




Introduction
Enterocytozoon bieneusi is the most important and prevalent microsporidian parasite recognized in human patients with AIDS1.
The diagnosis of intestinal infection due to E. bieneusi traditionally depended on visualization of the parasite by light and/or electron microscopy in fecal samples or biopsy specimens2. Polymerase chain reaction (PCR) techniques have been developed for the detection of E. bieneusi DNA in different biological samples, including fresh and fixed stool specimens, duodenal aspirates, sputum, bile and fresh biopsy samples3-5. In situ hybridization protocols have been described to identify E. bieneusi infections in formalin-fixed biopsy specimens6, but this technique is not feasible for a large number of samples. The goal of this study was to extend the previous use of the PCR assay to formalin-fixed duodenal biopsy samples, which are safer to handle and can readily be transported and stored for longer periods. Using the PCR technique in archived biopsy samples, should help to understand the role of E. bieneusi in gastrointestinal disease in human immunodeficiency virus (HIV)-positive patients, allowing the use of routinely processed materials in large-scale retrospective studies.
Methods
Ninety five cases were retrieved from the files of the Pathology Service, Hospital Dr. J. M. Penna, Buenos Aires, Argentina, from the period 1995 through 1997. All of the cases were adult HIV-infected patients and criteria for selection were chronic diarrhea of undiagnosed ethiology and CD4 cells count less than 300/μl. Blocks of 10% formalin-fixed paraffin-embedded archival duodenal biopsy specimens were cut in 10 mm sections, stained with hematoxylin-eosin and screened for the presence of organisms compatible with microsporidia. Spores could be detected microscopically in slides from eleven of the ninety five patients and one block of each one was employed for the PCR protocols.
Negative (five duodenal biopsy specimens of HIV-negative patients with diagnosis of ulcer) and positive controls (two samples with E. bieneusi infection confirmed by electron microscopy) were employed.
Three to four 15-μm sections were cut from the paraffin wax blocks. Deparaffinization and DNA preparation were carried out essentially following standard protocols7.
Four PCR protocols were employed: a single PCR, amplifying a 607 bp fragment of the E. bieneusi SSU-rRNA gene8, a double PCR, consisting of two consecutive single PCRs amplifying the 607 bp fragment, a nested PCR, where a 1,271 bp fragment of the E. bieneusi SSU-rRNA gene9 is amplified in the first round, followed by amplification of the 607 bp fragment in the second round8, and the PCR Eb.gc/Eb.gt, amplifying a 210 bp fragment of the internal trancribed spacer (ITS) of E. bieneusi rRNA genes4.
Reaction mixtures and cycle conditions were performed essentially as previously described4,8,9, employing 5 ml template DNA.
For PCR and double PCR, specific primers for E. bieneusi were employed (EBIE1F/R corresponding to nucleotides 295-315 and 901-881 of E. bieneusi SSU-rRNA)8; for nested PCR, generic primers for microsporidia were used in the first round (MICRO-F and 1492N4 R corresponding to nucleotides 1-21 and 1271-1249 respectively of E. bieneusi SSU-rRNA)9 and specific primers in the second round. For the fourth PCR assay, PCR Eb.gc/Eb.gt, a previously described primer set for the ITS of E. bieneusi was used (Eb.gc and Eb.gt corresponding to nucleotides 1-22 and 210-189, respectively)4.
Positive controls corresponding to plasmids in which the expected amplicon sequence had been cloned, were included in all the PCR reactions. All human samples were tested for the presence of PCR inhibitors in order to evaluate the DNA extraction steps. The experiment was carried out by spiking the samples with 0.03 ng of plasmid DNA containing the cloned amplicon of E. bieneusi for the first round of the PCR reactions5.
An aliquot of 10 μl of amplification products was resolved by electrophoresis on a 1.5% agarose gel and stained with ethidium bromide for visual analysis.
