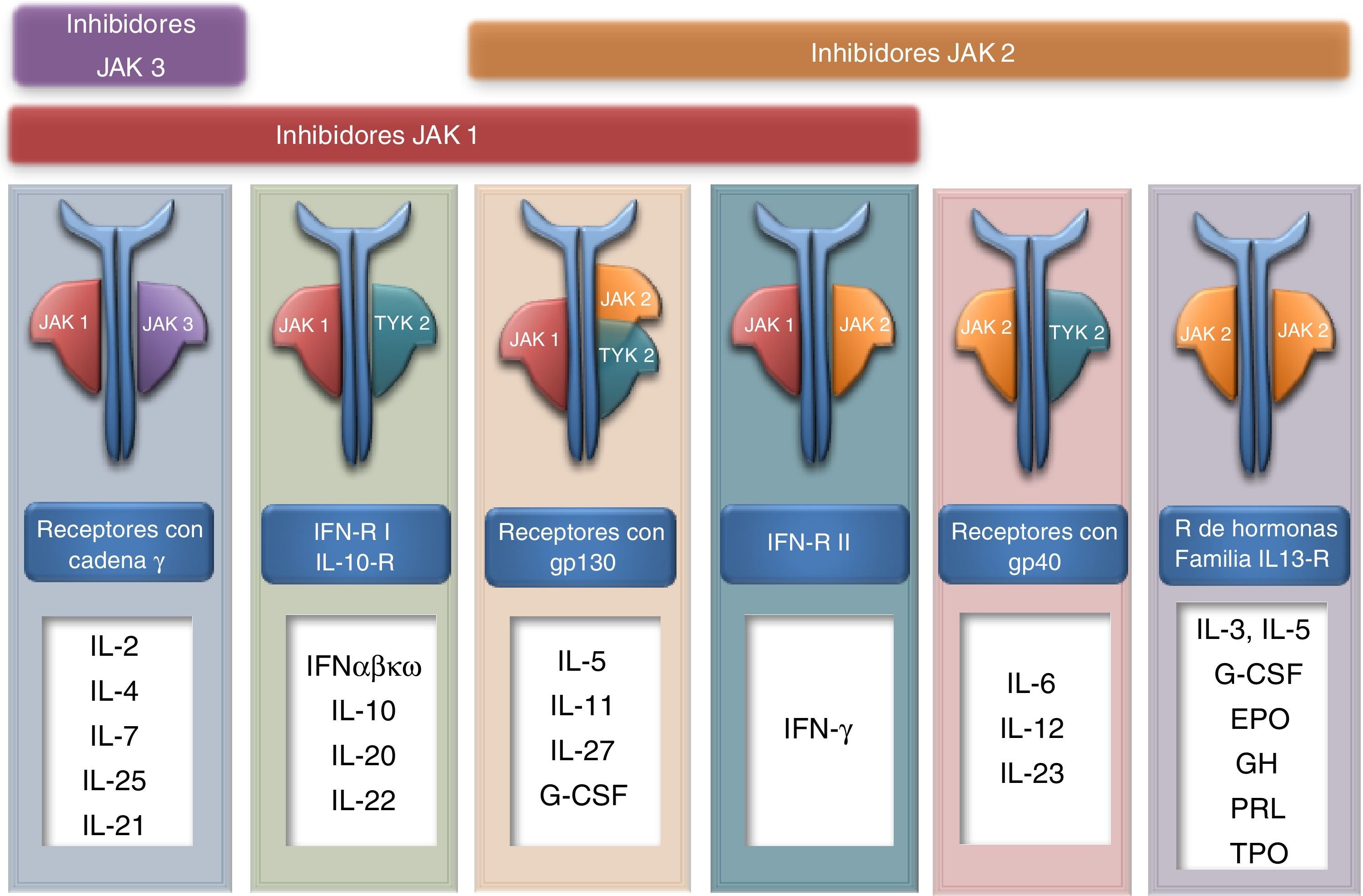
Inhibidores de las JAK quinasasLas quinasas de Janus (JAK) son una familia de 4 proteínas (JAK1, JAK2, JAK3 y TYK2) encargadas de la señalización intracelular de distintas citocinas (fig. 2). Están asociadas al dominio intracitoplasmático del receptor de citocinas por pares. La activación del receptor activa las JAK quinasas fosforilando a las proteínas del sistema STAT que se traslocan al núcleo donde modulan la transcripción de numerosos genes implicados en la respuesta inmune y diferentes funciones celulares como hematopoyesis, diferenciación, maduración y crecimiento76. Por tanto, la inhibición de este sistema además de un efecto inmunomodulador puede presentar otros posibles efectos no deseados (fig. 1) en función de la JAK quinasa bloqueada. Los fármacos anti-JAK son moléculas sencillas y por tanto carentes de inmunogenicidad. Al contrario que los biológicos, la inhibición es parcial y reversible, de otra forma el bloqueo completo probablemente supondría una toxicidad inaceptable.

