














Actualizar las recomendaciones sobre el tratamiento antirretroviral (TAR) para adultos infectados por el virus de la inmunodeficiencia humana tipo 1 (VIH-1).
MétodosEste documento ha sido consensuado por un panel de expertos de GeSIDA y de la Secretaría del Plan Nacional sobre el Sida tras revisar los resultados de eficacia y seguridad de ensayos clínicos, estudios de cohortes y de farmacocinética publicados en revistas biomédicas (PubMed y Embase) o presentados en congresos. La fuerza de cada recomendación y la gradación de su evidencia se basan en una modificación de los criterios de la Infectious Diseases Society of America.
ResultadosSe recomienda iniciar el TAR en los pacientes sintomáticos, las embarazadas, las parejas serodiscordantes con alto riesgo de transmisión, la hepatitisB que requiera tratamiento y la nefropatía por VIH. En los pacientes asintomáticos se recomienda si el número de linfocitos CD4+ es <500células/μl. Cuando este es >500células/μl puede considerarse su inicio en pacientes con hepatitisC crónica, cirrosis hepática, riesgo cardiovascular elevado, carga viral plasmática (CVP)>105copias/ml, proporción de linfocitos CD4+<14%, trastornos neurocognitivos o edad >55años. El objetivo del TAR es lograr una CVP indetectable. El TAR de inicio debe ser siempre una combinación de 3fármacos que incluya 2inhibidores de la transcriptasa inversa (ITI) análogos de nucleósidos y un ITI no-análogo de nucleósidos, un inhibidor de la proteasa potenciado con ritonavir, o un inhibidor de la integrasa. Se han consensuado diversos regímenes de TAR de inicio así como criterios específicos para el TAR en la infección aguda, pacientes con tuberculosis u otros episodios definitorios de sida, la mujer, el embarazo, la coinfección por VHB o VHC, la infección por VIH-2 y la profilaxis postexposición.
ConclusionesEste nuevo documento actualiza las recomendaciones previas respecto a cuándo y con qué regímenes iniciar el TAR, cómo monitorizarlo y qué hacer cuando fracasa o desarrolla toxicidad. Asimismo actualiza los criterios específicos del TAR ante la presencia de diversas comorbilidades y situaciones especiales.
This consensus document is an update of combined antiretroviral therapy (cART) guidelines for HIV-1 infected adult patients.
MethodsTo formulate these recommendations a panel composed of members of the GeSIDA/National AIDS Plan Secretariat (Grupo de Estudio de Sida and the Secretaría del Plan Nacional sobre el Sida) reviewed the efficacy and safety advances in clinical trials, cohort and pharmacokinetic studies published in medical journals (PubMed and Embase) or presented in medical scientific meetings. The strength of the recommendations and the evidence which support them are based on a modification of the criteria of Infectious Diseases Society of America.
ResultscART is recommended in patients with symptoms of HIV infection, in pregnant women, in serodiscordant couples with high risk of transmission, in hepatitisB co-infection requiring treatment, and in HIV nephropathy. cART is recommended in asymptomatic patients if CD4 is <500cells/μl. If CD4 are >500cells/μl cART should be considered in the case of chronic hepatitisC, cirrhosis, high cardiovascular risk, plasma viral load >100.000 copies/ml, proportion of CD4 cells <14%, neurocognitive deficits, and in people aged >55years. The objective of cART is to achieve an undetectable viral load. The first cART should include 2 reverse transcriptase inhibitors (RTI) nucleoside analogs and a third drug (a non-analog RTI, a ritonavir boosted protease inhibitor, or an integrase inhibitor). The panel has consensually selected some drug combinations, for the first cART and specific criteria for cART in acute HIV infection, in tuberculosis and other HIV related opportunistic infections, for the women and in pregnancy, in hepatitisB or C co-infection, in HIV-2 infection, and in post-exposure prophylaxis.
ConclusionsThese new guidelines update previous recommendations related to first cART (when to begin and what drugs should be used), how to monitor, and what to do in case of viral failure or adverse drug reactions. cART specific criteria in comorbid patients and special situations are similarly updated.
El tratamiento antirretroviral (TAR) evoluciona con tal rapidez que exige una frecuente actualización de sus recomendaciones. Desde que en 1995 la Secretaría del Plan Nacional sobre el Sida (SPNS) y su Consejo Asesor Clínico editaran las primeras «Recomendaciones de tratamiento antirretroviral en el adulto»1, este organismo, junto con el Grupo de Estudio de Sida (GeSIDA) de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC), han actualizado estas recomendaciones con periodicidad anual, publicándolas en la revista Enfermedades Infecciosas y Microbiología Clínica o en sus respectivas páginas web2. Otras instituciones y sociedades científicas elaboran y actualizan sus propias recomendaciones sobre el empleo de los fármacos antirretrovirales (FAR)3,4.
El objetivo de este documento es dar a conocer a la comunidad científica y a los profesionales que tratan a pacientes adultos infectados por el VIH el estado del arte del TAR para que puedan ajustar sus actuaciones terapéuticas. Se incluyen algunos aspectos del tratamiento y prevención que tanto GeSIDA como la SPNS, en colaboración con otras sociedades científicas, han elaborado in extenso, a las que se remite al lector interesado en el tema.
MetodologíaEl panel lo integran un grupo de clínicos expertos en el tratamiento de pacientes infectados por el VIH y en el uso de los FAR. Estos profesionales han sido seleccionados por la Junta Directiva de GeSIDA tras una convocatoria pública entre sus socios y por la SPNS y han aceptado participar voluntariamente. Cada miembro del panel ha emitido una declaración de conflicto de intereses. Los componentes del panel se han dividido en grupos formados por un redactor y 2consultores para actualizar cada capítulo de las recomendaciones. Tres miembros del panel actúan como coordinadores y 2 como redactores generales.
Para la actualización de estas guías cada redactor ha revisado los datos más relevantes de las publicaciones científicas (PubMed y Embase; idiomas: español, inglés y francés) o las comunicaciones de los congresos recientes de la materia (hasta el 30 noviembre de 2012). Con esta recopilación el redactor de cada grupo actualiza su capítulo y somete sus aportaciones a los consultores, consensuando las aportaciones. Cada capítulo se remite a los coordinadores y finalmente se ensamblan en un documento en el que las novedades se resaltan en amarillo. El documento se consensúa en una reunión presencial de los coordinadores y redactores. Si con posterioridad publicara información considerada relevante, se incluiría.
Tras ello, el documento se expone durante un periodo de tiempo en la web de las entidades promotoras (GeSIDA y SPNS) para que los profesionales, los pacientes o quien esté interesado pueda proponer matices o cambios que el panel puede o no aceptar a posteriori.
En este documento la fuerza de la recomendación y la gradación de las pruebas que la sustentan se basan en una modificación de los criterios de la Infectious Diseases Society of America (IDSA)5. Según estos criterios, cada recomendación debe ofrecerse siempre (A), en general (B) u opcionalmente (C), y ello basado en la calidad de los datos obtenidos a partir de uno o más ensayos clínicos aleatorizados con resultados clínicos o de laboratorio (I), de uno o más ensayos no aleatorizados o datos observacionales de cohortes (II), o de la opinión de expertos (III).
Se debe recordar que los datos sobre TAR cambian frecuentemente, por lo que los lectores deben consultar con regularidad otras fuentes de información. GeSIDA y la SPNS actualizarán estas recomendaciones periódicamente en función de la evolución de los conocimientos.
Principios generalesTras más de 20años de estudios clínicos con FAR efectuados en todos los estadios evolutivos de la infección por el VIH y utilizando los fármacos en distintas combinaciones, pueden establecerse los siguientes principios:
- 1.
El TAR se basa en combinaciones de al menos 3fármacos, lo que retrasa la progresión clínica (morbilidad e ingresos hospitalarios), reduce los costes y aumenta la supervivencia6. Se han establecido pautas eficaces con menos número de fármacos en esquemas de simplificación y rescate que no están autorizadas por las autoridades sanitarias como tratamiento de inicio.
- 2.
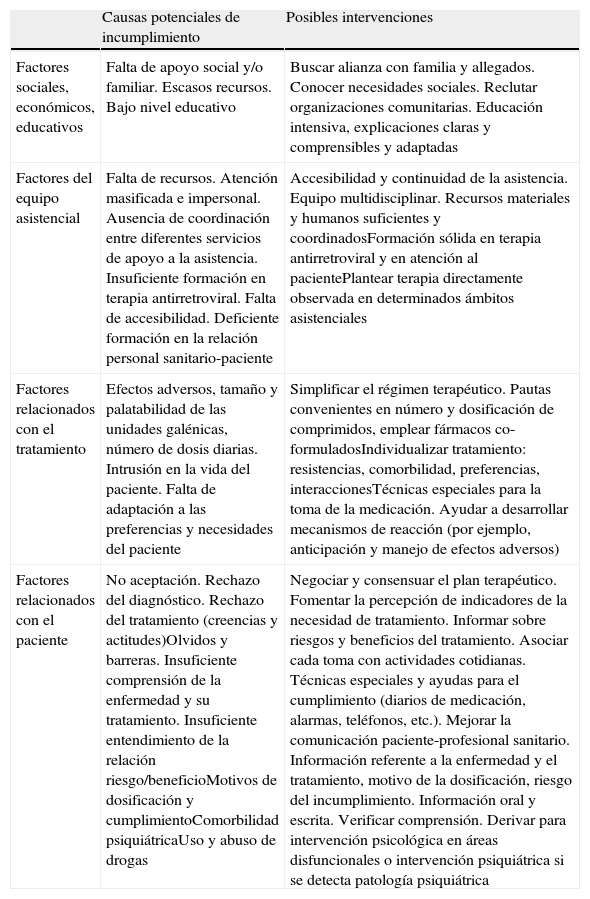
La adherencia al TAR desempeña un papel primordial en el grado y en la duración de la respuesta antiviral7. Por ello es imprescindible que cada centro hospitalario tenga una estrategia para mejorar el cumplimiento del TAR mediante una estrecha colaboración entre todos los profesionales implicados.
- 3.
La situación clínica, la cifra de linfocitos CD4 y la carga viral plasmática (CVP) son los elementos básicos para establecer las decisiones terapéuticas y monitorizar la efectividad del TAR. Los linfocitos CD4 y la CVP son los parámetros imprescindibles para la toma de decisiones. Ambos son factores predictores independientes de la progresión de la enfermedad y de la aparición de otras entidades que en principio no se creían relacionados con el VIH. Además, el nivel de CD4 indica el riesgo de padecer episodios oportunistas y señala el momento de iniciar las profilaxis de las infecciones oportunistas. Existe una buena correlación entre las respuestas virológica, inmunológica y clínica (restauración de la inmunidad celular, retraso en la progresión y aumento de supervivencia)8.
- 4.
El objetivo del tratamiento es reducir la CVP por debajo de los límites de detección (<50 copias/ml) y mantenerla suprimida el mayor tiempo posible.
- 5.
La aparición de resistencias es un fenómeno inevitable cuando el VIH continúa replicando bajo presión selectiva de fármacos. La detección de las mutaciones de resistencias por métodos genotípicos es muy útil en el fracaso virológico.
- 6.
Con las pautas actuales de TAR es posible cierta restauración cuantitativa y cualitativa del sistema inmune independientemente de la inmunodepresión de partida9,10. La recuperación es lenta y constante mientras el TAR sea efectivo y es más difícil a partir de un determinado grado de deterioro y en la edad avanzada.
- 7.
En enero de 2013 disponemos de 24FAR comercializados, pertenecientes a 6familias, lo que posibilita múltiples estrategias terapéuticas individualizadas.
- 8.
La toxicidad de los FAR a medio y a largo plazo es un factor limitante que obliga a buscar nuevas opciones terapéuticas manteniendo la potencia antiviral11.
- 9.
Hay diversas pautas de TAR que son similares en cuanto a potencia antirretroviral. La elección dependerá de factores como tolerabilidad y seguridad, adherencia, tratamientos previos, resistencias cruzadas, interacciones farmacológicas, disponibilidad y coste, así como las preferencias del médico o del paciente.
- 10.
La recomendación de iniciar el TAR cuando la cifra de CD4 desciende por debajo de un determinado nivel se debe fundamentalmente al temor a los efectos secundarios11, a las dificultades de adherencia7 y el riesgo de desarrollo de resistencias. También ha influido en esta actitud la imposibilidad de erradicar el VIH12 y de restaurar la respuesta inmunoespecífica frente a él13,14. Sin embargo, la mejoría gradual de las pautas de inicio en cuanto a tolerabilidad y simplicidad, la evidencia de un efecto negativo directo del VIH per se, así como el incremento de opciones de rescate tras un fracaso virológico han reabierto el debate sobre este tema y algunos expertos abogan por recomendaciones menos restrictivas para iniciar el tratamiento.
- 11.
La complejidad creciente del TAR implica que el cuidado de los pacientes debe efectuarse por personal especializado que tenga los conocimientos y los medios adecuados15.
- 12.
La prevención de la infección por el VIH es un aspecto fundamental que no debe olvidarse nunca en la práctica clínica y que debe introducirse de forma sistemática en la educación de los pacientes y de las personas de su entorno16.
Para el control del seguimiento y la evaluación del impacto de estas recomendaciones en la evolución clínica y respuesta al TAR de los pacientes seguidos en cada centro o por cada profesional se pueden utilizar algunos de los parámetros específicos incluidos en la publicación de GeSIDA sobre indicadores de calidad17.
Parámetros para guiar el tratamiento antirretroviralEl recuento de linfocitos CD4 y la CVP son los parámetros que se utilizan para indicar el TAR, monitorizar su eficacia y tomar decisiones respecto a cambios.
Existen otros parámetros que, aunque no intervienen directamente en el inicio del TAR, se deben tener en cuenta igualmente en la evaluación inicial o en el seguimiento, ya que pueden matizar decisiones terapéuticas (tabla 1).
Exploraciones complementarias en la valoración inicial y el seguimiento de los pacientes con infección por el VIH-1
| Valoración inicial |
| Exploración física completa |
| Presión arterial |
| Medidas antropométricas (talla, peso y cintura) |
| Hematimetría |
| Bioquímica plasmática (incluyendo glucemia, perfil hepático y lipídico) |
| Análisis elemental de orina y sedimento |
| Serología: VHB, VHC, VHA, lúes, CMV y Toxoplasma |
| Serología de Trypanosoma cruzi (si inmigrante de un país endémico)a |
| Estudio de poblaciones linfocitarias |
| Carga viral del VIH-1 |
| Estudio genotípico de resistencia |
| HLA-B*5701 |
| Mantoux (o quantiferón) y radiografía de tórax |
| Citología cervical uterina (en la mujer) |
| Citología anal (considerar) |
| Valoración de la fibrosis hepáticab |
| Previamente al inicio del TAR |
| Estudio genotípico de resistencia |
| Determinación de subtipos virales |
| Tropismo viral (si se prevé utilizar maraviroc en el TAR de primera línea) |
| Seguimiento (a las 4 semanas del inicio y cada 3-6 meses) |
| Hematimetría y bioquímica de rutina (con perfil lipídico) |
| Carga viral del VIH-1 y estudio de poblaciones linfocitarias |
| Estudio genotípico de resistencia si existe fracaso virológico |
| Tropismo viral (si existe fracaso virológico) |
En personas procedentes de áreas con alta prevalencia de infestación por Strongyloides stercoralis se puede considerar la realización de estudio serológico frente a este parásito, sobre todo si se sospecha infestación (por ejemplo, eosinofilia). En la evaluación de un paciente inmigrante, sin documentación de vacunación en su historia previa, puede considerarse la valoración de su estado vacunal o ser remitido a un centro adecuado para su valoración.
El número de linfocitos CD4+ es el marcador principal de riesgo de progresión clínica de la infección VIH-1 y necesidad de TAR. Un objetivo del TAR es la restauración inmunológica, y la forma más práctica de valorarlo es midiendo el incremento de los linfocitos CD4+, que es evidente en las primeras semanas de tratamiento18–20. Además del número de linfocitos CD4+, se restaura la respuesta proliferativa frente a mitógenos y antígenos memoria, pudiendo retirar las profilaxis de infecciones oportunistas19–22. Paralelamente al aumento de los linfocitos CD4+, hay una disminución de los linfocitos CD8+ y otros marcadores de activación del sistema inmune19,21.
El aumento de la cifra de linfocitos CD4+ es lento pero constante en el tiempo. No hay datos que definan cuál es la respuesta inmunológica adecuada. Se admite, según estudios de cinética celular, que durante el primer año debería existir un aumento mínimo de 50-100linfocitos CD4+/μl23. No es raro observar una discordancia entre respuesta virológica e inmunológica: pacientes que mantienen una cifra de linfocitos CD4+ estable o que disminuye a pesar de tener una CVP no detectable24–27. En esta situación puede existir carga viral detectable en el tejido linfático por un TAR subóptimo26. Sin embargo, esta linfopenia puede deberse a otras causas, como hipertensión portal o toxicidad farmacológica. En este sentido, se ha sugerido que en cirróticos se pueda usar el porcentaje de linfocitos CD4+ para la toma de decisiones28,29, aunque también en estos pacientes el número de linfocitos CD4+ ha demostrado ser el mejor predictor de riesgo30. Por otra parte, se ha comunicado la posibilidad de suspender la profilaxis frente a P.jiroveci en pacientes con <200linfocitos CD4+/μl si la CVP está indetectable, lo que orienta a que la supresión viral continuada debe ser necesaria para la reconstitución de la función inmune31.
En los pacientes asintomáticos deben medirse los linfocitos CD4+ cada 3-6meses, y ante un hallazgo que oriente a tomar una decisión terapéutica, debe repetirse en 3-4semanas32,33.
Recomendaciones- •
Se debe controlar el número de linfocitos CD4, ya que es el parámetro más importante para decidir el inicio del TAR. (A-I)
- •
Se aconseja la repetición del recuento de linfocitos CD4 antes de tomar la decisión de iniciar el TAR. (B-III)
El objetivo del TAR es suprimir la replicación viral de modo rápido y duradero. La CVP desciende rápidamente al inicio del TAR, y el nadir, que se alcanza a las 4-8semanas, se correlaciona con la duración de la respuesta34–37. Los pacientes con CVP muy elevadas pueden tardar hasta 24semanas en conseguir niveles inferiores a 50copias/ml38.
El objetivo de supresión de la CVP es conseguir una cifra inferior a 50copias/ml, cifra con la que se ha comprobado que no se seleccionan mutaciones39,40 y con la que la duración de la respuesta virológica es mucho mayor (frente a los que mantienen CVP entre 50 y 500copias/ml)35. En los pacientes con CVP controlada se han observado ocasionalmente brotes transitorios de viremia de bajo nivel (blips)41 que vuelve espontáneamente a ser indetectable sin ningún cambio en el tratamiento. La patogenia de los blips no está clara, y se ha sugerido que la activación inmune por infecciones intercurrentes estimularía las células con infección latente (reservorios)42. La mayoría de estudios no relacionan los blips con fracaso virológico43-46, aunque un pequeño porcentaje pueden desarrollarlo con aparición de mutaciones de resistencia47,48.
Los criterios de respuesta y fracaso virológicos son:
- •
Respuesta virológica. Descenso de la CVP>1log10 a las 4 semanas de TAR y CVP<50copias/ml a las 16-24semanas.
- •
Fracaso virológico. Cualquiera de las siguientes situaciones: a)CVP detectable a las 24semanas de TAR, o b)si tras alcanzar CVP indetectable (<50copias/ml) vuelve a ser detectable en 2determinaciones consecutivas.
Es conveniente medir la CVP a las 4semanas de inicio del TAR para comprobar la respuesta virológica y como medida indirecta de adherencia. Posteriormente la determinación se hará cada 3-6meses. Si la medida de la CVP se efectúa tras un proceso viral intercurrente o vacunación, puede haber brotes transitorios49.
Recomendaciones- •
Se debe conocer la CVP, ya que es un criterio secundario para el inicio del TAR, complementario al número de CD4. (A-II)
- •
La eficacia del TAR se controla mediante la CVP, que es el parámetro principal para su evaluación, definir su fracaso y tomar decisiones de cambio. (B-I)
- •
Debe utilizarse una técnica de determinación de CVP cuyo dintel mínimo de detección sea <50copias/ml y usar siempre la misma técnica. (A-I)
- •
Si se van a tomar decisiones terapéuticas en función de un resultado de la CVP, se debe confirmar con una segunda determinación. (A-II)
Las concentraciones plasmáticas de algunos FAR se correlacionan con su eficacia o toxicidad, por lo que se ha sugerido que la determinación de los niveles plasmáticos podría ser útil para optimizar su uso50.
Se conocen determinadas situaciones clínicas o factores que pueden inducir variaciones importantes en los niveles plasmáticos de los FAR, lo que justificaría su determinación51. Entre ellos están el sexo, la edad, el peso y la superficie corporal, los niveles de alfa1-glucoproteína y variaciones en las isoformas del citocromo P450, las interacciones medicamentosas, el embarazo, la insuficiencia hepática o la renal.
La monitorización de los niveles plasmáticos se limita a ITINN e IP, ya que la determinación de la forma activa de los ITIAN (intracelular) tiene una variabilidad inter e intrapaciente tan amplia que dificulta su uso clínico.
En los IP la determinación de los niveles ha perdido vigencia con respecto a su eficacia desde que se utilizan potenciados, aunque ha aumentado su valor para reducir la toxicidad. Los datos que relacionaban niveles plasmáticos de IP y eficacia se obtuvieron en los estudios de desarrollo donde se utilizaron en monoterapia52. Los estudios en regímenes de combinación han mostrado resultados dispares, variando según línea de tratamiento (sin terapia previa frente a pretratados), fármacos acompañantes (otros IP o ITINN) o potenciación con RTV53–59. En cuanto a toxicidad, se ha demostrado una relación entre niveles plasmáticos y algunos efectos secundarios como alteraciones gastrointestinales, hipertrigliceridemia y parestesias (RTV), alteraciones renales (IDV), hepatotoxicidad (NVP), colesterol total y triglicéridos (LPV/r)60–64. Igualmente hay datos que sugieren que los pacientes que alcanzan concentraciones más elevadas de EFV tienen mayor riesgo de síntomas neuropsiquiátricos65,66.
Las limitaciones del uso rutinario de niveles plasmáticos en la clínica diaria son múltiples: no existen estudios prospectivos que demuestren su utilidad en mejorar la eficacia, ni se dispone de rangos terapéuticos asociados a respuesta o reducción de reacciones adversas, y no se dispone de la técnica en la mayoría de los laboratorios.
Recomendaciones- •
La medición de niveles plasmáticos de los FAR no está recomendada de forma rutinaria para el manejo del paciente con infección por VIH-1. (C-III)
- •
La medición de niveles plasmáticos de los FAR podría ser de ayuda en el manejo de situaciones clínicas concretas (interacciones farmacológicas, TAR en trasplante de órgano, delgadez u obesidad mórbidas, embarazo, insuficiencia hepática o renal, etc.). (C-III)
La tasa de mutación espontánea de los retrovirus se estima en un nucleótido por cada 104 o 105 nucleótidos y copia de la cadena del ARN67–69. Por otro lado, la vida media de los linfocitos CD4 infectados que replican activamente es de un día, y se estima que la vida media del virus en plasma es de 6h70–73. La conjunción de esta alta tasa de error de la transcriptasa inversa y de la rápida renovación de la población viral produce una gran cantidad de variantes virales, que reciben el nombre de cuasi especies. El número de variantes genéticas distintas presentes en un momento dado en un individuo infectado se estima entre 5×105 y 5×1010.