Results
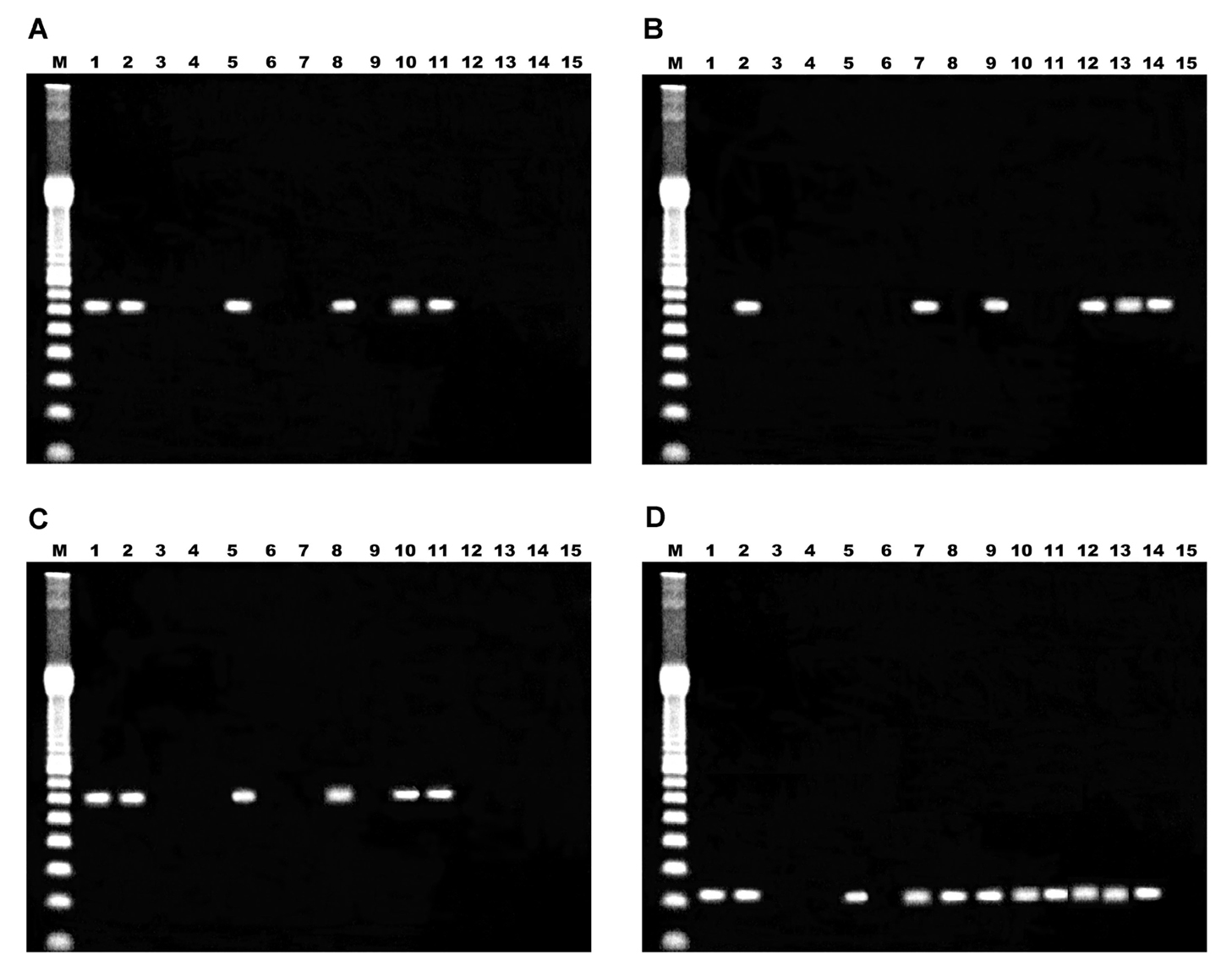
Amplification of the cloned amplicons of E. bieneusi by spiking during the first round of the PCR reactions was positive in all cases, thus indicating the absence of inhibitors. When the target sequence corresponded to the SSU rRNA gene, positive and negative control biopsy specimens yielded appropriate results (figs. 1A-C, lanes 1, 2 and 3). For one positive control, the double PCR gave a negative result. Amplicons of the right size were observed after single, double, and nested PCR (first and second round) when the plasmid control containing the target sequence was analyzed. For samples 1 to 11, amplicons of 607 bp were obtained by at least one method in nine cases, remaining two samples identified as negative (samples 2 and 11).The one-step PCR produced a correctly sized product only in four samples, and the result was reproduced when a nested PCR assay was performed (figs. 1A, 1C, lanes 5, 8, 10 and 11). No amplification products were visible by agarose gel electrophoresis after the first round of the nested PCR. However, these four biopsy specimens failed to show the amplification when a double PCR method was achieved (fig. 1B, lanes 5, 8, 10 and 11). In order to rule out an inhibitory effect of template excess in the second round of PCR, the products of the first round were diluted 1:10 and the second round was performed without changing conditions. An identical protocol was applied to the positive control that showed the same problem and after the dilution tests, the expected band of 607 bp was detected on the agarose gel (data not shown). For the remaining five samples, the fragment of the correct size was amplified only by double PCR, showing negative results when the one-step PCR or nested PCR were carried out (figs. 1A-C, lanes 7, 9, 12, 13 and 14).