Es un fármaco oral inhibidor reversible con afinidad por las JAK1 y JAK3. Tiene una vida media aproximada de 3h y una dosis de 5mg conlleva una inhibición del 50-60% de las citocinas dependientes (fig. 1), disminuyendo al 20-30% en el momento de la concentración valle76. Se ha demostrado su utilidad en la artritis reumatoide, psoriasis, dermatitis atópica y trasplante renal, incluso en fallo a biológico previo. Cuando se ha comparado con otros biológicos (con etanercept en psoriasis y con adalimumab en artritis reumatoide) ha mostrado una eficacia similar77,78.
En la CU la eficacia está más contrastada. El primer ensayo clínico que demuestra utilidad del fármaco es un estudio fase II que incluye a 194 pacientes aleatorizados en 4 grupos: placebo, 0,5, 3, 10 y 15mg administrados por vía oral cada 12h durante 8 semanas con un seguimiento de 4 semanas más. Un 30% habían recibido previamente fármacos anti-TNF y un 36% IMM. La respuesta clínica (objetivo principal del estudio) en la semana 8 era significativamente superior al placebo en el grupo de 15mg únicamente (42% vs. 78%; p<0,001); sin embargo, la remisión alcanzaba significación en los grupos de 3, 10 y 15mg (33, 48 y 41% vs. 10% respectivamente). Las lesiones endoscópicas también cicatrizaban (índice de Mayo=0) en un mayor número de pacientes tratados con tofacitinib (3mg: 18%, p=0,01; 10mg: 30%, 15mg: 27%; ambas p<0,001) que en los asignados a placebo (2%). En este estudio los IMM eran retirados al iniciar el ensayo.
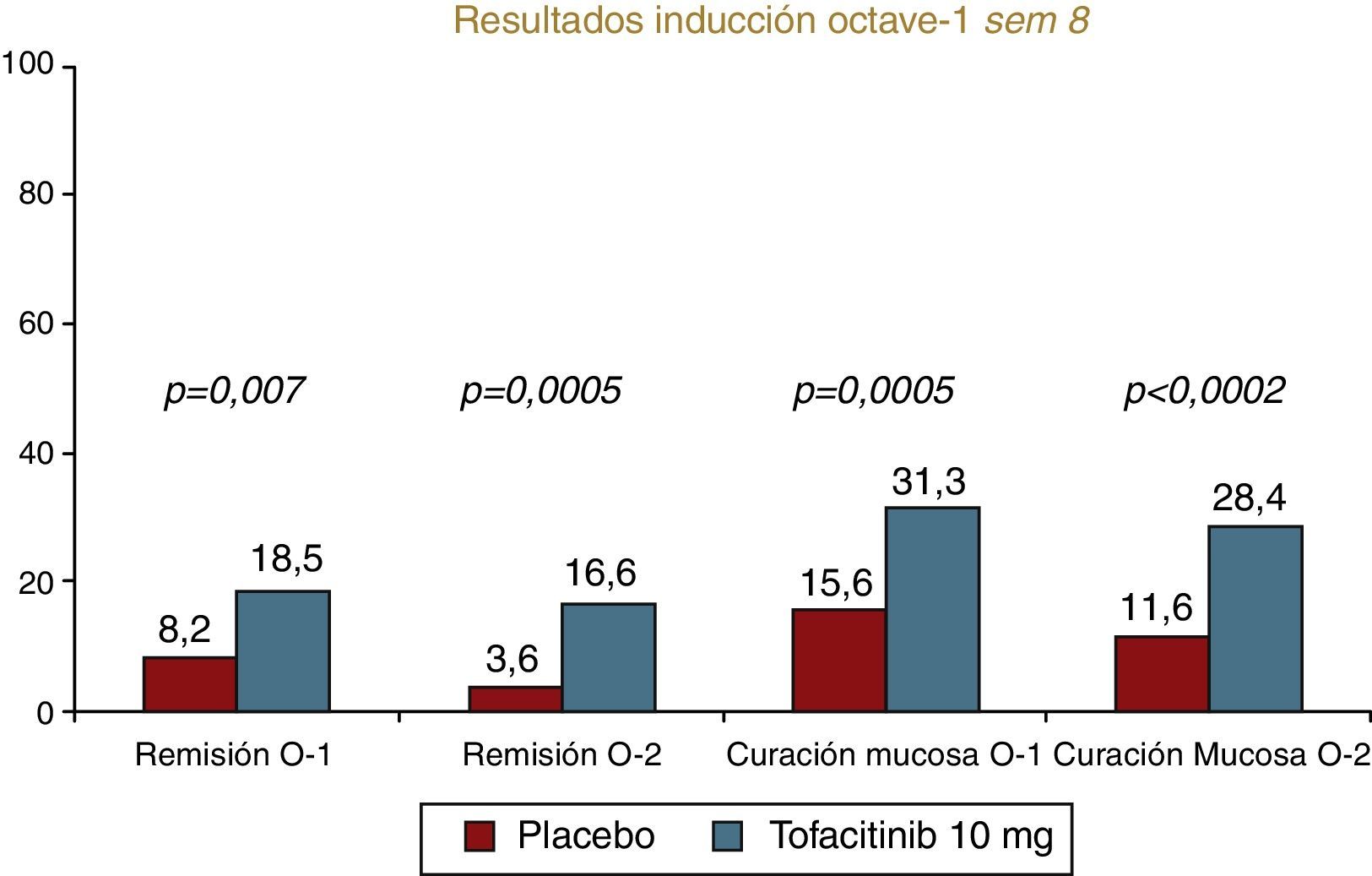