Las mutaciones que confieren resistencia pueden existir en estas cuasi especies, pero representan una proporción mínima de la población viral hasta que se ve sometida a la presión selectiva del TAR72. Así pues, con el tratamiento las variantes resistentes se convierten en la población dominante al cabo de semanas o meses si no se suprime la replicación viral74–76.
No todas las mutaciones tienen la misma importancia. Para cada FAR existen las llamadas mutaciones «principales», cuya presencia está estrechamente ligada a la aparición de resistencia y que reducen la eficiencia biológica del virus (fitness), y las «secundarias», que también contribuyen en menor medida a la resistencia, pero en general actúan modificando la capacidad replicativa viral.
Detección de resistencias del virus de la inmunodeficiencia humana tipo 1 a fármacos antirretroviralesLas variantes resistentes pueden detectarse mediante técnicas genotípicas o fenotípicas77. Las genotípicas detectan cambios específicos en el genoma de las enzimas diana de los fármacos (transcriptasa inversa, proteasa, integrasa, envuelta), mientras que las técnicas fenotípicas determinan la respuesta de la población viral mayoritaria a concentraciones crecientes de los fármacos. Ambas comparten limitaciones como la dificultad de detección cuando la población mutada es <20% de la población viral o la CVP es <1.000copias/ml77, aunque técnicas recientes van reduciendo ambas limitaciones. En el fracaso virológico las pruebas de resistencias deben realizarse durante el TAR78 activo, ya que la población viral resistente será sustituida por otra sensible a las pocas semanas de retirar los fármacos. Los resultados de estas pruebas se deben interpretar teniendo presentes los estudios previos de resistencia, la historia terapéutica y la adherencia.
Las técnicas genotípicas y fenotípicas tienen ventajas y desventajas que las hacen complementarias entre sí77. Las técnicas genotípicas son más sencillas, rápidas y accesibles para la mayor parte de laboratorios y permiten la detección de mutaciones centinela antes de que se detecten cambios de susceptibilidad en las pruebas fenotípicas. Su mayor limitación estriba en la dificultad de establecer una correlación genotipo-fenotipo y, sobre todo, en las dificultades de interpretación para algunos fármacos. Además, su aplicación en la práctica diaria requiere el conocimiento previo por parte del clínico de la influencia que tiene cada mutación en la eficacia de cada fármaco. Las técnicas fenotípicas tienen la ventaja de informar del efecto neto de las distintas concentraciones sobre la sensibilidad real de la cepa predominante a los FAR, hayan sido utilizados o no. Existe una buena correlación entre las 2técnicas existentes en la actualidad: Virologic Phenosense y Virco Antivirogram79. Sus mayores desventajas son el coste, la disponibilidad limitada y la demora en la obtención de resultados. Para superar estas desventajas se ha desarrollado el fenotipo virtual80, obtenido a partir de una base de datos que tiene miles de muestras analizadas por ambas técnicas. Ante un determinado genotipo el sistema busca todos los genotipos coincidentes en la base de datos y calcula el fenotipo medio de estos pacientes. En una modificación posterior (VircoType®) se añade información de predicción de la respuesta: proporción de pacientes con respuesta máxima o reducida y ausencia de respuesta (puntos de corte clínicos). Se ha demostrado que existe una buena correlación entre ambos métodos79,80, sobre todo con los ITINN y menos con los ITIAN. La correlación es bastante más deficiente en pacientes multitratados.
Numerosos trabajos han estudiado, en países desarrollados, la prevalencia de resistencias primarias en pacientes con infección aguda o crónica. Se sabe que la mayoría de las mutaciones pueden detectarse durante años y que su prevalencia ha aumentado, llegando a superar el 10%81–83. En España, en un estudio multicéntrico de pacientes con infección reciente se encontró que el 14% de las cepas tenían mutaciones primarias84, pero datos posteriores muestran una reducción de la prevalencia en los diagnósticos nuevos al 10%85,86. Y aún más recientemente, la prevalencia de transmisión de mutaciones de resistencia en población no tratada en el periodo 2004-2008 en los nuevos diagnósticos de la cohorte prospectiva de adultos de la red de Sida (CoRIS) se ha estimado en el 8,4%87. Actualizaciones posteriores de la cohorte han mostrado una disminución progresiva de la prevalencia global de transmisión de cepas con mutaciones de resistencia, siendo la del 2011 del 7,5%88,89. En pacientes en seguimiento durante años se debería considerar repetir el genotipado antes de iniciar el TAR ante la posibilidad de haber sufrido una reinfección con mutaciones de resistencia. En otro orden de cosas, y dado el incremento del porcentaje de pacientes con subtipos no-B que se diagnostican actualmente en España, se debe considerar la realización del subtipo viral a la vez que el estudio de resistencia87. Con respecto a la posibilidad de transmisión de virus con mutaciones de resistencia en la integrasa viral los datos actuales muestran la ausencia de las mismas, por lo que actualmente no se recomienda esta determinación en los nuevos diagnósticos90.
En España, una de las actividades de la plataforma de resistencias de la Red de Sida ha sido el establecimiento de una base de datos de secuencias de pacientes en fracaso virológico atendidos en numerosos centros hospitalarios españoles. Expertos de esta plataforma han elaborado un algoritmo de interpretación que se actualiza anualmente y que permite la predicción de la resistencia on line. Se puede acceder a dicho algoritmo a través de la página web de la Red de Investigación en Sida (www.retic-ris.net).
Una de las principales limitaciones de los estudios de resistencias consiste en que no son capaces de detectar mutaciones que no representan más del 15-20% de la población viral. Aunque se han desarrollado algunas tecnologías (PCR alelo específica, secuenciación de genomas individuales o secuenciación masiva de genomas únicos [UDS]) que permiten detectar mutaciones de resistencia en niveles de hasta el 0,1-1%, en la actualidad no están disponibles para su uso rutinario y no se conoce con precisión la utilidad de la detección de estas poblaciones minoritarias que se escapan al estudio genotípico convencional91–98. Sin embargo, las últimas Guías europeas sobre uso de pruebas de resistencia tampoco aconsejan su uso de forma rutinaria, alegando que son costosas, que no están disponibles en la mayoría de los laboratorios y que los controles de calidad no están resueltos99. De todas formas, en un metaanálisis se ha visto que existe una relación entre el número de variantes minoritarias detectadas, fundamentalmente mutaciones a los ITINN, y el riesgo de fracaso de tratamientos que incluyan EFV100.
Significado clínico de las resistencias del virus de la inmunodeficiencia humana tipo 1 a fármacos antirretroviralesCon el uso de la CVP en el seguimiento de los pacientes con infección por VIH-1 se ha evidenciado la relación entre aparición de mutaciones de resistencias y fracaso virológico. Sin embargo este fenómeno no es homogéneo para todos los fármacos, ya que es muy claro en los ITIAN e ITINN pero existen datos contradictorios con los IP, con los que se ha constatado fracaso virológico sin evidencia de mutaciones101. En este sentido se ha definido el fenómeno de resistencia celular, ya que se ha detectado la existencia de bombas de expulsión de los FAR en la membrana de los linfocitos y otras células. Se han descrito el MDRP-1 (glucoproteínaP) para los IP y la MDRP-4 para los ITIAN102. Se desconoce la relevancia clínica de estos hallazgos.
Los estudios prospectivos y aleatorizados que han utilizado las pruebas de resistencias para el manejo del fracaso virológico comparan la eficacia del cambio de TAR cuando se realiza según las distintas pruebas de resistencia (genotipo, fenotipo o fenotipo virtual) con o sin consejo de expertos o en función de la historia terapéutica previa y/o la experiencia clínica de los médicos103–111. Un metaanálisis de los primeros estudios comunicados puso de manifiesto que el uso del genotipo para diseñar el TAR de rescate frente al estándar (historia terapéutica y experiencia del médico) se asociaba con un control virológico significativamente mayor a los 3 y 6 meses112. Estas diferencias no se observaron en los estudios que compararon los métodos fenotípicos frente al manejo estándar112. Por otro lado, no se han detectado diferencias cuando se han comparado el fenotipo virtual y el fenotipo real110,111. Se requieren pues más datos para aclarar el papel de las pruebas fenotípicas.
Recomendaciones- •
Se deben estudiar las mutaciones de resistencia, ya que su conocimiento permite un mejor uso de los fármacos. (A-I)
- •
Se considera indicada la realización estudios genotípicos de resistencias en la práctica asistencial en las situaciones expuestas en la tabla 2. (B-II)
- •
No se recomienda el genotipado de la integrasa viral en pacientes naïve. (C-III)
- •
Se debe considerar la determinación de los subtipos virales, especialmente en pacientes inmigrantes o en caso de evolución clínica rápida. (C-III)
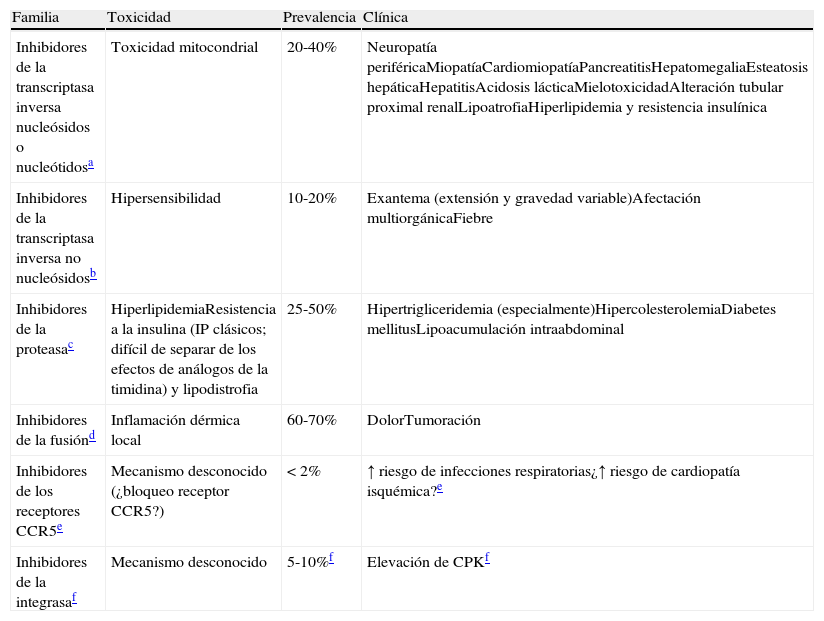
La reacción de hipersensibilidad (RHS) a ABC es un síndrome multiorgánico que se manifiesta con una combinación variable y de intensidad creciente de fiebre, mialgias, síntomas respiratorios, gastrointestinales o un exantema, pudiendo ser fatal en caso de continuar tomando el fármaco o reintroducirlo. Suele aparecer durante las primeras 6semanas de tratamiento, se presenta en el 5-8% de los pacientes que toman ABC y es la causa más frecuente de su discontinuación113. Se sabe que la RHS es más frecuente en la población blanca y se dispone de una prueba cutánea (parche) para su confirmación.
Estudios farmacogenéticos han identificado que la RHS ocurre en las personas portadoras del alelo HLA B*5701. En un ensayo en el que los pacientes se aleatorizaron a comenzar TAR con ABC o comenzarlo solo si el HLA*5701 era negativo, se valoró la aparición de la RHS por datos clínicos que se confirmaron con una prueba cutánea (confirmación inmunológica). La prevalencia del HLA B*5701 en esta cohorte era del 5,6%. La genotipificación del HLA B*5701 redujo la incidencia de sospecha clínica de RHS (3,4% frente al 7,8%) y la inmunológica (0% frente al 2,7%), siendo el valor predictivo negativo de esta prueba del 100%114. Se ha validado esta prueba en población negra, confirmando los resultados115.
El ABC no debe utilizarse en personas portadoras del alelo HLA B*5701. Si es negativo no se descarta la posibilidad de RHS, por lo que se debe informar y controlar los síntomas de estos pacientes respecto a la RHS cuando se inicia tratamiento con ABC.
Recomendaciones- •
Se deben determinar el HLA B*5701 a todos los pacientes en el momento del diagnóstico o cuando vayan a comenzar TAR con ABC. (A-I)
- •
Si el HLA B*5701 es positivo no se debe prescribir ABC. (A-I)
- •
Si el HLA B*5701 es negativo no se puede descartar completamente la RHS, por lo que se debe informar al paciente y vigilar su posible aparición. (A-I)
- •
Si se prescribe ABC sin conocer el HLA B*5701, se debe informar al paciente y estar alerta para detectar síntomas de la RHS. (C-III)
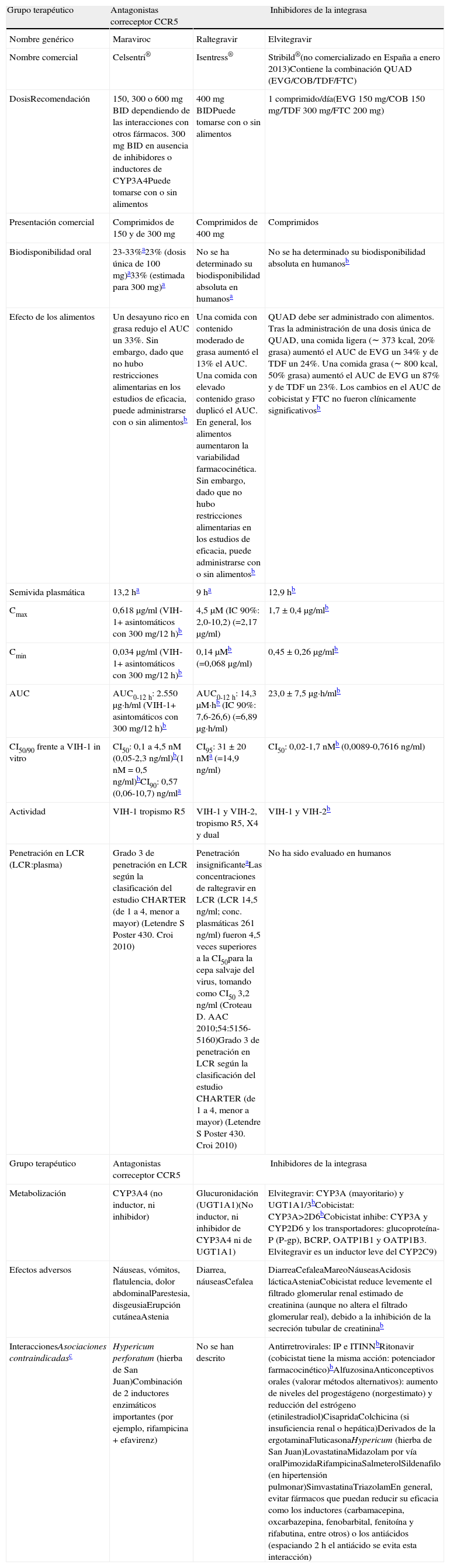
El VIH-1 entra en la célula diana por un mecanismo que incluye el reconocimiento del receptor CD4+, seguido de la unión a uno de los correceptores CCR5 o CXCR4 y la fusión de las membranas con paso del ARN del VIH-1 a la célula invadida. Los inhibidores del CCR5 (MVC, vicriviroc) son fármacos que bloquean este receptor impidiendo la entrada del VIH-1 en la célula116.
Tras la infección por el VIH-1 la mayoría de los pacientes albergan virus que usan el correceptor CCR5 (R5). Si no se inicia un tratamiento, el virus evoluciona a variantes que, en mayor o menor proporción, utilizan el correceptor CXCR4 (X4) o duales o mixtas (D/M) que pueden usar ambos correceptores. Este cambio se relaciona con un descenso de CD4 y aumento de la inmunodepresión, ya que se consideran más virulentas117. En los pacientes multitratados con CVP detectable las variantes X4 o D/M son más prevalentes, pudiendo llegar a superar el 50% entre los que tienen <100CD4/μl118,119.
Actualmente existe una técnica fenotípica para la detección del tropismo (Trofile®, Monogram Biosciences, EE.UU.) que se realiza en un solo centro (California, EE.UU.) y que llega a detectar la población X4 o D/M cuya proporción supere el 0,3% si la CVP es ≥1.000copias/ml (prueba ultrasensible)120. Este ensayo presenta limitaciones técnicas y logísticas que dificultan su utilización en la práctica clínica. Los ensayos genotípicos (secuenciación de la regiónV3) se presentan como una alternativa más económica, rápida y factible de desarrollar en cualquier laboratorio especializado de VIH-1 que cuente con tecnología para realizar estudios genotípicos. Los primeros estudios de correlación entre métodos genotípicos y Trofile® mostraron, en general, una baja sensibilidad de los primeros para la detección de variantes X4-trópicas121. Posteriormente, la introducción de mejoras en la interpretación de algunos algoritmos genotípicos y de estrategias de combinación de los mismos121–124 ha conseguido mejorar notablemente la sensibilidad de estos para detectar variantes X4-trópicas en comparación con el ensayo de Trofile®. En un análisis retrospectivo de los ensayos clínicos con MVC MOTIVATE y MERIT en pacientes pretratados y sin tratamiento previo, respectivamente, se ha demostrado que las herramientas genotípicas y el ensayo de Trofile® son comparables en la predicción de respuesta virológica a MVC125,126. Estos datos ponen de manifiesto la viabilidad de la utilización de métodos genotípicos para la determinación del tropismo viral en la práctica clínica, aunque el método genotípico no está homologado. En marzo de 2010 un grupo de investigadores nacionales, con experiencia en la determinación de tropismo viral, iniciaron una serie de reuniones que han culminado en la publicación de una revisión en la que se detallan las recomendaciones que este grupo estima adecuadas para la determinación genotípica del tropismo en la práctica clínica127, y también recientemente se han publicado las guías europeas para la realización del tropismo128. Las recomendaciones actuales respecto a la determinación del tropismo serían efectuarlo en todos los pacientes que hayan fracasado a cualquier línea de tratamiento y vayan a iniciar un tratamiento de rescate. En pacientes sin tratamiento previo se recomienda realizar la determinación del tropismo cuando se va a iniciar el TAR en determinadas situaciones clínicas (resistencias primarias, toxicidad a fármacos de primera línea, etc.) en las que un fármaco antagonista del receptor CCR5 pueda considerarse una buena opción terapéutica. La información del tropismo (tropismo R5/tropismo X4), incluyendo el valor del false positive rate (FPR), debe estar disponible rutinariamente junto con el estudio de resistencias con el fin de facilitar la selección del mejor tratamiento de rescate. Cuando se plantee el uso de MVC en pacientes con carga viral indetectable se recomienda la realización del tropismo a partir del ADN proviral. Entre las recomendaciones metodológicas se aconseja utilizar un FPR del 10% si solo se realiza una sola reacción de amplificación y secuenciación. La posibilidad del cambio de tropismo en pacientes que se mantienen suprimidos es muy baja, por lo que se asume el mismo tropismo en estos pacientes129,130.
Recomendaciones- •
El tropismo viral se debe conocer antes de iniciar el tratamiento con un fármaco inhibidor del receptor CCR5. (A-I)
- •
Se recomienda la determinación del tropismo del VIH-1 en todos los pacientes que hayan fracasado a cualquier línea de tratamiento y vayan a iniciar un tratamiento de rescate. (A-III)
- •
La información del tropismo (R5/X4) debe estar disponible rutinariamente junto con el estudio de resistencias a todos los antirretrovirales con el fin de facilitar la selección del tratamiento de rescate más óptimo. (A-III)
- •
En pacientes que vayan a iniciar el TAR se debe determinar el tropismo si se considera el MVC como una opción terapéutica. (B-III)
La primoinfección por el VIH-1 es sintomática en más de la mitad de los casos, pero puede pasar desapercibida ya que sus síntomas son los de una virosis común131–137. El cuadro clínico es similar a la mononucleosis infecciosa o a una meningoencefalitis viral135–137. Los síntomas y signos más comunes son fiebre, odinofagia, adenopatías de predominio laterocervical, mialgias, exantema, sudoración nocturna y artralgias138. Debe sospecharse en toda persona con síntomas compatibles, con o sin conducta de riesgo. Las determinaciones necesarias para el diagnóstico difieren de la infección crónica. Como en esta fase todavía no hay anticuerpos (período ventana) debe determinarse la CVP139, que se detecta a partir de la primera semana, precede a los síntomas y tiene una sensibilidad y especificidad del 100 y del 97%, respectivamente (debe ser una CVP alta, ya que si es <10.000copias/ml puede ser un falso positivo). Las pruebas de ELISA de cuarta generación tienen la capacidad adicional de detectar el antígeno p24 del VIH-1 en la misma reacción, acortando el período ventana en una semana. La CVP en la infección aguda suele estar muy elevada (>6log10) y se relaciona con la intensidad de las manifestaciones clínicas. La seroconversión se detecta 1-2semanas más tarde138. La técnica de Western blot, con la que se confirma el diagnóstico, puede ser negativa o indeterminada y deberá repetirse unas semanas después del comienzo de los síntomas para confirmar su positivización. La presencia o ausencia de la banda p31 indica que la infección lleva más o menos 90días de evolución136. El término infección aguda (diagnóstico antes de la seroconversión, menos de 30días) no debe confundirse con infección reciente, que es la que tiene menos de 6meses de evolución (menos de 180días)135–137.
Desde la descripción de los primeros casos de infección aguda se sabe que la progresión a sida es más rápida en los pacientes sintomáticos. En estudios de cohortes que han analizado la historia natural de la infección VIH-1 se ha evidenciado que la progresión a sida o muerte se asocia con factores iniciales de la infección, como la gravedad de la sintomatología en la infección aguda140 (mayor riesgo a mayor número de síntomas), cuantía del descenso inicial del número de linfocitos CD4+ (mayor riesgo si es <500células/μl)141, nivel de la CVP basal142 o a partir del cuarto mes (mayor progresión si el setpoint es >100.000copias/ml)140, a la cuantía de ADN proviral inicial (mayor progresión si es >3,4log10 copias/millón de células mononucleares en sangre periférica)141, a la infección por virus X4 o con tropismo D/M143–146, a las infecciones por más de un virus VIH-1147 y al perfil genético de los individuos infectados148,149. De todos estos factores, las características de la infección sintomática, la CVP, la cifra de linfocitos CD4+ y el tropismo viral son los parámetros que nos pueden ayudar a decidir el inicio de TAR en la práctica clínica.