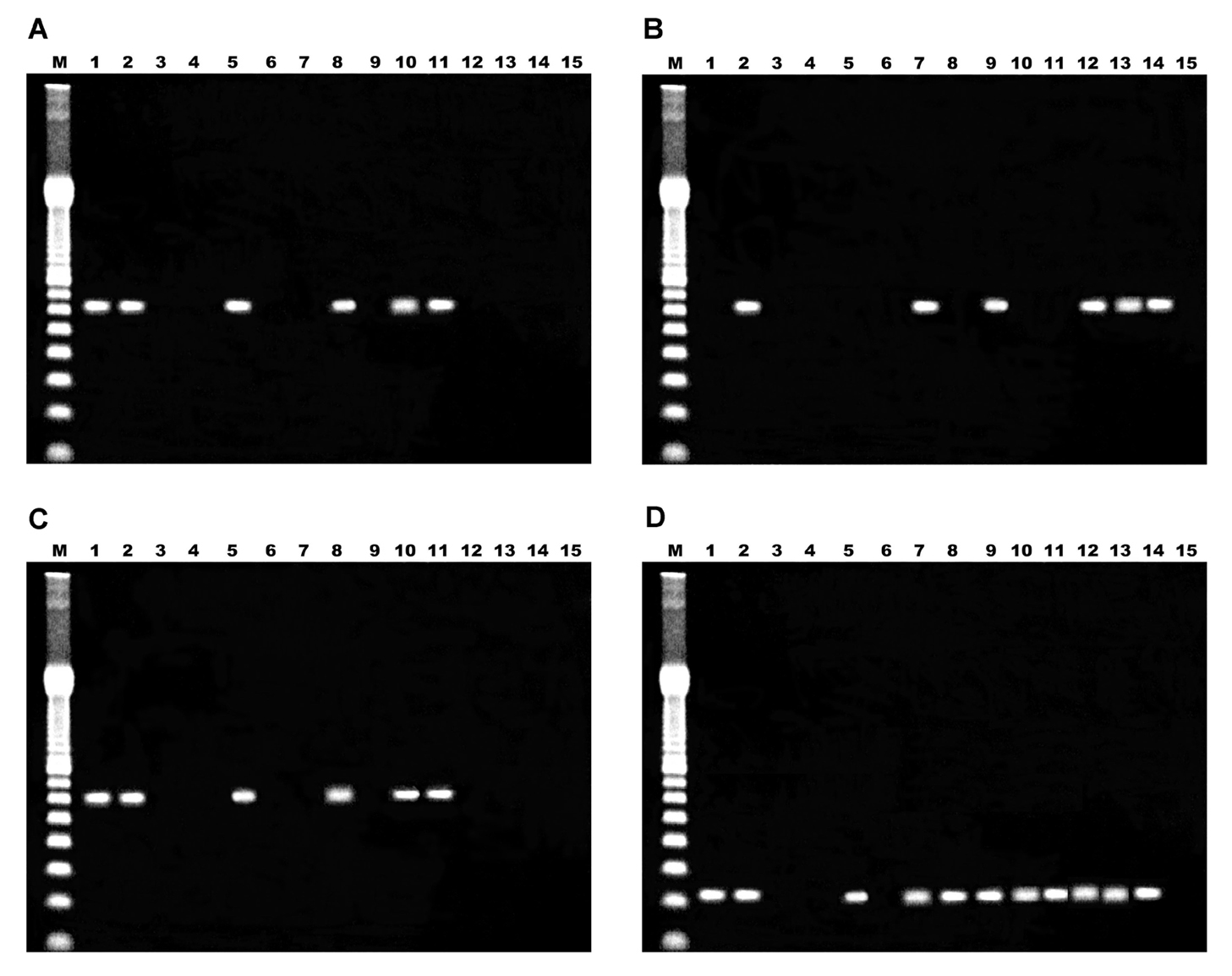
Figure 1. Analysis of amplification products by agarose gel electrophoresis. A) single PCR; B) double PCR; C) nested PCR; D) PCR Eb.gc/Eb.gt. Lane M, 100 bp ladder; lanes 1 and 2, positive controls; lane 3, one negative control; lane 4, reaction mixture; lanes 5 to 15, samples 1 to 11 respectively.
When the target sequence corresponded to ITS of the rRNA genes, positive and negative control biopsy specimens yielded appropriate results (fig. 1D, lanes 1, 2 and 3), with an amplicon size of 210 bp. For samples 1 and 3 to 10, amplicons of 210 bp were obtained, remaining samples 2 and 11 as negative.
Discussion
We report here a PCR methodology that allows the use of archival fixed and embedded tissue specimens as a source of DNA for diagnosis of infections due to E. bieneusi. Light microscopy of biopsy specimens do not allow the identification of the parasites to the species level1,6. In order to achieve an specific method, a set of pan-microsporidial primers was only employed in the first round of the nested PCR and two different sets of previously reported E. bieneusi specific primers were used in all the other assays4,8,9.
The applicability of the PCR assays presented in this report deserves further comment. The DNA sources were formalin-fixed paraffin wax embedded archival materials. Although these tissues can provide high molecular weight DNA sufficiently intact, it can be degraded resulting in a less robust amplification substrate compared with DNA from fresh tissues10. In this way, the results obtained by the four protocols have to be analyzed in conjunction. The nested PCR protocol was not useful for this kind of samples because of the large size of the expected first amplicon (1,271 bp). The results of the one-step single PCR procedure could not