Estos resultados son corroborados en 2 ensayos fase III recientemente comunicados de diseño idéntico, los estudios Octave 1 y Octave 279. Estos estudios incluyen a 596 y 541 pacientes, respectivamente, con fallo al tratamiento convencional o a anti-TNF (53-58%) que se aleatorizan a recibir 10mg de tofacitinib o placebo en una proporción 4:1. Los pacientes respondedores reciben 2 dosis de mantenimiento (5 o 10mg) o placebo durante 54 semanas. Únicamente se conocen los resultados tras la inducción, alcanzándose todos los objetivos planteados con diferencias significativas en remisión, respuesta y cicatrización mucosa (fig. 3). La mejoría del índice de Mayo es rápida con diferencias significativas ya en las semanas 2 y 4. Además, no hay diferencias de eficacia en función de la exposición previa o no a anti-TNF, por lo que constituye una alternativa interesante en este grupo de pacientes.

Globalmente, los efectos adversos en los ensayos clínicos son similares a los del grupo placebo, siendo los más frecuentes la gripe y la nasofaringitis80. Uno de los problemas asociados a este fármaco es la elevación del colesterol total a expensas tanto del colesterol LDL como del HDL; algo que se revierte al suspender el fármaco o realizando cotratamiento con estatinas81. También se han observado casos de leucopenia con las dosis de 10 y 15 mg80 y elevaciones de la CPK que motiva la retirada del fármaco en un 0,2% de los pacientes79. En cuanto a la predisposición a infecciones, es similar a la encontrada con biológicos excepto en las infecciones por los virus herpes (fundamentalmente herpes Zoster), que están aumentadas aproximadamente el doble, al menos en las series de artritis reumatoide82. Los efectos adversos (sobre todo algunos casos aislados de perforación intestinal) llevaron a la EMA a rechazar su autorización en 2013, aunque en la actualidad se está reevaluando su aprobación.