El TAR en la infección aguda puede tener ventajas e inconvenientes135,150. Las ventajas teóricas serían acortar la duración y la gravedad de los síntomas, suprimir la replicación viral, reducir el riesgo de transmisión del VIH-1 (muy elevado en particular en varones homosexuales)132,151–153, reducir la diversidad viral y el número de células infectadas (reservorio) y preservar o restaurar el sistema inmunitario y la inmunidad específica frente al VIH-1, tanto proliferativa (mediada por los linfocitos CD4+) como citotóxica (mediada por los linfocitos CD8+)131,154–158, lo que podría permitir el control inmunológico de la replicación viral, modificar la historia natural, disminuir el riesgo de progresión y mejorar el pronóstico. Para ello el TAR debería iniciarse en los primeros 30días de la infección, ya que a partir de entonces se produce el escape virológico a la respuesta VIH-específica celular y humoral159. Por el contrario, las principales desventajas del TAR en esta fase son la exposición a los FAR sin un beneficio clínico demostrado, su duración indefinida (ya que con las pautas de TAR que hasta ahora se han utilizado no se erradica la infección131 ni se restaura el tejido linfático asociado a mucosas160), el riesgo de efectos secundarios, de desarrollo de resistencias si el cumplimiento no es adecuado, la posible reducción de la calidad de vida y el tratamiento innecesario de los individuos no progresores.
En la actualidad el inicio del TAR durante la infección aguda o reciente aún es controvertido135,150,161–163, ya que su potencial beneficio a largo plazo es desconocido. La información disponible se ha obtenido de series pequeñas, generalmente sin grupo control, en países desarrollados, con pacientes infectados por el subtipoB del VIH-1 y con pautas de TAR similares a las utilizadas en la infección crónica. No se ha demostrado un beneficio clínico en términos de reducir la progresión a sida o muerte161, aunque en algún estudio se ha visto una mejor evolución inmunológica y virológica en pacientes tratados164–167 si se inicia el TAR durante la infección aguda por el VIH-1. Recientemente se ha publicado un estudio americano de cohortes168 que demuestra que durante los primeros 4meses que siguen a la infección por el VIH existe un aumento transitorio de la cifra de linfocitos CD4 y que tras alcanzar la cifra máxima posteriormente disminuye de forma progresiva, siendo la cifra de CD4 menor que la basal a partir de los 12meses de la infección. Este estudio demuestra que el máximo beneficio inmunológico del TAR se consigue cuando este se inicia en los primeros 4meses de la infección por el VIH (la proporción de pacientes que posteriormente alcanza una cifra de CD4>900células/μl fue del 64% vs. solo el 34% cuando el TAR se inició a partir de los 4meses de la infección; p<0,001)168.
En cualquier caso, si se decide iniciar TAR las pautas recomendadas son las mismas que en la infección crónica y la respuesta virológica a IP o NN es similar165,168–170. Sin embargo, en este escenario se podría considerar una pauta con 2ITIAN (preferentemente TDF/FTC) y un inhibidor de la integrasa (RAL y en un futuro con EVG/COBI o con DTG) por su mayor concentración en las secreciones genitales159 y la reducción más rápida de la CVP durante las primeras 4-8semanas de TAR en comparación con los IP o ITINN171,172, lo que podría facilitar la reducción de la transmisión del VIH171. Estudios preliminares con 4 o 5FAR, incluyendo inhibidores de la entrada y/o de la integrasa, no parecen ser más efectivas a las 48semanas en este escenario172,173. Como en la infección crónica, siempre debe efectuarse una prueba de resistencia, se vaya a iniciar TAR o no, por la posibilidad de transmisión de cepas resistentes. En pacientes con multirresistencia se debe realizar un tropismo viral porque se han descrito cepas con fenotipo dual R5/X4 con una rápida progresión a sida138–141. Dado que la prevalencia de cepas con resistencia a ITINN es mayor que a IP o inhibidores de la integrasa en pacientes con infección aguda o reciente84,174,175, si se decide iniciar el TAR y todavía no está disponible el resultado del estudio de resistencias es preferible comenzar con una pauta basada en IP. Por otra parte, se recomienda realizar un tropismo viral, ya que los pacientes con fenotipo viral no-R5 tienen una progresión a sida más rápida143–146. Con las pautas de TAR clásicas se ha observado que la prevalencia de lipodistrofia, dislipidemia y otros efectos adversos asociados al TAR es similar a la de los pacientes con infección crónica170,176–178.
Para evitar el TAR indefinido en este escenario y conseguir los objetivos mencionados previamente se han planteado diversas estrategias135,150,161–163: a)administrar el TAR durante un periodo limitado de tiempo; b)administrarlo de forma intermitente, a fin de potenciar la respuesta VIH-específica y controlar la replicación viral sin FAR; c)combinar el TAR con inmunosupresores (hidroxiurea, ciclosporina A, ácido micofenólico) o citoquinas (IL-2, interferón), y d)asociar TAR y vacunas terapéuticas. Sin embargo, ninguna de estas estrategias ha conseguido que el sistema inmune controle la replicación viral de forma sostenida en ausencia de TAR como en los controladores de élite, por lo que en la actualidad si se inicia el TAR durante la infección aguda probablemente se deba mantener de forma indefinida.
La eficacia del TAR durante un periodo variable se ha evaluado principalmente en estudios de cohortes en los que se han comparado pacientes con infección aguda o reciente tratados frente a no tratados. Los resultados de la mayoría de estos estudios no han conseguido demostrar un beneficio clínico, virológico ni inmunológico a las 48-144semanas de interrumpir el TAR, mientras que en otros solo una pequeña proporción de los pacientes tratados mostraban un setpoint de CVP más bajo, mayores cifras de linfocitos CD4+ o mantenían la respuesta inmunoespecífica frente al VIH-1168,179–190. La proporción de controladores de elite (CVP<50copias/ml) después de haber parado el TAR iniciado durante la infección aguda/reciente se ha estudiado en 2cohortes. En la cohorte CASCADE191 solo 11 (4%) de 259pacientes seroconversores no tuvieron un rebrote virológico a los 24meses de parar el TAR. La mediana de tiempo hasta el rebrote de la CVP (2determinaciones consecutivas de CVP>50copias/ml) en los 248pacientes restantes fue de 1,7meses. En la cohorte francesa PRIMO192, solo 14 (9%) de los 164pacientes que pararon el TAR después de haberlo iniciado durante la infección aguda por el VIH lo mantuvieron indetectable durante una mediana de 4,5años.
En el último año se han publicado 3ensayos clínicos193–195 que estudiaban el beneficio clínico, inmunológico o virológico de distintas estrategias de administración del TAR en la infección aguda por el VIH. El primero es el ensayo clínico SPARTAC193, un estudio multicéntrico y multinacional en el que se aleatorizó a 367pacientes con infección aguda o reciente a no recibir TAR (n=124), a recibirlo durante 12semanas (n=120) o durante 48semanas (n=123). La variable de análisis principal era inmunológica (tiempo hasta tener una cifra de linfocitos CD4+<350células/μl) o necesidad de iniciar TAR indefinido. La mediana de seguimiento de los pacientes fue de unos 3años. El estudio concluyó que el TAR de duración limitada no fue beneficioso. En comparación con la rama control sin TAR, el de 12semanas no se asoció a ningún beneficio y el TAR de 48semanas retrasó en algo más de un año el deterioro inmunológico o el reinicio de TAR, beneficiándose más aquellos pacientes que lo iniciaron en los primeros 90días de la infección por el VIH-1. Por otra parte, la interrupción del TAR no se acompañó de efectos secundarios ni de desarrollo de resistencias, y no comprometió la restauración inmunológica cuando los pacientes reiniciaron el TAR indefinido.
El segundo es el ensayo clínico PRIMO-SHM194, un ensayo multicéntrico realizado en los Países Bajos con un diseño similar al del SPARTAC, en el que 168pacientes con infección aguda o reciente por el VIH se aleatorizaron a recibir TAR durante 24 o 60semanas o a no recibir TAR. Los pacientes con una primoinfección grave (p.ej., meningitis) o que solo querían ser tratados se aleatorizaron a TAR durante 24 o 60semanas. Las variables de análisis principal eran la virológica (CVP a las 36semanas de finalizar el TAR o de la infección en el grupo control) o el tiempo sin TAR en el grupo control o tras pararlo en las 2ramas de TAR. Los criterios de inicio de TAR fueron inmunológicos (recuento de linfocitos CD4+ <350células/μl) o clínicos (sida). El número de pacientes aleatorizados a no recibir TAR, 24 o 60semanas de TAR fueron: 36, 62 y 66pacientes, respectivamente. Cuatro pacientes se perdieron durante el seguimiento. Las media (DE) de CVP a las 36 semanas sin TAR en las 3ramas del estudio fue de 4,8 (0,6), 4,0 (1,0) y 4,3 (0,9) log10/ml (p<0,001), aunque esta diferencia se perdió con el tiempo, siendo la CVP a las 144semanas similar en las 3ramas. La recuperación inmunológica fue mayor en los pacientes con TAR, pero la pendiente de caída del número de linfocitos CD4+ al parar el TAR fue similar a la de los pacientes que no recibieron TAR. La mediana (intervalo de confianza [IC] del 95%) de tiempo sin TAR en la rama control (no TAR) fue de 0,7 (0,0-1,8) años y en las ramas de 24 y 60semanas fue de 3,0 (1,9-4,2) y 1,8 (0,5-3,0) años, respectivamente (prueba de log-rank, p<0,001). Las conclusiones de este ensayo clínico fueron que el TAR durante la infección aguda/reciente por el VIH redujo transitoriamente el setpoint de CVP y difirió el reinicio del TAR indefinido durante la infección crónica por el VIH.
El tercero es el estudio Setpoint (ACTG A5217)195, un ensayo clínico aleatorizado en el que pacientes con infección reciente (<180días) fueron asignados a recibir TAR de forma inmediata durante 36semanas con TDF/FTC y LPV/r (rama inmediata) o a no recibir TAR (rama diferida). La variable de análisis principal fue la disminución del nivel de viremia en la semana 72 sin TAR (por lo tanto se compararon 36semanas de TAR seguidas de 36semanas sin TAR frente a 72semanas sin TAR). La variable secundaria fue la necesidad de iniciar TAR indefinido. El estudio fue interrumpido por el equipo de control de seguridad debido a que muchos de los pacientes de la rama de TAR diferido debieron iniciar TAR por criterios clínicos, inmunológicos (recuento de linfocitos CD4+<350células/μl) o virológicos (CVP>200.000copias/ml), y aunque se pudieran incluir los pacientes planeados no se podría comprobar si habría diferencias en el nivel de viremia a las 72semanas entre ambas ramas del estudio. Se incluyeron 130 de los 150participantes previstos. El equipo de control de seguridad del estudio basó la decisión de parar el estudio en que requirieron TAR indefinido 23 de los 64 (36%) pacientes asignados a la rama diferida frente a 7 de los 66 (11%) pacientes asignados a la rama inmediata, la mayoría por criterios inmunológicos. Los niveles de viremia (media [DE]) en la semana 72 en pacientes sin TAR en las ramas de TAR inmediato y diferido fueron de 3,99 (0,13) y 4,15 (0,13) log10/ml, respectivamente. Al comparar el ritmo de inicio de TAR indefinido en ambas ramas, marcando como tiempo 0 en la rama de TAR inmediato la semana 36, se observó que el TAR inmediato solo retrasó ligeramente el inicio de TAR indefinido (p=0,035). Por tanto, las conclusiones de estos 3ensayos clínicos son que en la práctica clínica, si se inicia TAR en la fase aguda o reciente de la infección por el VIH-1, este debe ser indefinido.
La administración de TAR intermitente con el fin de potenciar la respuesta VIH-1 inmunoespecífica para controlar la replicación viral no ha conseguido buenos resultados, siendo testimonial la proporción de pacientes que mantenían la respuesta inmune y el control de la replicación viral sin TAR a las 96semanas158,196. Además, esta estrategia de interrupción estructurada no está exenta de la aparición de resistencias197,198.
La estrategia de asociar al TAR citoquinas como interferón-pegilado o IL-2, a fin de disminuir el reservorio viral y mejorar la respuesta inmunitaria199–201, tampoco ha conseguido sus objetivos. Se han efectuado estudios con interferón pegilado202 o inmunosupresores (hidroxiurea, ciclosporina, ácido micofenólico) para reducir la activación del sistema inmune y controlar la replicación viral198,203–206. Algunos de estos estudios solamente han podido demostrar mayor toxicidad sin aumentar la eficacia.
Finalmente, las vacunas terapéuticas, asociadas o no a TAR, con el fin de restaurar o potenciar la respuesta inmunoespecífica frente al VIH-1 tampoco han mostrado mejores resultados. Se sabía que en modelos animales algunas vacunas habían sido satisfactorias207,208, pero los resultados de ensayos clínicos doble ciego en humanos han sido desalentadores209–211. Tanto el estudio QUEST209 (TAR frente a TAR más la vacuna ALVAC-HIV, y frente a TAR más la vacuna ALVAC-HIV más Remune®), como en el ensayo clínico que comparó diferentes dosis de una vacuna que usaba el virus de la viruela aviar con los genes gag/pol del VIH-1 insertados210, como en el estudio ACTG A5187 (TAR frente a TAR más vacuna VIH ADN)211, las respuestas virológica e inmunoespecífica fueron similares a las del grupo placebo.
Las vacunas preventivas tampoco parecen modificar la historia natural de la infección por el VIH en los pacientes que se infectan. En el ensayo clínico tailandés de ALVAC-VIH y AIDSVAX B/E212, que redujo la infección por el VIH en el 31%, la vacunación no alteró el curso clínico de la infección por el VIH después de la infección, al ser el tiempo hasta tener una cifra de CD4 <350células/μl o la necesidad de iniciar TAR similar en los grupos vacunado y placebo.
Por otra parte, también debería recomendarse el inicio del TAR en todas las indicaciones de inicio de TAR que sean independientes de la cifra de linfocitos CD4+ y que se describen en el apartado de la infección crónica por el VIH, y en las mujeres embarazadas que se infectan por el VIH durante el embarazo. (Aplicar el mismo nivel y fuerza de la recomendación.)
Finalmente, debido a lo anteriormente explicado, debe valorarse siempre el incluir a estos pacientes en protocolos de investigación o en ensayos clínicos que busquen la erradicación o la cura funcional del VIH213.
Recomendaciones- •
Se recomienda iniciar TAR de forma inmediata en los pacientes con infección aguda sintomática cuando: a)exista afectación neurológica (meningoencefalitis, síndrome de Guillain-Barré, etc.) o de cualquier otro órgano o sistema (hepatitis, miopericarditis, trombocitopenia, etc.); b)sea prolongada (>7días de duración); c)se acompañe de episodios clínicos B o C relacionados con la inmunodepresión, o d)se acompañe de una inmunodepresión celular avanzada (recuento de linfocitos CD4+<350/μl (B-II); o e)el paciente tenga un tropismo viral no-R5 o una CVP a los 3meses de la infección >100.000copias/ml (B-II). En los pacientes con una infección aguda asintomática o una con infección reciente por el VIH se recomienda iniciar TAR si cumplen los criterios d) o e). (B-II)
- •
Se debe considerar iniciar el tratamiento en los casos en que exista un alto riesgo de transmisión del VIH-1. (A-II)
- •
También debe recomendarse el inicio del TAR en las indicaciones de inicio de TAR que sean independientes de la cifra de linfocitos CD4+ y que se describen en el apartado de la infección crónica por el VIH y en las mujeres embarazadas que se infectan por el VIH durante el embarazo. (Aplicar el mismo nivel y fuerza de la recomendación.)
- •
Si se decide iniciar TAR se recomienda hacerlo con las mismas pautas preferentes para tratar la infección crónica por el VIH. Una pauta con 2ITIAN (preferentemente TDF/FTC) y un inhibidor de la integrasa (RAL) tendría la ventaja de una mayor concentración en las secreciones genitales y podría reducir más rápidamente la CVP durante las primeras 4-8semanas en comparación con los IP o ITINN, lo que podría facilitar la reducción de la transmisión del VIH. (B-III)
- •
Se debe efectuar siempre una prueba de resistencias y un tropismo viral al diagnóstico de la infección aguda o reciente, se vaya a iniciar TAR o no. (B-II)
- •
Si no se dispone del resultado del estudio de resistencias es preferible comenzar con una pauta basada en un IP/r hasta tener los resultados. (A-II)
- •
Si se inicia el TAR, este debe administrarse por tiempo indefinido. (A-I)
- •
En los pacientes sin criterios de TAR se recomienda reevaluarlos a partir de los 6meses, cuando la infección pasa a ser crónica. (A-I)
Los principales motivos para iniciar el tratamiento son la reducción de la morbimortalidad asociada a la infección por VIH-1, la mejoría de la calidad de vida, la restauración y preservación de la función del sistema inmunológico, la supresión completa y duradera de la replicación del VIH-1 y la prevención de la transmisión del virus. Es importante hacer una valoración individualizada del momento de inicio del TAR y de los fármacos que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones.
La disposición y la motivación del paciente es un factor crítico a la hora de tomar la decisión de cuándo empezarlo.
Cuándo iniciar el tratamiento antirretroviralSe dispone de combinaciones de FAR que bloquean de forma duradera la replicación viral en plasma y tejido linfático permitiendo la restauración, al menos parcial, del sistema inmunológico214–216. En ediciones previas de estas guías se hacía énfasis en la influencia en el balance riesgo/beneficio del TAR de la toxicidad a medio-largo plazo de los FAR, los problemas de adherencia, la aparición de resistencias, las interacciones medicamentosas y el impacto en la calidad de vida7. Aunque todos estos factores siguen siendo muy importantes, es justo reconocer que el número de opciones terapéuticas, la eficacia, la seguridad y la simplicidad de las combinaciones de antirretrovirales han aumentado marcadamente durante los últimos 6años217. Además, debido fundamentalmente al uso de IP/r, el problema de la resistencia a múltiples clases de FAR ha disminuido considerablemente.
Gracias al TAR se ha reducido drásticamente el riesgo de progresión y muerte de los pacientes con infección por el VIH-1. A pesar de todos estos avances, la esperanza de vida del paciente infectado por el VIH-1 en tratamiento está acortada con respecto a la de la población general8,218. Sin embargo, en los pacientes que han recibido TAR durante al menos 6años y han alcanzado una cifra de CD4>500células/μl, la supervivencia podría ser similar a la de la población general219.
El aumento de la eficacia, de la seguridad y de la simplicidad del TAR, junto con el reconocimiento de que aún existe margen de mejora en el aumento de la esperanza de vida del paciente infectado por el VIH-1, ha vuelto a plantear el debate sobre un inicio más temprano del TAR.
Riesgo de progresiónLa CVP y la cifra de linfocitos CD4+ son marcadores independientes de progresión de la infección por el VIH-1220,221.
Los resultados de diferentes ensayos clínicos demuestran claramente el beneficio del inicio del TAR en pacientes cuya cifra de linfocitos CD4+ es <350células/μl222,223.
El debate actual se centra en torno a si es necesario iniciar TAR con cifras de linfocitos CD4+>500células/μl. Los datos que sustentan este debate provienen de cohortes de pacientes en las que se ha evaluado la mortalidad, la progresión a sida, la incidencia de enfermedades no definitorias de sida, la recuperación inmunológica y la toxicidad del tratamiento en función de la cifra de linfocitos CD4+ al inicio del TAR. Los estudios de cohortes tienen importantes problemas metodológicos, como el sesgo de prescripción, que hacen que la calidad de la evidencia sea menor que la proveniente de ensayos clínicos aleatorizados.
Respuesta clínica (progresión a sida o muerte)Hay datos muy limitados sobre la comparación de la mortalidad y/o progresión a sida en pacientes que empiezan TAR por encima o por debajo de 350CD4/μl.
En un análisis conjunto de varias cohortes que incluyó más de 21.000pacientes, la progresión a sida o muerte fue significativamente mayor en los que iniciaban el TAR entre 251-350CD4/μl (HR: 1,28, IC95%: 1,04-1,57) que en los que lo empezaron entre 351-450. Igualmente en el primer grupo se observó una mayor mortalidad y aun mayor diferencia cuando se analizaba conjuntamente sida o muerte (HR: 1,13; IC95%: 0,80-1,60)9. Sin embargo, no hubo diferencias significativas cuando se compararon los estratos 351-450 y 451-550.
En la cohorte PISCIS el riesgo de progresión a sida tras inicio de TAR fue significativamente menor en los pacientes que lo iniciaron con cifras de CD4≥350células/μl que en los que empezaron entre 200-350CD4/μl (HR: 1,85; IC95%: 1,03 a 3,33), o <200CD4/μl (HR: 2,97; IC95%: 1,91-4,63)10.
En un subanálisis de los pacientes con >350linfocitos CD4+/μl del ensayo SMART que no estaban recibiendo TAR al inicio del estudio, los aleatorizados a demorar el TAR hasta alcanzar la cifra de 250linfocitos CD4+/μl tuvieron mayor riesgo de enfermedades oportunistas y/o muerte por causas no directamente relacionadas con sida que quienes iniciaron TAR inmediatamente con cifras de CD4≥350células/μl222.
Por último, en un análisis paralelo de 17.517pacientes con infección por VIH-1 asintomática en EE.UU. y Canadá (estudio NA-ACCORD) se estratificaron los pacientes en función de la cifra de linfocitos CD4+ (de 351 a 500células/μl o >500células/μl) en el momento de iniciar el TAR. En cada grupo se comparó el riesgo relativo de muerte de los pacientes que iniciaron tratamiento cuando la cifra de linfocitos CD4+ era superior a cualquiera de los 2puntos de corte (tratamiento precoz) con el de los pacientes que difirieron el tratamiento hasta que la cifra de linfocitos CD4+ cayó por debajo de dichos puntos de corte (tratamiento diferido). El primer análisis incluyó 8.362 pacientes, 2.084 (25%) que iniciaron tratamiento entre 351 y 500células/μl y 6.278 (75%) que difirieron el tratamiento. Tras ajuste por año de tratamiento, cohorte y características clínicas y demográficas se halló un incremento del riesgo de muerte del 69% comparando el grupo de tratamiento precoz con el de tratamiento diferido (RR en el grupo de TAR diferido: 1,69; IC95%: 1,26-2,26; p<0,001). En un segundo análisis con 9.155 pacientes, 2.220 (24%) iniciaron TAR precozmente (>500células/μl) y 6.935 (76%) difirieron el TAR. Se halló un incremento del riesgo relativo de muerte en el grupo de TAR diferido del 94% (RR: 1,94; IC95%: 1,37-2,79; p<0,001)224.
Por último, en un ensayo clínico, controlado, aleatorizado, abierto, llevado a cabo en Haití, se incluyeron 816 pacientes mayores de 18años a iniciar TAR (AZT+3TC+EFV) entre 200 y 350células CD4+/μl o diferirlo hasta 2semanas después del diagnóstico clínico de sida o que su cifra de linfocitos CD4+ descendiera por debajo de 200células/μl. El estudio fue interrumpido prematuramente tras un análisis interino planeado que mostró 6muertes en el grupo de 200 a 350células/μl frente a 23 en los pacientes que difirieron el TAR. Además, en el grupo de TAR diferido se diagnosticaron el doble de casos de tuberculosis que en el grupo de inicio inmediato223.