be improved by nested PCR. Better results were obtained by performing a second round of PCR amplification using the same primers (double PCR) with a target fragment of 607 bp. The amount of DNA could be another factor leading to an increased chance of false-negative results. The best results were obtained by employing the primer pair Eb.gc/Eb.gt, with a target sequence of 210 bp, showing that a shorter sequence is preferable as target in tissue specimens, which may have been fixed and embedded under inappropriate conditions.
Therefore, there is a number of cautions that have to be in mind applying PCR to material extracted from fixed histological blocks stored in archives: possible heterogeneity in fixation and preservation, template quality after extraction, target sequence length, amount of DNA10.
The ability to perform molecular biological analysis on fixed tissues from AIDS patients, allows access to a vast archival resource and simplifies transportation and decontamination of samples. The method described in this study could provide species-level diagnosis for archival specimens, simplify the storage conditions, allow its application on samples previously obtained for conventional pathology, be used in retrospective studies and contribute to the elucidation of the pathogenesis of Enterocytozoon bieneusi infection.
Acknowledgments
We thank Dr. Cecilia Di Risio, from the Gastroenterology Service of the Hospital "Dr. José María Penna" for performing the endoscopies, and Drs. Norman Pieniazek and Alexandre da Silva, from the Division of Parasitic Diseases, Center for Disease Control and Prevention, Atlanta, USA, for providing the primers for the nested PCR. We acknowledge Fernando Sodré, from the Universidade Estadual do Rio de Janeiro, Brazil, for critical comments and helpful discussions.