El perfil de paciente es amplio, aunque parece una buena opción ante fallo de anti-TNF, de momento en monoterapia y preferiblemente en pacientes sin comorbilidad cardiovascular por la hiperlipidemia que pueden ocasionar. La posición en el algoritmo terapéutico dependerá finalmente de que sea autorizado en Europa y de su coste, aunque con la introducción de los biosimilares y el precio en Estados Unidos probablemente se ubique tras fallo a anti-TNF.
En la EC, en un primer ensayo comparando 3 dosis de tofacitinib (1, 5 y 15mg) con placebo83, no se logran los objetivos del estudio, aunque con las dosis más altas hay descensos significativos de la PCR y la calprotectina, lo que justifica 2 nuevos ensayos fase IIb, uno en inducción (8 semanas de tratamiento) y otro en mantenimiento (26 semanas) con un mayor número de pacientes con las dosis de 10 y 15mg. Los resultados son similares en ambos grupos, con cifras de remisión y respuesta numéricamente más altas en los grupos de tofacitinib pero sin significación estadística, aunque con descensos en los biomarcadores que sí lo son84,85. Por tanto, este fármaco no ha demostrado su eficacia en la EC y en la actualidad no hay nuevos estudios en marcha, por lo que no es probable que se emplee en un futuro en esta indicación.
Otros inhibidores de JAK quinasasExisten numerosos fármacos en este grupo, entre los que destacan 2 con los que ya se han realizado estudios en EII. Ambos son inhibidores selectivos de la JAK1, el filgotinib (Gillead) y el ABT-494 (Abbvie). Con el primero de ellos se ha presentado recientemente un ensayo fase II (estudio Fitzroy)86 que incluye a 174 pacientes con EC y lesiones endoscópicas (SES-CD ≥7) de los cuales el 76% habían recibido anti-TNF previamente. Se administran 200mg de filgotinib diarios o placebo (3:1) durante 10 semanas, al cabo de las cuales se realiza una nueva aleatorización en 3 grupos: 100, 200mg y placebo, con un seguimiento de 20 semanas. Únicamente están disponibles los resultados de la inducción consiguiendo mejores resultados que placebo tanto en respuesta (60% vs. 41%; p<0,01) como en remisión (48% vs. 23%; p<0,05). No se detectaron más efectos adversos que con el grupo placebo; en particular no hubo leucopenia y solo se elevó el colesterol HDL. Existen en marcha ensayos en inducción y mantenimiento tanto en CU (NCT02914522) como en EC (NCT02914561) con sus correspondientes estudios de extensión (NCT02914535 y NCT02914600). De confirmarse estos resultados de eficacia y seguridad, este fármaco supondría una alternativa al menos tan eficaz como el tofacitinib, si no mayor, y con un perfil de seguridad mejor.
Reflexiones finalesTras varios años con múltiples fármacos ensayados en la EII sin eficacia, en la actualidad ya existen 2 alternativas a los anti-TNF disponibles y en un futuro próximo varias más. Esto por un lado mejora alguna de las características de los disponibles, como la administración oral de algunos de ellos o la ausencia de inmunogenicidad. Por otro lado, probablemente cambie nuestra estrategia de manejo global replanteándonos la utilización de un tercer o incluso un segundo anti-TNF. En este sentido, en reumatología el cambio de diana frente al cambio de fármaco anti-TNF consigue una mayor durabilidad en el control de la enfermedad87. Por último, y a la espera de las nuevas incorporaciones, queda pendiente definir perfiles de pacientes para cada fármaco que nos decanten por uno u otro, aunque probablemente la primera opción continúe siendo los anti-TNF por su menor coste.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónEl autor no ha recibido financiación para la elaboración y redacción de este artículo.
Conflicto de interesesEl autor ha recibido pagos por advisory o consejerías, actividad docente y asistencia a congresos por las siguientes empresas en los últimos 5 años: Abbvie, MSD, Shire, Faes, Kern, Takeda, como mis conflictos de intereses.