En el ensayo clínico multinacional HIV Prevention Trials Network (HPTN) 052 se incluyeron 1.763 parejas serodiscordantes en las que el miembro infectado por VIH-1 tenía una cifra de linfocitos CD4+ entre 350 y 550células/μl. Los miembros infectados fueron aleatorizados a recibir TAR de forma inmediata o a diferirlo hasta que la cifra de CD4+ descendiera por debajo de 250células/μl o apareciera un episodio relacionado con el VIH-1. Este ensayo clínico no solo pretendía investigar si el tratamiento del miembro infectado reducía la transmisión del VIH-1 al no infectado, sino también si el TAR precoz reducía el desarrollo de episodios clínicos en el infectado (tuberculosis pulmonar, infecciones bacterianas graves o episodio definitorio de estadio4 de la OMS). Durante el estudio desarrollaron un episodio clínico 105pacientes; 40 en el brazo de TAR precoz y 65 en el brazo de TAR diferido, con una reducción de episodios clínicos cerca del 40% en el grupo de TAR precoz (HR: 0,40; IC95%: 0,40-0,88; p=0,01)152.
En resumen, existen pruebas consistentes que indican que el riesgo de progresión y/o muerte es mayor cuando se inicia el TAR con cifras de CD4<350células/μl que cuando se inicia con cifras superiores.
Incidencia de enfermedades no relacionadas con el virus de la inmunodeficiencia humana tipo 1Recientemente los estudios de cohortes han puesto énfasis en el riesgo de aparición de enfermedades que hasta el momento no se habían considerado relacionadas con la inmunosupresión. Estas enfermedades son de tipo cardiovascular (infarto de miocardio, ictus), afectación de órganos (insuficiencia renal, hepatopatía descompensada) y aparición de cánceres no definitorios de sida.
Hay que resaltar que los pacientes que empiezan TAR con >200linfocitos CD4+/μl tienen la misma probabilidad de tener una enfermedad no relacionada con el VIH-1 que una entidad definitoria de sida225. Por lo tanto, es muy relevante investigar estrategias encaminadas a disminuir la incidencia de los 2tipos de complicaciones y no solo de las relacionadas clásicamente con la inmunosupresión. El TAR podría ejercer un efecto beneficioso sobre las complicaciones no relacionadas con sida mediante el control del estado pro-inflamatorio y la activación inmune que se asocian a la replicación viral persistente.
En la cohorte D:A:D226 y en la cohorte CASCADE227 se ha comunicado que existe una disminución progresiva del riesgo de enfermedades no relacionadas con el VIH-1 a medida que aumenta la cifra de células CD4+. Los pacientes que presentan el menor riesgo de desarrollar enfermedades no definitorias de sida son los que mantienen una cifra de linfocitos CD4+>350células/μl.
En el ensayo clínico SMART228 la incidencia de enfermedades no relacionadas con sida aumentó significativamente en el grupo de pacientes que suspendieron el TAR cuando la cifra de linfocitos CD4+ era <350células/μl. En el ensayo clínico FIRST225 también se ha puesto de manifiesto una relación entre la cifra de linfocitos CD4+ y el riesgo de desarrollar enfermedades no definitorias de sida, siendo menor el riesgo en los pacientes que mantuvieron la cifra de linfocitos CD4+ por encima de 350células/μl.
Subgrupos con mayor riesgo de progresiónEn diferentes estudios se ha observado que determinados subgrupos de pacientes, como los UDVP, los coinfectados por el VHC, los varones y los de edad avanzada, tienen mayor riesgo de mortalidad y/o progresión a sida independientemente de la cifra de linfocitos CD4+ al iniciar el TAR10,229–231. Aunque no se ha establecido claramente un corte de edad a partir del cual sea más beneficioso iniciar el TAR, los estudios observacionales sugieren que el riesgo de progresión está aumentado por encima de los 55-60años de edad232.
En un estudio con pacientes con un recuento de linfocitos CD4+ entre 200 y 350células/μl se observó una relación independiente entre el porcentaje de CD4+ al inicio del TAR y la supervivencia233. Tomando como referencia a los pacientes cuya proporción de linfocitos CD4+ era ≥15%, la supervivencia fue más corta en los que tenían porcentajes <5% (HR: 4,46) y entre el 5 y el 14% (HR: 2,43). Sin embargo, en la cohorte PISCIS el porcentaje de linfocitos CD4+ no fue un factor independiente asociado al riesgo de progresión10.
Respuesta inmunológicaHay datos contradictorios sobre si existe un límite en la reconstitución inmunológica de los pacientes que reciben TAR. Los trabajos al respecto llevados a cabo en la cohorte del Hospital Johns Hopkins y en la cohorte ATHENA233,234, ambas con una mediana de seguimiento de los pacientes de hasta 7años después del inicio de TAR, sugieren que la posibilidad de alcanzar cifras normales de linfocitos CD4+ depende de la cifra de partida de estos. En ambas cohortes, solo los pacientes que iniciaron el TAR con un cifra de linfocitos CD4+>6350células/μl se aproximaron a la normalización cuantitativa de CD4+; sin embargo, los datos de la cohorte Eurosida sugieren que se puede normalizar el número de linfocitos CD4+, independientemente del nadir alcanzado, si la replicación viral persiste suprimida por debajo de 50copias/ml de forma prolongada235. En el ensayo clínico HPTN 052, comentado anteriormente, la media de linfocitos CD4+ en la visita basal del estudio fue de 449células/μl en ambos brazos de tratamiento; sin embargo, en la visita de seguimiento del mes 45 (con casi 4años de seguimiento) la media era de 742células/μl en el grupo de TAR precoz (entre 350 y 550linfocitos CD4+/μl) y 400células/μl en el grupo de TAR diferido (<250 linfocitos CD4+/μl)152.
Toxicidad del tratamiento antirretroviralUn argumento para diferir el TAR es evitar la toxicidad asociada al uso de FAR. Sin embargo, en un estudio reciente con más de 2.000pacientes seguidos durante más de 3años se observó una menor incidencia de neuropatía periférica, anemia, insuficiencia renal y lipodistrofia en los que iniciaron el TAR con cifras de linfocitos CD4+>350células/μl que en los que lo empezaron entre 200 y 350células/μl236.
Tratamiento antirretroviral para reducir la transmisión del virus de la inmunodeficiencia humana tipo 1Estos aspectos se tratan más adelante en el capítulo «Prevención de la transmisión del virus de la inmunodeficiencia humana tipo 1».
Recomendaciones- •
El inicio del TAR debe basarse en los siguientes elementos: manifestaciones clínicas, número de linfocitos CD4+, CVP y presencia de comorbilidades. (A-II)
- •
En caso de infección sintomática (eventos clínicos B o C de la clasificación de los CDC de 2003) se recomienda iniciar el TAR en todos los casos. (A-I)
- •
Si la infección es asintomática el inicio del TAR se basa en el número de linfocitos CD4+, en la CVP o en determinadas comorbilidades o características del paciente (tabla 3):
- -
Si la cifra de linfocitos CD4+ es <500células/μl se recomienda iniciar el TAR. (A-I)
- -
Si el número de linfocitos CD4+ es >500células/μl se desconoce actualmente si es mejor iniciar el TAR o diferirlo; sin embargo debe ser recomendado en los pacientes con determinadas comorbilidades (cirrosis hepática, hepatitis crónica por VHC, CVP>105copias/ml, proporción de linfocitos CD4+<14%, edad≥55años, riesgo cardiovascular elevado y trastornos neurocognitivos, entre otras). (C-III)
Tabla 3.Indicaciones de TAR en pacientes asintomáticos con infección crónica por el VIHa
aSe recomendará siempre, independientemente de la cifra de linfocitos CD4+, en la mujer embarazada, en caso de parejas serodiscordantes con alto riesgo de transmisión, en la nefropatía por VIH y en la hepatitis B que requiera tratamiento.
cAlgunos expertos recomiendan iniciar TAR es este estrato de CD4+, mientras que otros lo recomendarían solo en determinadas situaciones: cirrosis hepática, hepatitis crónica por VHC, carga viral plasmática >105copias/ml, proporción de CD4+<14%, edad>55años, riesgo cardiovascular elevado y trastornos neurocognitivos.
- -
- •
En pacientes asintomáticos e independientemente del número de linfocitos CD4+, se recomendará inicio del TAR:
- -
En parejas serodiscordantes con alto riesgo de transmisión por vía sexual para reducir el riesgo. (A-I) En ningún caso ello debe suponer la abstención de otras medidas para impedir la transmisión del VIH-1. (A-II)
- -
En mujeres gestantes, para prevenir la transmisión maternofetal. (A-I)
- -
En la nefropatía VIH. (A-II)
- -
En la hepatitisB que requiere tratamiento. (A-II)
- -
- •
A pesar de las consideraciones previas, el inicio del TAR debe valorarse siempre individualmente. Antes de tomar la decisión de iniciarlo deben confirmarse la cifra de linfocitos CD4+ y de CVP. Además, debe prepararse al paciente, ofertando las distintas opciones, adaptando el esquema terapéutico al estilo de vida, a las comorbilidades, a posibles interacciones y valorando el riesgo de mala adherencia. (A-III)
El tratamiento de elección de la infección por el VIH-1 en el momento actual consiste en una combinación de al menos 3fármacos que incluyan 2ITIAN asociados a un IP/r, un ITINN o un InInt (tabla 4). Con la mayoría de estas combinaciones se puede conseguir una CVP<50copias/ml en >70% de los casos a las 48semanas237.
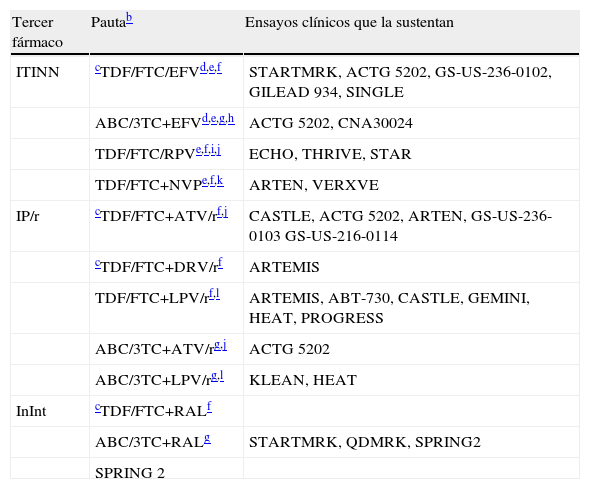
Combinaciones de tratamiento antirretroviral de inicioa
| Tercer fármaco | Pautab | Ensayos clínicos que la sustentan |
| ITINN | cTDF/FTC/EFVd,e,f | STARTMRK, ACTG 5202, GS-US-236-0102, GILEAD 934, SINGLE |
| ABC/3TC+EFVd,e,g,h | ACTG 5202, CNA30024 | |
| TDF/FTC/RPVe,f,i,j | ECHO, THRIVE, STAR | |
| TDF/FTC+NVPe,f,k | ARTEN, VERXVE | |
| IP/r | cTDF/FTC+ATV/rf,j | CASTLE, ACTG 5202, ARTEN, GS-US-236-0103 GS-US-216-0114 |
| cTDF/FTC+DRV/rf | ARTEMIS | |
| TDF/FTC+LPV/rf,l | ARTEMIS, ABT-730, CASTLE, GEMINI, HEAT, PROGRESS | |
| ABC/3TC+ATV/rg,j | ACTG 5202 | |
| ABC/3TC+LPV/rg,l | KLEAN, HEAT | |
| InInt | cTDF/FTC+RALf | |
| ABC/3TC+RALg | STARTMRK, QDMRK, SPRING2 | |
| SPRING 2 |
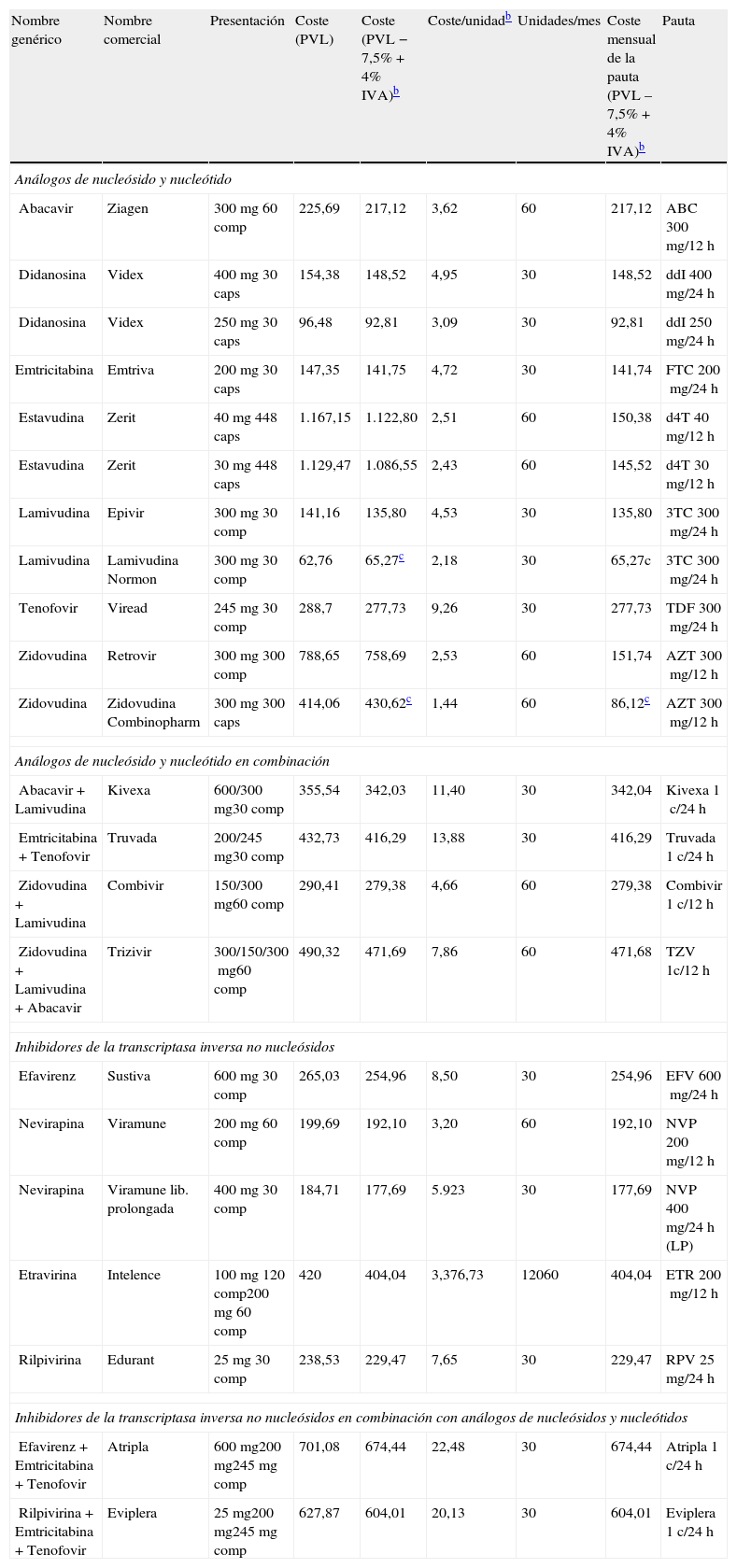
En otro apartado de estas guías se tratan aspectos de precio y de costes de los diferentes regímenes terapéuticos. Simultáneamente con las guías se publica un artículo en el que se hace un análisis formal de coste/eficacia de las pautas recomendadas como preferentes.
Ordenado por tercer fármaco. Se recomienda el uso de preparados que combinen fármacos a dosis fijas. No existe en la actualidad suficiente información que permita considerar como equivalentes terapéuticos a FTC y 3TC, por lo que el uso de uno u otro fármaco en los regímenes seleccionados depende fundamentalmente de la experiencia disponible en su uso conjunto con los otros fármacos de la combinación.
Los comentarios reflejan aspectos que se deben considerar en la elección de régimen, pero no pretenden ser una guía exhaustiva de las precauciones a tomar en el uso de los fármacos. Para mayor información se recomienda revisar el texto del documento, así como las fichas técnicas de los fármacos.
Evitar en mujeres que planean quedarse embarazadas y en pacientes con alteraciones neuropsiquiátricas no estabilizadas. Usar con precaución en pacientes que realicen tareas peligrosas si presentan síntomas de somnolencia, mareos y/o trastornos de la concentración.
Es preciso realizar previamente un estudio genotípico que descarte mutaciones de resistencia a ITINN.
Usar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal. Contraindicado si FG<30ml/min. El uso combinado de IP/r y TDF incrementa particularmente el riesgo de nefrotoxicidad.
En el estudio ACTG 5202 ABC/3TC se asoció con una mayor frecuencia de fracasos virológicos que TDF/FTC en los pacientes con CVP>100.000copias/ml.
La elección de una u otra familia tiene ciertas ventajas sobre otras: a)interacciones farmacológicas (de menos a más): InInt, ITINN, IP/r; b)mayor barrera genética: IP/r, y c)menor coste: ITINN (además, el momento idóneo del uso de los ITINN de primera generación es el tratamiento inicial, ya que en las pautas de rescate tienen menos actividad que otras familias de FAR).
Recomendación- •
Puede utilizarse la combinación de 2ITIAN+un ITINN, 2ITIAN+un IP/r, o 2ITIAN+InInt como tratamiento de inicio (los fármacos preferentes se detallan más adelante) (A-I) (tabla 4).
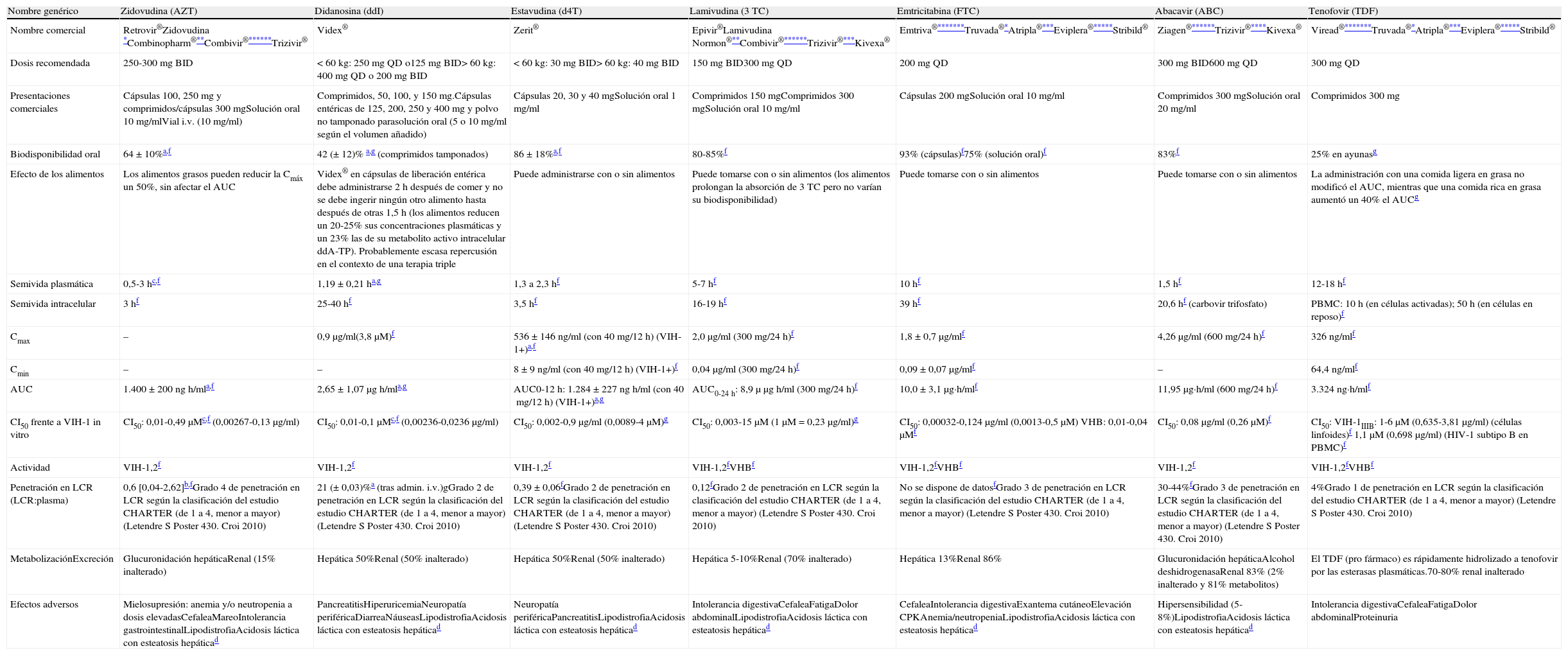
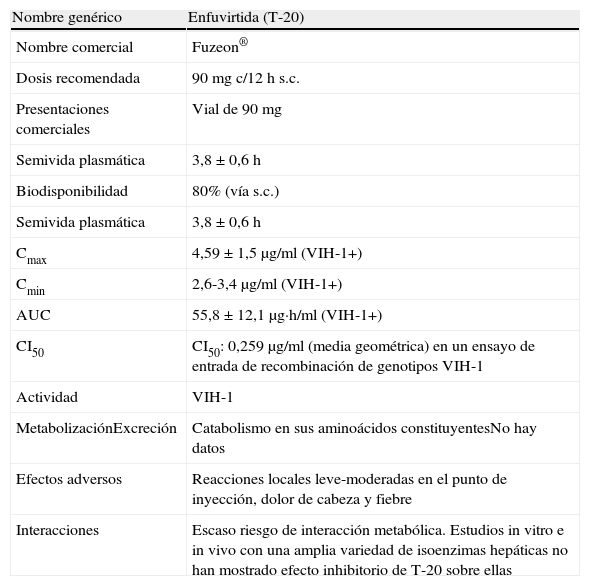
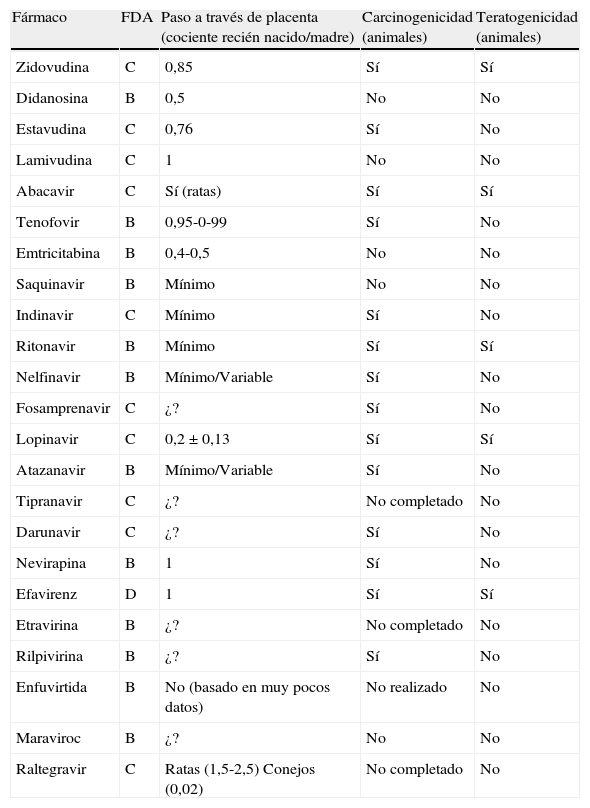
En España están comercializados 6ITIAN: zidovudina (AZT), didanosina (ddI), estavudina (d4T), lamivudina (3TC), emtricitabina (FTC) y abacavir (ABC). También se dispone de un análogo de nucleótido, tenofovir (TDF). A efectos prácticos, la abreviatura ITIAN incluye también al TDF en esta guía. Las principales características de los ITIAN se describen en la tabla 54,238,239.
Inhibidores de la transcriptasa inversa análogos de nucleósido y de nucleótido
| Nombre genérico | Zidovudina (AZT) | Didanosina (ddI) | Estavudina (d4T) | Lamivudina (3TC) | Emtricitabina (FTC) | Abacavir (ABC) | Tenofovir (TDF) |
| Nombre comercial | Retrovir®Zidovudina *Combinopharm®**Combivir®******Trizivir® | Videx® | Zerit® | Epivir®Lamivudina Normon®**Combivir®******Trizivir®***Kivexa® | Emtriva®*******Truvada®*Atripla®***Eviplera®*****Stribild® | Ziagen®******Trizivir®****Kivexa® | Viread®*******Truvada®*Atripla®***Eviplera®*****Stribild® |
| Dosis recomendada | 250-300mg BID | <60kg: 250mg QD o125mg BID>60kg: 400mg QD o 200mg BID | <60kg: 30mg BID>60kg: 40mg BID | 150mg BID300mg QD | 200mg QD | 300mg BID600mg QD | 300mg QD |
| Presentaciones comerciales | Cápsulas 100, 250mg y comprimidos/cápsulas 300mgSolución oral 10 mg/mlVial i.v. (10mg/ml) | Comprimidos, 50, 100, y 150mg.Cápsulas entéricas de 125, 200, 250 y 400mg y polvo no tamponado parasolución oral (5 o 10mg/ml según el volumen añadido) | Cápsulas 20, 30 y 40mgSolución oral 1mg/ml | Comprimidos 150mgComprimidos 300mgSolución oral 10mg/ml | Cápsulas 200mgSolución oral 10mg/ml | Comprimidos 300mgSolución oral 20mg/ml | Comprimidos 300mg |
| Biodisponibilidad oral | 64±10%a,f | 42 (±12)% a,g (comprimidos tamponados) | 86±18%a,f | 80-85%f | 93% (cápsulas)f75% (solución oral)f | 83%f | 25% en ayunasg |
| Efecto de los alimentos | Los alimentos grasos pueden reducir la Cmáx un 50%, sin afectar el AUC | Videx® en cápsulas de liberación entérica debe administrarse 2h después de comer y no se debe ingerir ningún otro alimento hasta después de otras 1,5h (los alimentos reducen un 20-25% sus concentraciones plasmáticas y un 23% las de su metabolito activo intracelular ddA-TP). Probablemente escasa repercusión en el contexto de una terapia triple | Puede administrarse con o sin alimentos | Puede tomarse con o sin alimentos (los alimentos prolongan la absorción de 3TC pero no varían su biodisponibilidad) | Puede tomarse con o sin alimentos | Puede tomarse con o sin alimentos | La administración con una comida ligera en grasa no modificó el AUC, mientras que una comida rica en grasa aumentó un 40% el AUCg |
| Semivida plasmática | 0,5-3hc,f | 1,19±0,21ha,g | 1,3 a 2,3hf | 5-7hf | 10hf | 1,5hf | 12-18hf |
| Semivida intracelular | 3hf | 25-40hf | 3,5hf | 16-19hf | 39hf | 20,6hf (carbovir trifosfato) | PBMC: 10h (en células activadas); 50h (en células en reposo)f |
| Cmax | – | 0,9μg/ml(3,8μM)f | 536±146ng/ml (con 40mg/12h) (VIH-1+)a,f | 2,0μg/ml (300mg/24h)f | 1,8±0,7μg/mlf | 4,26μg/ml (600mg/24h)f | 326ng/mlf |
| Cmin | – | – | 8±9ng/ml (con 40mg/12h) (VIH-1+)f | 0,04μg/ml (300mg/24h)f | 0,09±0,07μg/mlf | – | 64,4ng/mlf |
| AUC | 1.400±200ngh/mla,f | 2,65±1,07μgh/mla,g | AUC0-12h: 1.284±227ngh/ml (con 40mg/12h) (VIH-1+)a,g | AUC0-24h: 8,9μ μgh/ml (300mg/24h)f | 10,0±3,1μg·h/mlf | 11,95μg·h/ml (600mg/24h)f | 3.324ng·h/mlf |
| CI50 frente a VIH-1 in vitro | CI50: 0,01-0,49μMc,f (0,00267-0,13μg/ml) | CI50: 0,01-0,1μMc,f (0,00236-0,0236μg/ml) | CI50: 0,002-0,9μg/ml (0,0089-4μM)g | CI50: 0,003-15μM (1μM=0,23μg/ml)g | CI50: 0,00032-0,124μg/ml (0,0013-0,5μM) VHB: 0,01-0,04μMf | CI50: 0,08μg/ml (0,26μM)f | CI50: VIH-1IIIB: 1-6μM (0,635-3,81μg/ml) (células linfoides)f 1,1μM (0,698μg/ml) (HIV-1 subtipo B en PBMC)f |
| Actividad | VIH-1,2f | VIH-1,2f | VIH-1,2f | VIH-1,2fVHBf | VIH-1,2fVHBf | VIH-1,2f | VIH-1,2fVHBf |
| Penetración en LCR (LCR:plasma) | 0,6 [0,04-2,62]b,fGrado 4 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 21 (±0,03)%a (tras admin. i.v.)gGrado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 0,39±0,06fGrado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 0,12fGrado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | No se dispone de datosfGrado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 30-44%fGrado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 4%Grado 1 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) |
| MetabolizaciónExcreción | Glucuronidación hepáticaRenal (15% inalterado) | Hepática 50%Renal (50% inalterado) | Hepática 50%Renal (50% inalterado) | Hepática 5-10%Renal (70% inalterado) | Hepática 13%Renal 86% | Glucuronidación hepáticaAlcohol deshidrogenasaRenal 83% (2% inalterado y 81% metabolitos) | El TDF (pro fármaco) es rápidamente hidrolizado a tenofovir por las esterasas plasmáticas.70-80% renal inalterado |
| Efectos adversos | Mielosupresión: anemia y/o neutropenia a dosis elevadasCefaleaMareoIntolerancia gastrointestinalLipodistrofiaAcidosis láctica con esteatosis hepáticad | PancreatitisHiperuricemiaNeuropatía periféricaDiarreaNáuseasLipodistrofiaAcidosis láctica con esteatosis hepáticad | Neuropatía periféricaPancreatitisLipodistrofiaAcidosis láctica con esteatosis hepáticad | Intolerancia digestivaCefaleaFatigaDolor abdominalLipodistrofiaAcidosis láctica con esteatosis hepáticad | CefaleaIntolerancia digestivaExantema cutáneoElevación CPKAnemia/neutropeniaLipodistrofiaAcidosis láctica con esteatosis hepáticad | Hipersensibilidad (5-8%)LipodistrofiaAcidosis láctica con esteatosis hepáticad | Intolerancia digestivaCefaleaFatigaDolor abdominalProteinuria |
| Nombre genérico | ZidovudinaAZT | DidanosinaddI | Estavudinad4T | Lamivudina3tC | EmtricitabinaFTC | AbacavirABC | TenofovirTDFe |
| InteraccionesAsociaciones no recomendables o contraindicadasa# | EstavudinaEvitar la asociación con TPV/r por disminución de los niveles de zidovudina, a no ser que no se disponga de otros análogosInhibidores proteasa VHC: boceprevir y telaprevir (aumento del riesgo de anemia) | AlopurinolEstavudina±hidroxiureaRibavirinaEstavudinaTenofovir (si la asociación fuera imprescindible, ajustar dosis de didanosina) | ZidovudinaDidanosina±hidroxiurea | EmtricitabinaCotrimoxazol (dosis altas; a dosis profilácticas NRAD) | Lamivudina | Evitar la asociación con tipranavir/ritonavir por disminución de los niveles de abacavir, a no ser que no se disponga de otros análogos | AdefovirAtazanavir no potenciadoDidanosina (si la asociación fuera imprescindible, ajustar dosis de didanosina)Debe evitarse el uso concomitante o reciente de tenofovir con nefrotóxicos (p. ej., aminoglucósidos, anfotericina B, foscarnet, ganciclovir, pentamidina, vancomicina, cidofovir o interleucina-2). Si fuera inevitable, se debe controlar semanalmente la función renal. |
| Terapia con 3 análogos: didanosina + estavudina + abacavir, abacavir + lamivudina + tenofovir y didanosina + lamivudina + tenofovir (aumento del riesgo de fracaso virológico) |
Eviplera® (aprobado su uso en España en enero de 2013): asociación a dosis fijas de tenofovir 300mg, emtricitabina 200mg y rilpivirina 25mg.
Stribild® (todavía no comercializado en España en enero de 2013): asociación a dosis fijas de tenofovir 300mg, emtricitabina 200mg, elvitegravir 150mg, cobicistat 150mg.
No se han incluido en todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en castellano) y www.hiv-druginteractions.org (en inglés). Debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
Tenofovir se elimina mayoritariamente por vía renal y no actúa como sustrato, inductor o inhibidor del citocromo P-450, por lo que no se espera que tenga interacciones relevantes de carácter metabólico.
Por las consideraciones que se detallan en los párrafos que siguen, los miembros de este panel consideran como combinaciones de ITIAN de elección las formadas por TDF/FTC y por ABC/3TC y combinaciones alternativas a TDF+3TC, ddI+3TC, AZT/3TC, ddI+FTC, AZT+ddI. No se recomiendan las combinaciones d4T+ddI por toxicidad, TDF+ddI por toxicidad y menor eficacia240–242, AZT+d4T por antagonismo, FTC+3TC por tener similar perfil de resistencias y pocos beneficios clínicos.
Las combinaciones TDF/FTC, ABC/3TC y ddI+3TC pueden administrarse una vez al día; de ellas, las 2primeras se presentan coformuladas en un solo comprimido.
Combinaciones de inhibidores de la transcriptasa inversa análogos a nucleósido de elecciónCombinaciones con tenofovir/emtricitabina (o lamivudina)En el estudio Gilead 903 con EFV como tercer fármaco, la combinación TDF/FTC fue más eficaz y menos tóxica (toxicidad mitocondrial, neuropatía periférica, lipoatrofia, dislipidemia) que 3TC+d4T243. En el estudio Gilead 934, con EFV como tercer fármaco, la combinación TDF/FTC también resultó más eficaz, menos tóxica (anemia, dislipidemia, lipoatrofia) y se asoció con mayor ganancia de CD4 que AZT/3TC244.
Combinaciones con abacavir/lamivudinaEn el estudio CNA30024 con EFV como tercer fármaco, ABC+3TC BID tuvo una eficacia similar a AZT/3TC; sin embargo el incremento de linfocitos CD4+ fue mayor con ABC+3TC245. En el estudio CNA30021, con EFV como tercer fármaco, se demostró la no-inferioridad de ABC QD frente a ABC BID en combinación con 3TC QD246. En el estudio ABCDE, con EFV como tercer fármaco, ABC/3TC fue más eficaz y produjo menos lipoatrofia que 3TC+d4T247.
Combinaciones con tenofovir/emtricitabina frente a combinaciones con abacavir/lamivudinaEl estudio HEAT fue un ensayo clínico doble ciego con 688pacientes sin TAR previo que comparó ABC/3TC (600/300mg) QD con TDF/FTC (300/200mg) QD, ambos en combinación con LPV/r en cápsulas de 800/200mg QD248. La proporción de pacientes con CVP<50copias/ml en la semana 48 fue del 68% en el grupo ABC/3TC y del 67% en el grupo TDF/FTC, y la proporción de pacientes con CVP<50copias/ml en la semana 96 fue del 60 y del 58%, respectivamente, con lo que se pudo demostrar la no-inferioridad de ABC/3TC frente a TDF/FTC en ambos momentos. Además, la eficacia de ambas combinaciones fue similar en pacientes con CVP basal ≥100.000copias/ml o con cifra de linfocitos CD4+<50células/μl. La frecuencia de fracaso virológico a las 96semanas fue del 14% en ambos grupos y se documentó retirada prematura del estudio por efectos adversos en el 6% en ambos grupos. En la semana 96 la mediana del incremento de linfocitos CD4+ fue de 250células/μl en el grupo de ABC/3TC y de 247células/μl en el grupo de TDF/FTC.
El estudio ACTG 5202 fue un ensayo clínico factorial que comparó ABC/3TC y TDF/FTC (a doble ciego) en 1.857pacientes sin TAR previo. Los pacientes fueron aleatorizados además a recibir ATV/r o EFV (de forma abierta)249. En este estudio, la variable principal de eficacia fue el tiempo desde la aleatorización hasta el fracaso virológico, definido como: a)una CVP≥1.000copias/ml en la semana 16 o entre las semanas 16 y 24, o b)una CVP≥200copias/ml en o después de la semana 24. El comité de seguridad decidió abrir el ciego para los pacientes con CVP basal ≥100.000copias/ml tras observar diferencias en eficacia entre los 2grupos de ITIAN. Tras una mediana de seguimiento de 60semanas, entre los 797pacientes con CVP≥100.000copias/ml el tiempo hasta el fracaso virológico fue más corto en el brazo de ABC/3TC que en el brazo de TDF/FTC (HR: 2,33; IC95%: 1,46-3,72; p<0,001), con 57 fracasos virológicos (14%) en el grupo de ABC/3TC frente a 26 (7%) en el grupo de TDF/FTC. Además, el tiempo hasta el primer efecto adverso de grado 3-4 fue más corto en el grupo de ABC/3TC que en el grupo de TDF/FTC. No se documentaron diferencias significativas entre los grupos con respecto al incremento de linfocitos CD4+ en la semana 48. El estudio ACTG 5202 demostró, por tanto, que entre los pacientes con CVP basal ≥100.000copias/ml tanto el tiempo hasta el fracaso virológico como el tiempo hasta el primer efecto adverso de grado 3-4 fueron más cortos en los pacientes que recibieron ABC/3TC que en los que recibieron TDF/FTC. En los pacientes con CVP basal <100.000copias/ml no hubo diferencias en eficacia virológica entre ABC/3TC y TDF/FTC, independientemente de que se administraran con ATV/r o EFV250,251.
El estudio ASSERT es un ensayo clínico abierto en el que se compararon los perfiles de seguridad de TDF/FTC y ABC/3TC (ambos administrados con EFV) en pacientes HLA-B*5701-negativos. Los cambios de filtrado glomerular fueron similares en ambas ramas, mientras que la excreción urinaria de beta-2-microglobulina y de proteína unida al retinol (retinol-binding protein) fue significativamente mayor en la rama de TDF/FTC. En este estudio lograron una CVP<50copias/ml a las 48semanas más pacientes aleatorizados a TDF/FTC (137/193; 71%) que aleatorizados a ABC/3TC (114/192; 59%) (diferencia: 11,6%; IC95%: 2,2-21,1)252.
Se ha publicado un metaanálisis de 12 ensayos clínicos que han comparado ABC/3TC (1.769pacientes) con TDF/FTC (3.399pacientes) formando parte de pautas de TAR de inicio en combinación con IP/r. En los ensayos que utilizaron LPV/r, ATV/r y FPV/r como tercer fármaco la frecuencia de supresión virológica a las 48semanas fue significativamente inferior para ABC/3TC que para TDF/FTC (68,8% frente al 76,1%; p= 0,0015). En los pacientes con CVP<100.000copias/ml la eficacia fue menor para ABC/3TC que para TDF/FTC (70,1% frente al 80,6%; p= 0,0161), mientras que para los pacientes con CVP>100.000copias/ml la diferencia no alcanzó la significación estadística (67,5% frente al 71,5%; p=0,0523)253.
Reacción de hipersensibilidad a abacavirEn el pasado, el principal inconveniente que limitaba el uso de ABC era el desarrollo de una reacción de hipersensibilidad (RHS) que afectaba al 5-8% de los pacientes expuestos al fármaco durante las primeras 6semanas de tratamiento254. El ensayo clínico PREDICT demostró que la incidencia de RHS a ABC puede disminuir drásticamente mediante la genotipificación del HLA-B*5701114. En este ensayo clínico la prueba de genotipificación del HLA-B*5701 tuvo un valor predictivo negativo del 100% para descartar la RHS a ABC confirmada mediante una prueba cutánea. No obstante, la ficha técnica del fármaco refleja que el 3,4% de los sujetos con un estatus HLA negativo que recibieron ABC desarrollaron una RHS (no confirmada).
Seguridad cardiovascular de abacavirEn la cohorte D:A:D se observó que el uso reciente (pero no el acumulado) de ABC (definido como estar recibiéndolo en el momento actual o haberlo suspendido durante los últimos 6meses) se asoció con un incremento de 1,9veces en el riesgo de padecer un infarto de miocardio (comparado con los pacientes que no han utilizado recientemente ABC). Esta asociación persistía después del ajuste por otros factores de riesgo. El aumento del riesgo de infarto de miocardio fue más relevante desde el punto de vista clínico en los pacientes que ya tenían un riesgo cardiovascular alto según la ecuación de Framingham. En este estudio también se encontró un incremento significativo del riesgo de infarto de miocardio asociado al uso reciente de ddI, si bien la magnitud de la asociación fue menor255. En el estudio SMART también se observó una asociación entre uso de ABC y riesgo de enfermedad cardiovascular en el grupo de pacientes aleatorizados a recibir TAR de manera continua; en este estudio, el riesgo de infarto de miocardio fue más evidente en los pacientes con factores de riesgo o con alteraciones electrocardiográficas en el momento basal256. La asociación entre uso de ABC y riesgo de infarto de miocardio también se ha observado en un estudio de cohorte nacional llevado a cabo en Dinamarca en el que se incluyó a todos los pacientes que recibieron TAR desde 1995 a 2005257. En un estudio de casos y controles efectuado en el seno de la French Hospital Database on HIV se observó que la exposición reciente a ABC se asociaba con mayor riesgo de infarto agudo de miocardio pero que tal asociación desaparecía en los pacientes que no usaban cocaína o drogas por vía intravenosa258.
La FDA ha comunicado los resultados de un metaanálisis de 26ensayos clínicos aleatorizados efectuados en adultos entre 1996 y 2010 (16patrocinados por el fabricante, 5por los ACTG y 5por centros académicos). En dichos ensayos clínicos se documentaron 46infartos, de los que 24 ocurrieron en 5.028sujetos aleatorizados a pautas con ABC y 22 en 4.840pacientes aleatorizados a pautas sin ABC. No se observó una asociación significativa entre infarto agudo de miocardio y ABC (Mantel-Haenszel OR: 1,02; IC95%: 0,56-1,84)259. Del mismo modo, en una revisión de 52ensayos clínicos con ABC esponsorizados por GlaxoSmithKline se revisó la información de 14.174pacientes, de los que 9.639habían recibido ABC (7.485personas-año de seguimiento) y 4.672 no lo habían recibido (4.267personas-año de seguimiento). Ambos grupos eran comparables en cuanto a características demográficas y variables relacionadas con el VIH-1, lípidos y glucosa. La frecuencia de infarto de miocardio fue similar entre los expuestos (2,09×1.000personas-año) y los no expuestos a ABC (2,57×1.000personas-año)260. Tampoco se ha observado que el tratamiento con ABC aumente el riesgo de infarto en un metaanálisis de ensayos clínicos llevado a cabo por investigadores independientes261, ni en una revisión de los ensayos clínicos llevados a cabo en el seno de los ACTG262.
Combinaciones de inhibidores de la transcriptasa inversa análogos a nucleósidos alternativasCombinación de zidovudina/lamivudinaLa combinación de AZT/3TC ha sido bien evaluada en el estudio ACTG-384, de diseño factorial, en el que se investigaron la eficacia y la tolerabilidad de distintas combinaciones de ITIAN cuando se asocian a un tercer fármaco (ITINN o IP). Desafortunadamente el diseño factorial se frustró por el hecho de que las combinaciones de ITIAN no fueron independientes del efecto del tercer fármaco. Sin embargo, a partir de los datos de este estudio se pueden hacer 2observaciones con respecto a las combinaciones de ITIAN: 1)desde el punto de vista de la eficacia virológica es mejor empezar con AZT/3TC+EFV que con d4T+ddI+EFV o con AZT/3TC+NFV, y 2)d4T+ddI resultó más tóxica que AZT/3TC263,264.
Combinación de didanosina + lamivudinaLa combinación de ddI+3TC ha sido bien evaluada en el estudio GeSIDA 3903 con EFV como tercer fármaco265. En dicho estudio la combinación de ddI+3TC en pauta QD fue no-inferior desde el punto de vista de la eficacia a AZT/3TC. La discontinuación del tratamiento por efectos adversos fue menor en el grupo de ddI+3TC, así como la toxicidad hematológica. No hubo diferencias entre ambas pautas en cuanto a la recuperación inmunológica ni en la prevalencia de lipoatrofia y/o lipoacumulación, valoradas por criterios del investigador.
Resumen de los datos de ensayos y cohortesLa combinación TDF/FTC es más eficaz y tiene un menor riesgo de lipoatrofia que la combinación AZT/3TC.
La combinación ABC/3TC tiene una eficacia similar a AZT/3TC con menor riesgo de lipoatrofia, y ha demostrado la no-inferioridad frente a la pauta TDF/3TC cuando ambas se administran con LPV/r.
Es posible que el riesgo de fracaso virológico sea mayor con la pauta ABC/3TC que con la pauta TDF/3TC en pacientes con CVP elevada cuando ABC/3TC se administra con EFV o ATV/r.
No se han realizado grandes estudios de la combinación TDF+3TC con un IP/r como tratamiento inicial, pero sí de TDF/FTC (con ATV/r, FPV/r, LPV/r, DRV/r y SQV/r). No existe experiencia de ensayos clínicos de la combinación ABC/3TC con NVP, pero sí con ATV/r, LPV/r, FPV/r266–268.
La combinación ddI+3TC asociada a EFV no es inferior a AZT/3TC+EFV265. No hay datos sobre el riesgo de lipoatrofia medida por DEXA de la combinación ddI+3TC.
No existe experiencia de las combinaciones ddI+FTC con NVP ni con IP.
La combinación d4T+3TC ha demostrado su eficacia en múltiples estudios, pero produce más alteraciones del metabolismo de los lípidos, lipodistrofia y neuropatía periférica que la combinación TDF+3TC.
Recomendaciones sobre inhibidores de la transcriptasa inversa análogos a nucleósidosCombinaciones preferentes- •
Las combinaciones de ITIAN de elección para regímenes de inicio son TDF/FTC o ABC/3TC. (A-I) Se recomienda el uso de nucleósidos coformulados. (A-I)
- •
La combinación TDF/FTC debe utilizarse con precaución en pacientes con insuficiencia renal. (B-II)
- •
La combinación ABC/3TC debe ser empleada con precaución en pacientes con CVP elevada (>100.000copias/ml), especialmente si el tercer fármaco es un ITINN. (A-I)
Las combinaciones de 3ITIAN han demostrado eficacia virológica e inmunológica en varios estudios. Existe una coformulación con la asociación AZT/3TC/ABC que permite su administración en forma de un comprimido BID, pauta atractiva desde el punto de vista de la adherencia.
Combinación de zidovudina/lamivudina/abacavirEn el estudio ACTG A5095 se compararon la eficacia y la seguridad de 3pautas de inicio: AZT/3TC/ABC (Trizivir®), AZT/3TC+EFV y AZT/3TC/ABC+EFV. A las 48semanas la proporción de pacientes con CVP<200copias/ml (ITT) era del 74% en el brazo con 3ITIAN y del 89% en los otros 2brazos, y por este motivo el comité de seguridad recomendó que el grupo de AZT/3TC/ABC se interrumpiese, continuando el estudio de forma ciega con los pacientes en los brazos que contenían EFV269. A los 3años de seguimiento la pauta AZT/3TC/ABC+EFV no demostró ser superior a la pauta AZT/3TC+EFV270. En el estudio CNA3005, el TAR de inicio con AZT/3TC/ABC resultó equivalente a AZT/3TC+IDV para alcanzar una CVP<400copias/ml a las 48semanas; sin embargo, en los pacientes con CVP>100.000copias/ml el régimen con ABC fue inferior al de IDV para conseguir CVP<50copias/ml271.
Combinación de zidovudina/lamivudina/abacavir + tenofovirEn un estudio multicéntrico español no comparativo en el que se inició TAR a pacientes con 4ITIAN (Trizivir®+TDF), con seguimiento hasta la semana96, la proporción de pacientes con CVP<50copias/ml fue del 63% (ITT) y del 87% (OT). Los resultados fueron mejores si la CVP era <5log10 o la cifra de linfocitos CD4+ era >250células/μl272. En el estudio TIMS se comparó la combinación de 4ITIAN (Trizivir®+TDF) frente a AZT/3TC+EFV en 113pacientes. Tras 48semanas de seguimiento, la proporción de pacientes con CVP<50copias/ml fue del 67% (ITT) en el grupo de 4ITIAN y del 67% en el grupo de AZT/3TC+EFV273.
Otras pautas basadas en 3 inhibidores de la transcriptasa inversa análogos a nucleósidosEn el estudio CLASS se comparó la eficacia virológica de una pauta basada en ABC/3TC en combinación con EFV, APV/r o d4T. En la semana48 la pauta con EFV resultó superior a APV/r y a d4T (proporción de pacientes con CVP<50copias/ml: 76, 59 y 62%, respectivamente, con un análisis por ITT)274.
En el estudio ESS30009, la respuesta virológica de TDF/3TC/ABC administrado QD fue inferior a ABC/3TC+EFV. En los casos con fracaso virológico, todas las cepas de los pacientes del brazo de TDF/3TC/ABC tenían la mutación M184V y más de la mitad tenían la K65R, que puede reducir la susceptibilidad a TDF y ABC275.
En un estudio piloto con ddI+3TC+TDF, el 91% de los pacientes tuvieron un fracaso virológico (descenso de la CVP≤2log10 en la semana12). La mutación M184I/V se detectó en el 95% de los pacientes, y el 50% tenían también la K65R.
Comparando la eficacia y la tolerabilidad de d4T+ddI+ABC frente a SQV+RTV (400/400mg BID)+AZT+3TC frente a NFV+NVP+AZT+3TC, se estudiaron 180 pacientes con una mediana de linfocitos CD4+ de 161células/μl y de CVP de 5log10. A las 48semanas, la proporción de pacientes con CVP<20copias/ml fue inferior en el grupo de 3ITIAN que en los otros 2 (43, 62 y 69%, respectivamente) y los efectos secundarios fueron más frecuentes en este grupo276.
Inhibidores de la transcriptasa inversa no nucleósidosEn España hay 3ITINN comercializados (nevirapina, efavirenz, etravirina) y otro cuya comercialización ha sido aprobada el 29 de enero de 2013 (rilpivirina). Sus principales características se describen en la tabla 6238,239. Son inductores de algunas isoenzimas del citocromo P450, pudiendo interaccionar con otros fármacos. EFV se administra en pauta QD (un comprimido de 600mg/día) y NVP se puede administrar tanto en pauta QD (400md/día) como BID (200mg/12h), aunque durante los primeros 14días se administra un comprimido al día. ETR se administra en BID (200mg/12h) o QD (400mg/24h). ETR no está aprobada por la EMA para TAR de inicio. La dosis aprobada de RPV es 25mg (un comprimido) una vez al día.
Inhibidores de la transcriptasa inversa no nucleósidos
| Nombre genérico | Nevirapina | Efavirenz | Etravirina | Rilpivirina |
| Nombre comercial | Viramune® | Sustiva®*Atripla® | Intelence® | Edurant®**Eviplera® |
| Dosis recomendada | 200mg QD×14 díasseguidas de 200mg BID | 600mg QD | 200mg BID (dispersable en agua) | 25mg QD |
| Presentaciones comerciales | Comprimidos 200mgSuspensión 10mg/ml | Cápsulas 50, 100 y 200mg y comprimidos 600mg | Comprimidos 100 y 200mg | Comprimidos 25mg |
| Biodisponibilidad oral | 93±9%b,e (comprimidos)91±8%b (solución oral) | 22% (dosis única de 600mg con comida de alto contenido graso)e17% (dosis única de 600mg con comida normal)e | No se ha determinado su biodisponibilidad absoluta en humanose | No se ha determinado su biodisponibilidad absoluta en humanose |
| Efecto de los alimentos | Se puede administrar con o sin alimentos (al administrar nevirapina con un desayuno rico en grasas, el grado de absorción fue comparable al observado en condiciones de ayuno). | Evitar las comidas ricas en grasa, pues la biodisponibilidad aumenta un 50% y podría aumentar la toxicidad | Debe administrarse con alimentos. El AUC y la Cmax de la formulación usada en estudios de faseiii (F060) fueron un 51 y un 44% menores cuando se administró en ayunas, y un 25 y 38% menores tras un desayuno alto en fibra (en comparación con un desayuno estándar). Las diferencias en las concentraciones de ETR administrado tras una comida con alto contenido lipídico, tras un desayuno estándar o tras un croissant, no fueron clínicamente significativas | Debe administrarse con alimentos. En ayunas su biodisponibilidad fue un 40% menor y solamente con una bebida nutricional rica en proteínas fue un 50% menore |
| Semivida plasmática | 25-30h (tras dosis múltiples)e45h tras una dosis única. La diferencia se debe a la autoinducción de su propio metabolismo | 40-55h (tras dosis múltiples)e52-76h tras una dosis única. La diferencia se debe a la autoinducción de su propio metabolismof | 30-40he | 45he |
| Cmax | 5,74μg/ml (5,00-7,44) (200mg/12h)e | 4,07μg/mlf12,9±3,7μM (VIH-1+)b,e | – | – |
| Cmin | 3,73μg/ml (3,20-5,08) (200mg/12h)e | 5,6±3,2μM (1,77μg/ml) (VIH-1+)b,e | 296,74±377,52ng/ml (VIH-1+)b,f | 79±35 ng/ml (VIH-1+)b,f |
| AUC | 109,0μg·h/mle (96,0-143,5) (200mg/12h)e | 184±73μM·h (58,14μg·h/ml)b,e | 4.531,53±4.543,69ng·h/ml (VIH-1+)b,e | 2.235±851ng·h/ml (VIH-1+)b,f |
| CI50/90 frente a VIH-1 in vitro | CI50: 0,063μMe (0,017μg/ml) | CI90-95: 0,00014-0,0021μg/ml (0,00046-0,0068μM)e | CI50: 0,9-5,5nM (0,39-2,39ng/ml)e | CI50: 0,73nM (0,27ng/ml)e |
| Actividad | VIH-1e | VIH-1e | VIH-1e | VIH-1e |
| Penetración en LCR (LCR:plasma) | 45%±5%eGrado 4 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | 0,69% (0,26-1,19%)e. Las concentraciones de EFV en LCR exceden la CI50 para la cepa salvaje del virus: LCR/plasma: 0,005 (IQR 0,0026-0,0076; n=69). CSF/CI50: 26 (IQR 8-41), tomando CI50=0,51ng/ml (Best BM. J. Antimicrob. Chemother. (2010) doi: 10.1093/jac/dkq434)Grado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | No disponibleeGrado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | No ha sido evaluado en humanosEn investigación: (ClinicalTrials.gov NCT01562886) |
| MetabolizaciónExcreción | Hepática CYP3A4(inducción)Renal 80% (3% inalterado)Heces 10% | Hepática CYP3A4(inducción-inhibición)Renal 34% (1% inalterado)Heces 16-61% | Hepática:- Inducción de CYP3A4;- Inhibición: 2C9, 2C19- GlucuronidaciónRenal: <1,2%Excrección fundamentalmente por heces | Hepática CYP3A4e(Probablemente no modifique los niveles de otros fármacos eliminados por el citocromo P-450 a la dosis de 25mg/24h)eRenal 6% (<1% inalterado)e |
| Efectos adversos | ExantemaAumento de transaminasas y hepatitis aguda | ExantemaSíntomas neuropsiquiátricosAumento de las transaminasasTeratogenicidad en monos | Exantema | Náuseas (9%), mareos (8%), pesadillas (8%), cefalea (6%), insomnio (5%), diarrea (5%)Prolongación del intervalo QTc en el ECG a dosis supraterapéuticas. A la dosis aprobada de 25mg/día el efecto sobre el QTc no es clinicamente relevantee |
| InteraccionesAsociaciones contraindicadasa | Anticonceptivos oralesAtazanavir/ritonavirClaritromicina (para tratamiento de MAC, valorar azitromicina)EfavirenzEtravirinaFosamprenavir no potenciadoHypericum (hierba de San Juan)Inhibidores proteasa VHC: boceprevir y telaprevir (por ausencia de datos)KetoconazolItraconazolRifampicinaRilpivirinaSaquinavir(como único IP)cVoriconazol | Anticonceptivos oralesAstemizolAtazanavir (considerar ATV/r 400/200mg c/24h)Inhibidores proteasa VHC: boceprevirCarbamacepinaCisapridaClaritromicina (para tratamiento de MAC, valorar azitromicina)Deriv. ergotaminaEtravirinaFosamprenavir no potenciadoHipericum (hierba de San Juan)ItraconazolKetoconazolMidazolamNevirapinaPimozidaPosaconazolRilpivirinaSaquinavir no potenciadodTerfenadinaTriazolamVoriconazol (evitar/ajustar dosis) | CarbamacepinaClaritromicina (para tratamiento de MAC, valorar azitromicina)Dexametasona (considerar alternativa a dexametasona, especialmente en uso crónico)Diacepam (considerar alternativa a diacepam)EfavirenzFenitoínaFenobarbitalHypericum (hierba de San Juan)ItraconazolKetoconazolNevirapinaRifampicinaRilpivirinaTipranavir/ritonavirIP no potenciados | CarbamacepinaDexametasona por vía sistémica (excepto dosis única)EfavirenzEtravirinaEsomeprazol, FenitoínaFenobarbitalHierba de San Juan o hipérico (Hypericum perforatum)Inhibidores proteasa VHC: ausencia de datos con boceprevirLansoprazolNevirapinaOmeprazol, OxcarbacepinaPantoprazolRabeprazolRifabutinaRifampicinaRifapentina |
No se han incluido en todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en castellano) y www.hiv-druginteractions.org (en inglés). Debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
Saquinavir (Invirase®) puede asociarse con NVP si se combina con ritonavir (SQV/RTV 1.000/100mg c/12h), en cuyo caso datos preliminares han mostrado buenos resultados.
Saquinavir puede asociarse con la dosis habitual de EFV si se combina con ritonavir (1.000/100-200mg c/12h).
En el ensayo ARTEN se compararon la eficacia y la seguridad de NVP administrada BID o QD frente a ATV/r, ambos combinados con TDF/FTC277. La variable primaria de evaluación fue una CVP<50 copias/ml en 2visitas consecutivas antes de la semana 48. Se aleatorizaron 569pacientes y lograron el objetivo primario el 66,8% de los tratados con NVP y el 65,3% de los tratados con ATV/r (diferencia: 1,9%; IC95%: −5,9-9,8%). Ninguno de los 28pacientes con fracaso virológico del brazo de ATV/r seleccionó cepas de VIH-1 con mutaciones de resistencia, mientras que ello ocurrió en 29 de los 44pacientes con fracaso virológico del brazo de NVP. Se documentó una frecuencia similar de efectos adversos graves (9,6% con NVP y 8,8% con ATV/r), aunque las retiradas motivadas por efectos adversos resultaron más frecuentes con NVP que con ATV/r (13,6% frente al 3,6%, respectivamente). En comparación con los pacientes tratados con ATV/r, los que recibieron NVP experimentaron un mayor incremento de colesterolHDL y apolipoproteínaA1, un menor incremento en la cifra de triglicéridos y un menor incremento en el índice colesterol total/colesterolHDL.
Los estudios OCTANE comparan el tratamiento con NVP frente a LPV/r, ambos con TDF/FTC, en mujeres de varios países africanos con una cifra de linfocitos CD4+<200células/μl. La variable principal fue el tiempo desde la aleatorización hasta la muerte o fracaso virológico (definido como descenso de CVP<1log10 en la semana 12 o CVP>400copias/ml en la semana 24). En el OCTANE1 se aleatorizaron 234mujeres que habían recibido al menos una dosis de NVP en los 6meses previos para prevención de la trasmisión madre-hijo. El estudio se interrumpió a las 66semanas de seguimiento al documentarse que LPV/r era más eficaz que NVP y con menos efectos adversos278. El OCTANE2 se designó para demostrar equivalencia (definida como IC95% de la HR comprendidos entre 0,5 y 2,0) entre las 2ramas. Se aleatorizaron 500mujeres (249 a NVP y 251 a LPV/r) que no habían tomado previamente NVP. Las características basales eran similares. El seguimiento medio fue de 118semanas. Se perdieron 14mujeres de la rama de NVP y 6 de la de LPV/r. El objetivo del estudio lo consiguieron 42 (17%) mujeres de la rama de NVP y 50 (20%) del LPV/r (HR: 0,85; IC95%: 0,56-1,29), cumpliendo, pues, el criterio de equivalencia (análisis por ITT). Se analizaron por separado el fracaso virológico y la mortalidad (15% frente al 17% y 2% frente al 3%) de las ramas con NVP y LPV/r. Globalmente 93mujeres discontinuaron el tratamiento (70 [28%] en la rama de NVP y 23 [9%] en la de LPV/r). Lo hicieron por efectos adversos relacionados con el fármaco 35 (14%) de las tratadas con NVP y ninguna de las de LPV/r279.
En el ensayo Combine se comparó la eficacia de NVP frente a NFV280. Es de destacar que este ensayo no tenía suficiente poder estadístico para evaluar la equivalencia entre ambas pautas. El número de pacientes incluidos con CVP elevada (>100.000copias/ml) fue bajo y no se observaron diferencias entre NVP y NFV. En una recopilación de diversos estudios con NVP en pacientes sin TAR previo el 83% de los que tenían CVP>100.000copias/ml presentaban CVP indetectable a los 6meses281.
Pautas con efavirenzEn el estudio ACTG 5202 se compararon la eficacia, la seguridad y la tolerabilidad de ABC/3TC o TDF/FTC (doble ciego) en combinación con ATV/r o EFV (en abierto). Tal y como se comentó anteriormente, el comité de seguridad decidió abrir el ciego en los pacientes con CVP>100.000copias/ml tras documentar un tiempo más corto hasta el fracaso virológico en el brazo de ABC/3TC que en el brazo de TDF/FTC, hallazgo que fue comunicado en una primera publicación249. Posteriormente los autores han comunicado la comparación entre ATV/r (463pacientes con ABC/3TC y 465 con TDF/FTC) y EFV (465pacientes con ABC/3TC y 464 con TDF/FTC)251. Los investigadores compararon los tiempos hasta el fracaso virológico (eficacia), hasta el primer efecto adverso grado 3 o 4 (seguridad), y hasta el cambio o discontinuación de ATV/r o EFV (tolerabilidad). La eficacia virológica resultó similar en los tratados con ATV/r que en los tratados con EFV, independientemente que recibieran ABC/3TC o TDF/FTC (HR para el tiempo hasta el fracaso virológico: 1,13 [IC95%: 0,82-1,56] y 1,01 [IC95%: 0,70-1,46], respectivamente). Entre los pacientes que experimentaron fracaso virológico, la aparición de cepas con mutaciones de resistencia fue significativamente menos frecuente en los pacientes tratados con ATV/r que entre los tratados con EFV, independientemente de la pareja de ITIAN que estuvieran recibiendo. El tiempo hasta el primer evento de seguridad y el primer evento de tolerabilidad fue significativamente más largo para los pacientes con ATV/r que para los pacientes con EFV cuando la pareja de ITIAN era ABC/3TC, pero no hubo diferencias en seguridad ni tolerabilidad entre ATV/r y EFV cuando la pareja de ITIAN era TDF/FTC. Los autores concluyeron que ATV/r y EFV tienen una actividad antiviral similar cuando se combinan con ABC/3TC o TDF/FTC251. Se han comunicado resultados de un subestudio metabólico (cambios en grasa y densidad mineral ósea) que se exponen en capítulos posteriores.
El estudio ACTG 5142 es un ensayo clínico aleatorizado cuyo objetivo era valorar la eficacia y la tolerancia de 3regímenes de tratamiento: LPV/r+2ITIAN, EFV+2ITIAN y LPV/r+EFV. Se incluyeron 753pacientes con una mediana de CD4 de 182células/μl y CVP de 100.000copias/ml. Los puntos finales de valoración fueron: 1)Fracaso virológico: a)fracaso temprano: imposibilidad de reducir la CVP>1log10 o rebrote antes de la semana32, y b)fracaso tardío: imposibilidad de suprimir la CVP por debajo de 200copias/ml o rebrote después de la semana32. 2)Finalización del régimen: fracaso virológico o suspensión relacionada con la toxicidad. En la semana96 la proporción de pacientes sin fracaso virológico fue del 67, del 76 y del 73% para los grupos de LPV/r+2ITIAN, EFV+2ITIAN y LPV/r+EFV, respectivamente. En este corte, la proporción de pacientes con CVP<200copias/ml (ITT) fue del 86, del 93 y del 92% para cada rama de tratamiento (p=0,041, LPV frente a EFV), y con CVP<50copias/ml fue del 77, del 89 y del 83%, respectivamente (p=0,003; LPV frente a EFV). El incremento de linfocitos CD4+ fue mayor en los brazos que contenían LPV/r frente al brazo de EFV (p=0,01 frente a EFV+2ITIAN). Los datos de resistencias indican que en caso de fracaso virológico es más probable que aparezcan resistencias a 2clases de fármacos en el grupo de EFV+2ITIAN que en los grupos de LPV/r, mientras que la hipertrigliceridemia fue más frecuente en los pacientes con LPV/r+EFV. En resumen, el estudio ACTG 5142 demuestra que tanto la eficacia virológica como el tiempo hasta el fracaso virológico son mejores con la pauta de EFV+2ITIAN y el incremento de los linfocitos CD4+ es mayor en los brazos con LPV/r282.
En el estudio DMP-006 se ha demostrado que EFV, combinado con AZT/3TC, tiene una mayor eficacia virológica que IDV+AZT/3TC283. En el estudio ACTG 384 se demostró que la combinación de EFV+AZT/3TC es más eficaz que NFV+AZT/3TC y que NFV+ddI+d4T263. En el ensayo FOCUS se demostró que una pauta con EFV+2ITIAN era más eficaz y menos tóxica que una pauta con SQV/r (1.600/100 QD)+2ITIAN284. En el estudio BMS AI424-034 se demostró que ATV (400mg QD)+AZT/3TC fue tan eficaz como EFV+AZT/3TC; sin embargo, los resultados de este estudio son difíciles de interpretar por un error en la determinación de la CVP relacionado con el procesamiento y con el tipo de tubos empleados para el transporte de muestras285. Como ya se ha comentado previamente, el estudio ACTG-A5095 demostró que las combinaciones de fármacos de 2familias (EFV+AZT/3TC o EFV+AZT/3TC/ABC) son más eficaces que la combinación de 3ITIAN (AZT/3TC/ABC)269. En el ensayo clínico CLASS se compararon d4T, EFV y APV/r, cada uno en combinación con ABC/3TC; el brazo con EFV+ABC/3TC fue el que logró mayor eficacia virológica definida como la proporción de pacientes con CVP<50copias/ml en las semanas 24 y 48 del estudio274.
En un estudio aleatorizado, doble ciego, controlado con placebo, se comparó ETR 400mg QD con EFV 600mg QD (ambos en combinación con 2ITIAN) en pacientes sin tratamiento previo. Se aleatorizaron 79pacientes en el brazo de ETR y 78 en el de EFV, respectivamente. El objetivo primario fue evaluar la tolerabilidad neuropsiquiátrica en la semana12. Como objetivo secundario se planteaba observar la eficacia virológica de ambos tratamientos a las 48semanas. Los efectos adversos fueron significativamente más frecuentes en los pacientes que tomaban EFV que en los que tomaban ETR. A las 48semanas el 76% de los pacientes con ETR y el 74% de EFV presentaban una carga viral indetectable (CVP<50copias/ml). Estos resultados sugieren que ETR en dosificación QD podría ser una opción como primer tratamiento. Sin embargo, ETR no dispone de indicación en este tipo de pacientes. Serán necesarios más estudios con un mayor número de pacientes286.
La combinación de EFV/FTC/TDF como TAR de inicio también ha sido evaluada en los estudios GS-US-236-0102 y SINGLE. El estudio 102 es un ensayo clínico aleatorizado y doble ciego que comparó 2regímenes administrados en pastilla única: EVG/COBI/FTC/TDF y EFV/FTC/TDF. La variable primaria de evaluación fue la proporción de pacientes con CV<50copias/ml en semana48 por un análisis ITT, según el algoritmo Snapshot de la FDA. Se trataron 700pacientes y lograron el objetivo primario 305/348 (87,6%) de los tratados con EVG/COBI/FTC/TDF y 296/352 (84,1%) de los tratados con EFV/FTC/TDF (diferencia: 3,6%; IC95%: −1,6-8,8%), confirmándose la no-inferioridad de la pauta experimental frente al comparador. El tratamiento con EVG/COBI/FTC/TDF produjo significativamente más nauseas pero menos mareos, sueños anormales, insomnio y exantema. No hubo, sin embargo, diferencias significativas en lo que respecta a retirada del tratamiento por efectos adversos entre ambos brazos de tratamiento. El incremento en la creatinina sérica en la semana48 fue mayor en los tratados con EVG/COBI/FTC/TDF que en los tratados con EFV/FTC/TDF (mediana [RIQ]: 13 [5-20] μmol/l frente a 1 [−6-8] μmol/l; p<0,001), lo que probablemente se debe a cambios en la secreción tubular de creatinina y no a un efecto directo sobre el filtrado glomerular287. En el estudio SINGLE se comparó la combinación coformulada de EFV/FTC/TDF con una combinación del inhibidor de la integrasa experimental dolutegravir (DTG), ABC y 3TC (DTG+ABC/3TC). El diseño fue doble ciego, y la variable primaria de evaluación fue la proporción de pacientes con CVP<50copias/ml en la semana48 por un análisis ITT, según el algoritmo Snapshot de la FDA, con un margen de no-inferioridad del −10% y con tests pre-especificados para superioridad. Se trataron 833pacientes y lograron el objetivo primario el 88% de los tratados con DTG+ABC/3TC y el 81% de los tratados con EFV/FTC/TDF (diferencia: 7,4%; IC95%: 2,5-12,3%), confirmándose no solo la no-inferioridad sino también la superioridad de DTG+ABC/3TC sobre EFV/FTC/TDF. La proporción de fracaso virológico fue similar en ambos brazos (aproximadamente el 4%). La proporción de interrupciones del tratamiento por efectos adversos fue, sin embargo, mayor en el brazo de EFV/FTC/TDF (10%) que en el brazo de DTG+ABC/3TC (2%). La detección de mutantes resistentes tras el fracaso virológico fue muy baja en ambos brazos de tratamiento, particularmente en el brazo de DTG+ABC/3TC, en el que no se detectaron mutaciones de resistencia al inhibidor de la integrasa en ningún caso288.
Pautas con efavirenz frente a pautas con nevirapinaEl estudio 2NN fue un ensayo clínico aleatorizado y abierto en el que se comparó la eficacia y la tolerancia de EFV, NVP 400mg QD, NVP 200mg BID y EFV+NVP, combinados todos con d4T/3TC289. Se incluyeron 1.216pacientes con una mediana de linfocitos CD4+ de 190células/μl y de CVP de 4,7log10 copias/ml. Se consideró fracaso de tratamiento el fracaso virológico (descenso de CVP<1log10 en la semana12 o 2determinaciones de CVP>50copias/ml a partir de la semana24 o una CVP≥50 copias/ml en la semana48), la progresión clínica a estadioC de los CDC o muerte y el cambio de tratamiento. En la semana 48 la proporción de pacientes con fracaso de tratamiento fue del 43,6% en el grupo de NVP QD; del 43,7% en el de NVP BID; del 37,8% en el de EFV, y del 53,1% en el de NVP+EFV. La diferencia del 5,9% (IC95%: −0,9-12,8) entre las ramas con NVP BID y EFV no fue significativa y no pudo demostrarse la equivalencia dentro de los límites del 10%. No hubo diferencias entre los grupos con NVP (QD o BID). El fracaso de tratamiento fue más frecuente en la rama NVP+EFV que en la de EFV (15,3%; p=0,0003), pero no hubo diferencias significativas respecto al grupo de NVP QD (9,5%; p=0,05). Tampoco hubo diferencias entre las distintas ramas con respecto a la proporción de pacientes con CVP<50copias/ml: 70% con NVP QD, 65% con NVP BID, 70% con EFV y 62,7% con EFV+NVP (ITT). En el subgrupo con CVP elevada (>100.000copias/ml) el fracaso de tratamiento fue del 19,9% en la rama de NVP QD, del 15,8% en la de NVP BID, del 8,2% en la de NVP+EFV y del 5,9% en la de EFV (p=0,004). El incremento de linfocitos CD4+ fue similar en las 4ramas. Los efectos adversos fueron más frecuentes en el grupo de NVP+EFV y la toxicidad hepatobiliar fue más frecuente en el de NVP QD que en los otros. Se registraron 25muertes, de las que 2 se atribuyeron a NVP. Como conclusión de este estudio se puede decir que la eficacia fue similar en los 3brazos que contenían un ITINN y que la eficacia de la combinación NVP+EFV es inferior a la que contiene solamente EFV.
A la hora de valorar los resultados de este estudio debe tenerse en cuenta que en el diseño se especificó que sería clínicamente significativa una diferencia de fracaso terapéutico inferior al 10% entre las 2pautas (semana48). Los resultados indicaron, sin embargo, que no podía descartarse una diferencia mayor ya que, según el IC95%, la eficacia de EFV sobre NVP puede superar el 10%. En un análisis de sensibilidad en el que solo se incluyeron los pacientes que tomaron la medicación, la proporción de pacientes con éxito terapéutico fue significativamente mayor en el grupo de EFV que en el de NVP BID.
Resumen sobre ensayos de inhibidores de la transcriptasa inversa no nucleósidosSe ha demostrado que pautas con EFV o NVP son más eficaces que pautas con 3ITIAN. Igualmente se ha demostrado que una pauta con EFV es más eficaz que las basadas en algunos IP (IDV, NFV, SQV/r, APV/r, LPV/r). ATV/r es no-inferior a EFV. Sin embargo, no se ha demostrado en ningún ensayo clínico que NVP sea más eficaz que un IP, pero sí no inferior a ATV/r. Por último, la comparación entre los 2ITINN no ha permitido obtener conclusiones definitivas.
Consideraciones sobre la elección de un régimen con nevirapina o efavirenz- 1)
EFV ha demostrado su eficacia en pacientes con CVP>100.000copias/ml o muy inmunodeprimidos (50-100linfocitos CD4/μl)290,291.
- 2)
EFV está disponible para su uso en coformulación junto a TDF/FTC en un único comprimido de administración una vez al día.
- 3)
EFV está contraindicado en el primer trimestre de la gestación (riesgo de teratogenicidad) y debería evitarse en mujeres que quieran quedarse embarazadas o que no utilicen métodos anticonceptivos seguros. EFV puede producir mareos, trastornos de la concentración y somnolencia, por lo que se deberá informar a los pacientes y recomendarles que, en presencia de estos síntomas, eviten tareas peligrosas como conducir o usar máquinas pesadas. Asimismo debería evitarse en pacientes con antecedentes psiquiátricos graves. EFV es inductor del metabolismo de metadona y puede producir síndrome de abstinencia.
- 4)
NVP ha demostrado no-inferioridad con respecto a ATV/r, incluso con pacientes con CVP>100.000copias/ml, y ha demostrado una eficacia similar a EFV (ambos combinados con 3TC/d4T) en el estudio 2NN.
- 5)
NVP puede producir exantema cutáneo, con o sin fiebre y síntomas seudogripales. Se han descrito episodios hepáticos graves e incluso fatales durante las primeras semanas de tratamiento, por lo que NVP debe administrarse con precaución en pacientes con hepatopatía crónica y transaminasas elevadas (contraindicada si las transaminasas están por encima de 5veces el límite superior de la normalidad). Los episodios hepáticos son más frecuentes en el TAR de inicio en mujeres con un recuento de linfocitos CD4+>250células/μl (11% frente al 0,9%) o en varones con >400 linfocitos CD4+/μl (6,3% frente al 1,2%). NVP también induce el metabolismo de metadona y puede producir síndrome de abstinencia.
- 6)
Existe muy escasa experiencia en cuanto a eficacia y tolerabilidad de la combinación ABC/3TC+NVP. Además, tanto ABC como NVP pueden presentar reacción de hipersensibilidad.
El estudio ECHO es un ensayo clínico de faseiii, doble ciego, que incluyó a pacientes adultos sin TAR previo y sin mutaciones de resistencia en el estudio genotípico basal. Los participantes fueron aleatorizados en proporción 1:1 (estratificando según CV) a recibir RPV: 25mg QD, con comida (n=346) o EFV: 600mg QD (n=344) en ayunas y por la noche. Como pareja de análogos se administró TDF/FTC coformulado. El estudio se diseñó para demostrar la no-inferioridad de RPV frente a EFV, tomando como variable primaria de eficacia la respuesta confirmada definida como CVP<50copias/ml en la semana48 mediante un análisis por ITT según el algoritmo TLOVR. El margen inferior del intervalo de confianza de la diferencia fue de −12%292. El estudio THRIVE, de diseño similar al ECHO, se diferencia únicamente de este en que los investigadores podían seleccionar 3diferentes parejas de ITIAN. En este estudio se incluyeron 340pacientes en el brazo de RPV (204TDF/FTC, 101AZT/3TC y 35ABC/3TC) y otros 340pacientes en el brazo de EFV (202TDF/FTC, 103AZT/3TC y 33ABC/3TC); se permitieron los cambios entre parejas de ITIAN solo por razones de intolerancia y siempre guiados por estudios de resistencia genotípicos293. En el estudio ECHO lograron una respuesta confirmada 287 (83%) pacientes del brazo de RPV y 285 (83%) del brazo de EFV292; en el subgrupo de pacientes con CVP basal <100.000copias/ml los porcentajes de respuesta fueron del 90% en el brazo de RPV y del 83% en el de EFV; en el subgrupo de CVP basal comprendida entre 100.000 y 500.000copias/ml: 79% con RPV y 83% con EFV, y en el de CVP basal >500.000copias: 62% con RPV y 81% con EFV. En el estudio THRIVE lograron una respuesta confirmada 291 (86%) de los pacientes tratados con RPV y 276 (82%) de los tratados con EFV293, poniéndose de relieve en ambos estudios la no-inferioridad de RPV frente a EFV. En el subgrupo de pacientes con CVP basal <100.000copias/ml los porcentajes de respuesta fueron del 91% en el brazo de RPV y del 84% en el de EFV; en el subgrupo de CVP basal comprendida entre 100.000 y 500.000copias/ml: 80% con RPV y 82% con EFV, y en el de CVP basal >500.000copias: 77% con RPV y 69% con EFV. No hubo diferencias significativas en el incremento de la cifra de linfocitos CD4+ entre RPV y EFV en ninguno de ambos estudios.
En el estudio ECHO, la interrupción del tratamiento por efectos adversos fue del 2% para los tratados con RPV y del 8% para los tratados con EFV292. En el estudio THRIVE, la interrupción del tratamiento por efectos adversos fue del 4% para los tratados con RPV y del 7% para los tratados con EFV293. En ambos estudios se documentó una frecuencia significativamente menor de efectos adversos de grado 2-4 con RPV que con EFV, incluyendo entre ellos exantema, mareos y alteraciones del sueño o pesadillas. Los incrementos en las cifras de triglicéridos, de colesterol total, de colesterolLDL y de colesterolHDL fueron menores en los tratados con RPV que en los tratados con EFV; sin embargo, no se observaron diferencias en los cambios del cociente colesterol total/colesterolHDL entre ambos brazos de tratamiento. Hubo un incremento pequeño, pero significativo, de la creatinina sérica en el brazo de RPV, lo que probablemente se debe a cambios en la secreción tubular de creatinina y no a un efecto directo sobre el filtrado glomerular. Por otra parte, se observó una mayor frecuencia de respuesta anormal de cortisol a la hormona adrenocorticotropa en los tratados con RPV en comparación con los tratados con EFV, que no se consideró clínicamente relevante.
En el estudio ECHO la frecuencia de fracaso virológico (definido como la ausencia de CV<50copias/ml antes de la semana 48 o el rebrote de la viremia al menos en 2ocasiones consecutivas tras haber logrado una CVP<50copias/ml) fue del 11% para los tratados con RPV y del 4% para los tratados con EFV en el análisis ITT-TLOVR292. En el estudio THRIVE, la frecuencia de fracaso virológico fue del 7% para los tratados con RPV y del 5% para los tratados con EFV293. El análisis conjunto de los datos de los estudios ECHO y THRIVE (planificado de antemano por los investigadores) confirmó los datos de eficacia y seguridad de los 2estudios individuales. Los porcentajes de respuesta confirmada en los pacientes con CVP basal <100.000copias/ml fueron: 90% en el brazo de RPV y 84% en el de EFV (diferencia: 6,6; IC95%: 1,6-11,5); en los pacientes con CVP basal entre 100.000 y 500.000copias/ml: 80% con RPV y 83% con EFV (diferencia: −3,1; IC95%: −9,8-3,7) y en los que tenían una CVP>500.000copias/ml: 70% con RPV y 76% con EFV (diferencia: −6,0; IC95%: −20,4-8,3). También mostró que la frecuencia de fracaso virológico era mayor en los tratados con RPV que en los tratados con EFV, principalmente en los pacientes con CVP alta (>100.000copias/ml) o cumplimiento subóptimo del TAR294. Así, se documentó fracaso virológico en 72/686 (9%) de los pacientes tratados con RPV y en 39/682 (5%) de los tratados con EFV. En los pacientes con CV basal ≤100.000copias/ml la proporción de pacientes con fracaso virológico fue similar en los tratados con RPV (19/368; 4%) que en los tratados con EFV (16/330; 3%). Sin embargo, en pacientes con CV>100.000copias/ml hubo más fracasos virológicos en los tratados con RPV (53/318; 17%) que en los tratados con EFV (23/352; 7%)294. El análisis conjunto de los estudios de resistencia de los 2ensayos clínicos reveló que tras el fracaso virológico ocurrió la aparición de mutaciones de resistencia a ITIAN (especialmente M184I y M184V) con más frecuencia en los tratados con RPV (42/62;68%) que en los tratados con EFV (9/28;32%)295. La aparición de mutaciones de resistencia a ITINN tras el fracaso resultó similar en los tratados con RPV (39/62; 63%) que en los tratados con EFV (15/28; 54%). Tras el fracaso con EFV la mutación más frecuente fue la K103N, mientras que tras el fracaso con RPV las mutaciones más frecuentes fueron la E138K (que confiere resistencia a ETR) y la K101E (que afecta a todos los ITINN)295.
En el ensayo clínico STaR se compararon 2pautas de TAR basadas en regímenes de pastilla única en pacientes infectados por el VIH sin TAR previo: RPV/TDF/FTC (n=394) frente a EFV/TDF/FTC (n=392). La aleatorización se estratificó por un valor de CVP basal mayor o menor de 100.000copias/ml. La variable primaria de eficacia fue la proporción de pacientes con CVP<50copias/ml en la semana48 según el algoritmo Snapshot de la FDA, con un margen inferior de no-inferioridad del 12%. La proporción de pacientes que lograron la variable primaria de eficacia fue la siguiente: a)en la totalidad de los pacientes: 86% con RPV y 82% con EFV; diferencia: 4,1% (IC95%: −1,1-9,2); b)grupo de pacientes con CVP≤100.000copias/ml: 89% con RPV y 82% con EFV; diferencia: 7,2% (IC95%: 1,1-13,4); c)grupo de pacientes con CVP>100.000copias/ml: 80% con RPV y 82% con EFV; diferencia: −1,8% (IC95%: −11,1-7,5). Por tanto, se demostró la no-inferioridad de RPV/TDF/FTC frente a EFV/TDF/FTC en la población total y en los pacientes con CVP>100.000copias/ml y la superioridad de RPV/TDF/FTC frente a EFV/TDF/FTC en los pacientes con CV≤100.000copias/ml. En un análisis no contemplado en el diseño inicial se observó que la respuesta en pacientes con CVP comprendida entre 100.000 y 500.000copias/ml fue del 83% con RPV y del 82% con EFV, mientras que en aquellos con CVP>500.000copias/ml fue del 72% con RPV y del 80% con EFV. Se documentaron los siguientes fracasos virológicos: a)en la totalidad de los pacientes: 8% con RPV y 6% con EFV; b)grupo de pacientes con CVP≤ 100.000copias/ml: 5% con RPV y 3% con EFV; c)grupo de pacientes con CVP entre 100.000 y 500.000copias/ml: 10% con RPV y 9% con EFV; d)pacientes con CVP>500.000copias/ml: 25% con RPV y 16% con EFV. Se detectaron mutaciones de resistencia a FAR en el 4% de los pacientes tratados con RPV y en el 1% de los tratados con EFV. Se detectaron mutaciones de resistencia a ITINN en el 4% de los pacientes tratados con RPV y en el 1% de los tratados con EFV. Se detectaron mutaciones a ITIAN en el 4% de los pacientes tratados con RPV y en el 0,3% de los tratados con EFV. Los pacientes tratados con RPV en relación con los tratados con EFV tuvieron una menor frecuencia de retirada del tratamiento por efectos adversos (2,5 y 8,7%, respectivamente; p<0,001), una menor incidencia de efectos adversos del SNC (30 y 51%, respectivamente; p<0,001) y una menor frecuencia de efectos adversos psiquiátricos (16 y 38%, respectivamente; p<0,001)296.
Recomendaciones sobre inhibidores de la transcriptasa inversa no nucleósidos- •
En general se recomienda EFV frente a NVP, por los resultados de los diferentes estudios. (C-III)
- •
EFV está contraindicado durante el primer trimestre de la gestación. Se recomienda considerar otras opciones en mujeres que no utilicen métodos anticonceptivos eficaces. Asimismo, se debe evitar en pacientes que realicen tareas peligrosas si presentan síntomas de somnolencia, mareos y/o trastornos de la concentración. (B-III)
- •
Está contraindicado el uso de NVP en mujeres con cifras de linfocitos CD4+>250células/μl y en varones con cifras de linfocitos CD4+>400células/μl. (A-II)
- •
En pacientes con CV>100.000copias/ml el riesgo de fracaso virológico es mayor en los pacientes tratados con RPV que en los tratados con EFV. (A-I)
En España se han comercializado 9IP: saquinavir (SQV), indinavir (IDV), ritonavir (RTV), nelfinavir (NFV), amprenavir (APV) —que se ha sustituido por su profármaco fosamprenavir (FPV)—, lopinavir (LPV), atazanavir (ATV), tipranavir (TPV) y darunavir (DRV). TPV/r está aprobado por la EMA solamente en pacientes pretratados. Las principales características de los IP se muestran en la tabla 7a,b. Los IP son inductores e inhibidores del citocromo P450 y frecuentemente pueden originar interacciones farmacológicas. La elección final del IP se basará en datos de eficacia, tolerabilidad, farmacológicos, posología y farmacocinética. Las características farmacodinámicas de los IP se potencian si se administran con una dosis mínima de RTV. LPV se administra en comprimidos coformulados con el RTV.
Inhibidores de la proteasa: indinavir, ritonavir, saquinavir, nelfinavir y fosamprenavir
| Nombre genérico | Indinavir | Ritonavir | Saquinavir | Nelfinavir | Fosamprenavir |
| Nombre comercial | Crixivan® | Norvir® | Invirase® | Viracept® | Telzir® |
| DosisRecomendación | 800mg TIDIDV/r 800/100 BIDSi no potenciado, debe tomarse con el estómago vacío (restricción dietética)Ingesta abundante de líquidos no carbónicos | Como potenciador de otros IP: 100 o 200mg con cada dosis de IPComo IP (600mg BID) se desaconseja | SQV/r 1.000/100 BIDTomar con comida grasa | 750mg TID o1.250mg BIDTomar con comida grasa | FPV/r 700/100mg c/12hCon o sin alimentos |
| Presentación comercial | Cápsulas 200 y 400mg | Solución oral 80 mg/mlCápsulas 100mgComprimidos 100mg | Comprimidos 200 y 500mg | Comprimidos 500mgPolvo (1cuch 1g=50mg de NFV) | Comprimidos 700mgSuspensión oral 50mg/ml |
| Biodisponibilidad oral | 65% (IC90%, 58-72%), tras dosis única de 800mge | No se ha determinado su biodisponibilidad absoluta en humanose | 4% (1-9%)d en ausencia de RTVeNo se ha determinado su biodisponibilidad absoluta de SQV potenciado con RTV en humanose | No se ha determinado su biodisponibilidad absoluta en humanose | No se ha determinado su biodisponibilidad absoluta en humanose |
| Efecto de los alimentos | No potenciado y en presencia de alimentos grasos el AUC se reduce un 77%, por lo que debe administrarse en ayunas o con una comida ligera de bajo contenido graso. Cuando se administra potenciado con ritonavir puede tomarse junto con las comidasf | En presencia de alimentos se reduce el AUC entre un 21 y un 23%fEl sabor amargo de Norvir solución puede enmascararse si se mezcla con un batido de chocolatee | En voluntarios sanos, el AUC aumentó más de 6 veces al tomarlo con una comida grasa, en comparación con su ingesta en ayunas. Se recomienda tomar saquinavir dentro de las 2h después de comerf | En presencia de alimentos aumenta el AUC entre 2 y 5 veces (aumento mayor cuanto mayor contenido calórico), por lo que nelfinavir debe administrarse con comidaf | Comprimidos: la administración con alimentos no modificó significativamente el AUC, por lo que puede ingerirse con o sin alimentosSolución oral: en presencia de alimentos el AUC se redujo un 28% en comparación con su ingesta en ayunas. En adultos, se recomienda tomar la solución en ayunas y en los niños con alimentos porque mejora la tolerabilidadf |
| Semivida plasmática | 1,8 ± 0,4hb,e | 5h (100mg/24h)e5h (100mg/12h)e4h (200mg/24h)e8h (200mg/12h)e | 7he | 3,5-5he | 7,7h (FPV)e15-23h (FPV/r)e |
| Cmax | 11.144 (IC90%: 9.19-13.512) nM (=7,9μg/ml) con IDV 800mg/8he19.001 (IC90%: 17.538-20.588) nM (=13,5μg/ml) con IDV/r 800/100mg/12he | En VIH-1+:0,84±0,39μg/mlb (100mg/24h)e0,89μg/ml (100mg/12h)3,4±1,3μg/mlb (200mg/24h)g4,5 ± 1,3μg/mlb (200mg/12h)e | En VIH-1+:5.208 ng/ml (1.536-14.369)d (SQV/r 1000/100mg/12h con comida rica en grasa)e | 3,0 ± 1,6μg/ml (750mg/8h)e4,0 ± 0,8μg/ml (1.250 mg/12h)e | FPV/r 700/100mg c/12h: 6,08μg/ml (IC95%: 5,38-6,86)eFPV/r 1.400/100mg c/24h: 7,93μg/ml (IC95%: 7,25-8,68)fFPV/r 1.400/200mg c/24h: 7,24μg/ml (IC95%: 6,32-8,28)f |
| Cmin | 211 (IC90%: 163-274) nM (0,15μg/ml), con IDV 800mg/8hg2.274 (IC90%: 1.701-3.042) nM (=1,6μg/ml, con IDV/r 800/100mg/12he | En VIH-1+:0,08 ± 0,04μg/mlb (100mg/24h)e0,22μg/ml (100mg/12h)0,16 ± 0,10μg/mlb (200mg/24h)e0,6 ± 0,2μg/mlb (200mg/12h)eLas Cmin descienden con el tiempo, posiblemente debido a la inducción enzimática, pero parecen estabilizarse al final de 2semanase | En VIH-1+:1.179ng/ml (334-5.176)d (SQV/r 1000/100mg/12h con comida rica en grasa)e | 1,4 ± 0,6μg/ml con la dosis de la mañana y 1,0 ± 0,5μg/ml con la de la tarde (750mg/8h)e2,2 ± 1,3μg/ml con la dosis de la mañana y 0,7 ± 0,4μg/ml con la de la tardee (1.250mg/12h)e | FPV/r 700/100mg c/12h: 2,12μg/ml (IC95%: 1,77-2,54)eFPV/r 1.400/100mg c/24h: 0,86μg/ml (IC95%: 0,74-1,01)fFPV/r 1.400/200mg c/24h: 1,45μg/ml (IC95%: 1,16-1,81)f |
| AUCc | AUC0-8h 27.813nM·h IC90%: 22.185-34.869) (=19.803ng·h/ml)(con IDV 800mg/8he116.067nM·h (IC90%: 101.680-132.490) (=82.639ng·h/ml)(con IDV/r 800/100mg/12he | En VIH-1+: AUC24h 6,6 ± 2,4μg·h/mlb (100mg/24h)eAUC12h 6,2μg·h/mlb (100mg/12h)eAUC24h 20,0 ± 5,6μg·h/mlb (200mg/24h)eAUC12h 21,92 ± 6,48μg·h/mlb (200mg/12h)e | En VIH-1+: AUC0-12h 34.926ng·h/ml (11.826-105.992)d (SQV/r 1.000/100mg/12h con comida rica en grasa)e | AUC24h 43,6 ± 17,8μg·h/ml (750mg/8h)eAUC24h 52,8 ± 15,7μg·h/ml (1.250mg/12h)e | FPV/r 700/100mg c/12h: AUC0-12h 39,6 (IC95%: 34,5-45,3) μg·h/mleFPV/r 1.400/100mg c/24h: AUC0-24h 66,4μg·h/ml (IC95%: 61,1-72,1)fFPV/r 1.400/200mg c/24h: AUC0-24h 69,4μg·h/ml (IC95%: 59,7-80,8)f |
| CI50/90/95 frente a VIH-1 in vitro | CI95: 50-100nMe (=35-71ng/ml) | – | CI50: 1-10nMe (=0,76-7,6ng/ml)CI50 ajustada al suero (50% suero humano) 25-250nM (=19,2-192ng/ml)CI90: 5-50nMe(=3,8-38ng/ml) | CI95: 58nM (7-111nM)d,e (=38,5ng/ml) | CI50 entre 0,012 y 0,08μM en células con infección aguda y 0,41μM en células con infección crónicae |
| Actividad | VIH-1.2e | VIH-1,2, aunque en la actualidad se utiliza como potenciador farmacocinéticoe | VIH-1,2e | VIH-1,2e | VIH-1,2e,f |
| Penetración en LCR (LCR:plasma) | No hay datoseIDV grado 3 e IDV/r grado 4 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | Grado 1 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | Insignificante con Invirase 600mg/8heSQV ± RTV: grado 1 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | Grado 1 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | Penetración insignificanteeGrado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) |
| Metabolización | CYP3A4 | CYP3A4 | CYP3A4 | CYP3A4 | CYP3A4 |
| Efectos adversos | NefrolitiasisIntolerancia gastrointestinalHiperbilirrubinemiaHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos | (En dosis reducidas la prevalencia de los efectos adversos es muy baja)Intolerancia gastrointestinal(vómitos, diarrea)Parestesias oralesHepatitisHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos | Intolerancia gastrointestinal (diarrea)Cefalea↑ transaminasasHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos | DiarreaHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos | Intolerancia gastrointestinal (diarrea)Exantema autolimitado (usar con precaución en pacientes con alergia conocida a las sulfamidas)CefaleaHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos |
| InteraccionesAsociaciones contraindicadasa | Alfuzosina (IDV/r)Alprazolam (IDV ± RTV)Amiodarona (IDV ± RTV)AstemizolAtazanavirCisapridaColchicina (si insuficiencia renal o hepática)Cloracepato (IDV/r)Clozapina (IDV/r)Deriv. ergotamina (IDV ± RTV)Dextropropoxifeno (IDV/r)Diacepam (IDV/r)Encainida (IDV/r)Estazolam (IDV/r)Flecainida (IDV/r)Fluracepam (IDV/r)HalofantrinaHypericum (hierba de San Juan)Inhibidores proteasa VHC: boceprevir y telaprevir (por ausencia de datos)Lovastatina (IDV ± RTV)LumefantrinaMeperidina (IDV/r)Midazolam oral(IDV ± RTV)Pimozida (IDV ± RTV)Piroxicam (IDV/r)Propafenona (IDV/r)Quinidina (IDV/r)Rifampicina (IDV ± RTV)SalmeterolSildenafilo (hipertensión pulmonar)Simvastatina (IDV ± RTV)TerfenadinaTriazolam(IDV ± RTV) | AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolCisapridaCloracepatoClozapinaColchicina (si insuficiencia renal o hepática)DextropropoxifenoDiacepamDeriv. ergotaminaDisulfiramEncainidaEstazolam, ÉxtasisFlecainidaFluracepamFluticasonaHalofantrinaHypericum (hierba de San Juan)Inhibidores de la proteasa VHC: solo está permitido el uso de ATV/r con telaprevirLovastatinaLumefantrinaMeperidinaMetanfetaminaMidazolam oralPimozidaPiroxicamPropafenonaQuinidinaSalmeterolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTriazolamVoriconazolZolpidem | Ajo, suplementos (utilizar SQV/r)AlfentaniloAlfuzosinaAmiodaronaAmitriptilinaAstemizolAtazanavir/ritonavirCarbamacepinaCisapridaClaritromicinaClozapinaColchicina (si insuficiencia renal o hepática)DapsonaDexametasonaDeriv. ergotaminaDisopiramidaEfavirenzEritromicinaFenitoínaFenobarbitalFenotiazinasFentaniloFlecainidaFluticasonaHalofantrinaHaloperidolHypericum (hierba de San Juan)ImipraminaInhibidores de la proteasa VHC: boceprevir y telaprevir (por ausencia de datos)Lidocaína(sistémica)Lopinavir/ritonavirLovastatinaLumefantrinaMetadonaMidazolam oralNevirapinaOmeprazol y afinesPentamidinaPimozidaPropafenonaQuinidinaRifampicinaSalmeterolSildenafiloSimvastatinaSotalolTadalafiloTerfenadinaTioridazinaTrazodonaTriazolamVardenafiloVoriconazolZiprasidonaMedicamentos que prolonguen el intervalo QT o PR | AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolBupropiónCarbamazepinaCisapridaColchicina (si insuficiencia renal o hepática)Deriv. ergotaminaFenobarbitalHalofantrinaHypericum (hierba de San Juan)Inhibidores de la proteasa VHC: boceprevir y telaprevir (por ausencia de datos)LovastatinaLumefantrinaMidazolam oralOmeprazol y afinesPimozidaQuinidinaRifampicinaSalmeterolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTriazolam | AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolCisapridaColchicina (si insuficiencia renal o hepática)Deriv. ergotaminaFlecainida (FPV/r)HalofantrinaHypericum (hierba de San Juan)Inhibidores de la proteasa VHC: telaprevir contraindicado y evitar boceprevir por ausencia de datos.LumefantrinaLopinavir/rLovastatinaMidazolam oralPimozidaPropafenona (FPV/r)QuinidinaRifampicinaSalmeterolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTriazolamVoriconazol (FPV/r) |
No se han incluido en todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en castellano) y www.hiv-druginteractions.org (en inglés). Debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
Información procedente de la ficha técnica europea. EPARS: European Public Assessment Reports [consultado 14 Nov 2012]. Disponble en: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/medicines/medicines_landing_page.jspμrl=menus/medicines/medicines.jsp∣=
Inhibidores de la proteasa: lopinavir/r, atazanavir, darunavir, tipranavir
| Nombre genérico | Lopinavir | Atazanavir | Darunavir | Tipranavir |
| Nombre comercial | Kaletra® | Reyataz® | Prezista® | Aptivus® |
| DosisRecomendación | 400/100mg c/12h800/200mg/24hTomar con o sin comida | 300/100mg c/24h o 400mg c/24hNota: En un estudio se observó que tan solo el 38% de los pacientes tratados con ATV no potenciado alcanzaron una Cmin>150ng/ml.Tomar con comida | 800/100mg/24he600/100mg c/12h (en pacientes pretratados)Tomar con comida | TPV/r 500/200mg c/12hTomar con comida |
| Presentación comercial | Comprimidos 200/50mgComprimidos 100/25mgSolución oral 80/20mg/ml | Cápsulas 150, 200 y 300mg | Comprimidos 75, 150, 400 y 600mg | Cápsulas 250mg |
| Biodisponibilidad oral | No se ha determinado su biodisponibilidad absoluta en humanosf | 68% (57-80%)h | 37% (DRV solo, dosis única 600mg)82% (DRV/r 600/100mg/12h)f | No se ha determinado su biodisponibilidad absoluta en humanosg |
| Efecto de los alimentos | Solución: debe administrarse con alimentosf. En comparación con la administración en ayunas, la administración con alimentos de contenido moderado en grasa aumentó un 80% el AUC y los alimentos ricos en grasa la aumentaron un 130%gComprimidos: pueden administrarse con o sin alimentosf | ATV: 400mg/24h: en comparación con la administración en ayunas, la administración con alimentos de contenido ligero en grasa aumentó un 70% el AUC y los alimentos ricos en grasa la aumentaron un 35%gATV/r: 300/100mg/24h: en comparación con la administración en ayunas, la administración con alimentos de contenido ligero en grasa aumentó un 33% el AUC y los alimentos ricos en grasa prácticamente no la modificarong | Se recomienda administrarlo con alimentos. Los alimentos aumentaron un 30% el AUC, sin que influyera el tipo de alimentog | Los alimentos mejoran la tolerabilidad, por lo que TPV/r debe administrarse con alimentosf |
| Semivida plasmática | 5-6hf | 6,5h (ATV 400mg/24h) (VIH-1+)g12h (ATV/r 300/100mg/24h) (VIH-1+)f | 15h (DRV/r 600/100mg/12h)f | 5,5h en mujeres y 6h en hombres (TPV/r 500/200mg/12h)f |
| Cmax | 12,3±5,4μg/ml (400/100mg/12h)b,f9,8±3,7μg/ml (400/100mg/12h) (VIH-1+)b,f11,8±3,7μg/ml (800/200mg/24h) (VIH-1+)b,g | En VIH-1+: 4.466ng/ml (ATV/r 300/100mg/24h)f3.152±2.231ng/ml (ATV 400mg/24h)g | – | En VIH-1+: 94,8±22,8μM en mujeres (TPV/r 500/200mg/12h)f77,6 ± 16,6μM en hombres (TPV/r 500/200mg/12h)f |
| Cmin | 8,1±5,7μg/ml (400/100mg/12h)b,f7,1 ± 2,9μg/ml (400/100mg/12h) (VIH-1+)b,g3,2 ± 2,1μg/ml (800/200mg/24h) (VIH-1+)b,g | En VIH-1+: 654ng/ml (ATV/r 300/100mg/24h)f273 ± 298bng/ml (ATV 400mg/24h)g | En VIH-1+: 2.282 ± 1.168bng/ml (DRV/r 800/100mg/24h)g3.578 ± 1.151b ng/ml (DRV/r 600/100mg/12h)g | En VIH-1+: 41,6 ± 24,3μM en mujeres (TPV/r 500/200mg/12h)f35,6 ± 16,7μM en hombres (TPV/r 500/200mg/12h)f |
| AUC | AUC0-12h: 113,2 ± 60,5μg·h/ml (400/100mg/12h)b,fAUC0-12h: 92,6 ± 36,7μg·h/ml (400/100mg/12h) (VIH-1+)b,gAUC0-24h: 154,1 ± 61,4μg·h/ml (800/200mg/24h) (VIH-1+)b,g | En VIH-1+: AUC0-24h: 44.185ng·h/ml (ATV/r 300/100mg/24h)fAUC0-24h: 22.262 ± 20.159b ng·h/ml (ATV 400mg/24h)g | En VIH-1+: 93.026 ± 27.050b ng·h/ml (DRV/r 800/100mg/24h)g124.698 ± 32.286b ng·h/ml (DRV/r 600/100mg/12h)g | En VIH-1+: AUC0-12h: 851 ± 309μM·h en mujeres y 710 ± 207μM·h en hombres (TPV/r 500/200mg/12h)f |
| CI50/90 frente a VIH-1 (in vitro) | CI50 frente a VIH-1IIIB en células MT4 fue 17nM (10,6μg/ml) en ausencia de suero humano y 102nM (63,75μg/ml) en presencia de suerofCI50 frente a varios aislados clínicos de VIH-1 en ausencia de suero 6,5nM (4,6μg/ml)f | CI50: 0,002-0,004μg/ml (2,6-5,3nM)g | CI50: 1,2-8,5nM (0,7-5,0ng/ml)f | CI50: 0,03-0,07μM (18-42ng/ml)fCI90: 0,07-0,18μM (42-108ng/ml)(La actividad antiviral de tipranavir disminuye 3,75 veces de media en presencia de suero humano)f |
| Actividad frente a VIH-1 | VIH-1,2f | VIH-1,2f | VIH-1,2f | VIH-1,2f |
| Penetración en LCR (LCR:plasma) | Grado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | LCR/plasma: 0,0021-0,0226d,gATV ± RTV: grado 2 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010)ATV/r: LCR: 10,3 (<5-21ng/ml)c en comparación con 1.278 (525-2.265ng/ml)cATV: LCR 7,9 (6,6-22 ng/ml)c con ATV, en comparación con 523 (283-1.344ng/ml). Concentraciones en LCR bajas (<1%) y muy variables (Best BM. AIDS 2009;23(1):83-87) | Grado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | No hay estudiosfGrado 1 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) |
| Metabolización | CYP3A4 | CYP3A4 | CYP3A4 | CYP3A4 |
| Efectos adversos | Intolerancia gastrointestinal(vómitos, diarrea)CefaleaAsteniaHiperglucemiaDislipidemiaLipodistrofiaPosible aumento del sangrado en hemofílicos | HiperbilirrubinemiaIntolerancia gastrointestinal (diarrea)CefaleaNefrolitiasisLos estudios disponibles a las 48 semanas no muestran alteraciones lipídicas relevantesATV/r: Dislipemia levePosible aumento del sangrado en hemofílicos | Intolerancia gastrointestinal (vómitos, diarrea)CefaleaAsteniaDislipidemia leveErupción cutánea, que suele ser moderada y autolimitada (usar con precaución en pacientes con alergia conocida a las sulfamidas)Posible aumento del sangrado en hemofílicos | Intolerancia gastrointestinal (diarrea)Alteraciones del SNC (vértigo, dificultad de concentración, enlentecimiento, cambios de humor)En combinación con RTV, aumento de triglicéridos y transaminasasSe han descrito 14 casos de hemorragia intracraneal, 8 de los cuales fueron mortales, entre 6.840 pacientes incluidos en ensayos clínicos. La mayoría tenían factores de riesgo |
| InteraccionesAsociaciones contraindicadasa | AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolCisapridaColchicina (si insuficiencia renal o hepática)Deriv. ergotaminaEncainidaÉxtasisFlecainidaFluticasona inh.FosamprenavirHalofantrinaHypericum (hierba de San Juan)Inhibidores proteasa VHC: boceprevir y telaprevirLovastatinaLumefantrinaMetanfetaminaMidazolam oralPimozidaPropafenonaQuinidinaRifampicinaSalmeterolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTipranavir/ritonavirTriazolamVardenafiloVoriconazol | En general, los inductores del CYP3A4 (ATV ± RTV)AlfuzosinaAstemizolBosentán con ATV no potenciadoCisapridaColchicina (si insuficiencia renal o hepática)Deriv. ergotaminaEfavirenz (considerar ATV/r 400/200mg c/24h en naïve; evitar en pretratados)Fluticasona inh. (ATV/r)HalofantrinaHypericum (hierba de San Juan)IndinavirInhibidores proteasa VHC: boceprevir (la coadministración podría considerarse en casos puntuales en pacientes con carga viral del VIH indetectable y ausencia de sospecha de resistencias, empleando ATV potenciado)IrinotecanLovastatinaLumefantrinaMidazolam oralNevirapinaOmeprazol y afinesPimozidaQuinidinaRifampicinaSalmeterolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTriazolamVoriconazol (ATV/r)Evitar fármacos que puedan prolongar el intervalo QT | AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolBudesonida inh.CisapridaColchicina (si insuficiencia renal o hepática)Deriv. ergotaminaÉxtasisFenobarbitalFenitoínaFluticasona inh.HalofantrinaHypericum (hierba de San Juan)Inhibidores proteasa VHC: boceprevir y telaprevirLidocaína sistémicaLopinavir/ritonavirLovastatinaMetanfetaminaMidazolam oralPimozidaQuinidinaRifampicinaSalmeterolSaquinavirSertindolSildenafilo (hipertensión pulmonar)SimvastatinaTadalafilo (hipertensión pulmonar)TerfenadinaTriazolamVoriconazol | Abacavir (evitar a menos que no se disponga de otros análogos)AlfuzosinaAmiodaronaAnticonceptivos oralesAstemizolAtazanavir/ritonavirBosentánCisapridaColchicinaDeriv. ergotaminaEncainidaEstatinasEtravirinaÉxtasisFlecainidaFluticasona inh.Fosamprenavir/ritonavirHalofantrinaHypericum (hierba de San Juan)Inhibidores proteasa VHC: boceprevir y telaprevir (por ausencia de datos)Lopinavir/ritonavirLovastatinaLumefantrinaMetanfetaminaMetoprololMidazolam oralOmeprazol y afines (reducción de eficacia de omeprazol; si imprescindible, aumentar dosis)PimozidaPropafenonaQuinidinaRifampicinaSalmeterolSaquinavir/ritonavirSertindolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTolterodinaTriazolamVoriconazolZidovudina (evitar a menos que no se disponga de otros análogos) |
No se han incluido en todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en castellano) y www.hiv-druginteractions.org (en inglés). Debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
En pacientes naïve y en pacientes adultos con TAR previo sin mutaciones asociadas a resistencia frente a DRV y que tienen una CVP<100.000copias/ml y un recuento de linfocitos CD4+≥100células/μl (ficha técnica de darunavir 400mg).
Información procedente de la ficha técnica europea. EPARS: European Public Assessment Reports [consultado 14 Nov 2012]. Disponible en: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/medicines/medicines_landing_page.jspμrl=menus/medicines/medicines.jsp∣=
Información procedente de la ficha técnica americana (fuentes: FDA and First Data Bank, Inc) [consultado 14 Nov 2012]. Disponible en: http://www.rxlist.com/drugs/alpha_a.htm
hGuardiola JM, Soriano V, editores. Tratamiento de la infección por VIH-SIDA. Fármacos y combinaciones, 10.a ed. Barcelona: Publicaciones Permanyer; 2007.
Se entiende por IP potenciado la coadministración de un IP con dosis reducidas de RTV (IP/r). RTV tiene un potente efecto inhibidor del citocromo P450, que inhibe el metabolismo del segundo IP, mejorando su perfil farmacocinético y el cociente Cmin/CI50 y reduciendo el riesgo de aparición de resistencias. Además, al potenciar un IP se reduce el número de comprimidos, la frecuencia de dosis y las restricciones dietéticas, todo lo cual favorece la adherencia. En la tabla 7 aparecen las combinaciones más importantes de IP y sus dosificaciones basadas en estudios farmacocinéticos.
Estudios que han comparado distintos inhibidores de la proteasaM98-863El M98-863 es un ensayo clínico aleatorizado, doble ciego, multicéntrico, que comparó LPV/r (400/100mg BID; n=326) frente a NFV (750mg TID; n=327) junto a d4T/3TC. En la semana60 se observó mejor respuesta virológica (ITT) en los pacientes tratados con LPV/r (CVP<50copias/ml, 64 y 52%, respectivamente; p=0,001)297.
Abbott 418En el ensayo clínico Abbott 418 se evaluaron la eficacia y la tolerancia de LPV/r BID frente a LPV/r QD combinados con TDF/FTC. Se incluyeron 190pacientes con una mediana de CD4 de 214-232células/μl y de CVP de 4,6-4,8log10 en cada grupo. A la semana48 la proporción de pacientes con CVP<50copias/ml fue similar (70 y 64% según pauta, LPV/r QD o LPV/r BID, análisis ITT)298.
KLEANEl estudio KLEAN comparó FPV/r (700/100mg BID) con LPV/r (300/100mg BID en cápsulas), ambos con ABC/3TC coformulados, en 887pacientes. En la semana48 la proporción de pacientes con CVP<50copias/ml (ITT) fue del 66% en el grupo de FPV/r y del 65% en el de LPV/r, demostrando la no-inferioridad de FPV/r frente a las cápsulas de LPV/r. No hubo diferencia significativa en cuanto a tolerancia digestiva (náuseas, vómitos, diarrea) ni alteraciones lipídicas266.
APV30001 - NEATEn el estudio NEAT se evaluaron la eficacia y la tolerancia de FPV (1.400mg BID) frente a NFV, ambos en combinación con ABC/3TC. Se aleatorizaron 166pacientes a FPV y 83 a NFV. A las 48 semanas, la proporción de pacientes con CVP<400copias/ml fue mayor en el grupo de FPV que en el de NFV (66 y 51%; ITT). En el grupo de CVP elevada (>100.000copias/ml) la proporción fue del 67 y del 35%, respectivamente (p<0,05)299.
APV30002 - SOLOEl SOLO es un ensayo clínico aleatorizado en el que compararon 322pacientes tratados con FPV/r QD (1.400/200mg) con 327pacientes con NFV, ambos asociados a ABC+3TC. La mediana de CD4 fue de 170células/μl y la CVP de 4,8log10. El 20% de los pacientes tenían <50CD4/μl. A las 48 semanas no hubo diferencias (ITT) en la proporción de pacientes con CVP<400copias/ml (69% FPV/r frente 68% NFV) ni con CVP<50copias/ml (55 frente al 53%). Sin embargo, la proporción de pacientes que presentaron fallo virológico fue superior en la rama de NFV (17%) que en la de FPV/r (7%)300.
COL100758El estudio COL100758 compararon FPV/r (1.400/100mg) y FPV/r (1.400/200mg), ambos QD, junto con ABC/3TC coformulados. Se incluyeron 115pacientes. A las 48semanas la proporción de pacientes con CVP<50copias/ml (ITT) fue del 79% (FPV/r 100mg) y del 63% (FPV/r 200mg); p=0,061. La adherencia fue mejor en el grupo de 100mg de RTV267. En voluntarios sanos se ha demostrado que los niveles plasmáticos de FPV (1.400mg QD) no difieren si se potencia con 100 o 200mg de RTV301.
ARTEMISEl estudio ARTEMIS comparó DRV/r (800/100mg QD) frente a LPV/r (BID y QD) en 689pacientes que recibieron además TDF/FTC coformulados. La dosificación de LPV/r fue variable: el 77% recibieron el LPV/r BID, el 15% QD y, además, el 7% cambiaron de BID a QD durante el estudio. El 15% recibieron LPV/r en cápsulas, el 2% en comprimidos y el 83% cambiaron de cápsulas a comprimidos durante el estudio. A las 48semanas la proporción con CVP<50copias/ml (ITT) fue del 84% en el grupo de DRV/r y del 78% en el de LPV/r (IC95%: −0,3-11,2; p=0,062), demostrando la no-inferioridad de DRV/r frente a LPV/r. Los pacientes tratados con DRV/r presentaron menos diarrea de grado 2-4 que los tratados con LPV/r (4% frente al 10%) y las elevaciones lipídicas fueron menores (triglicéridos y colesterol total)302. A las 96 semanas, el 79% de los pacientes en la rama de DRV/r y el 71% en la LPV/r tenían CVP<50copias/ml, confirmando no solo la no-inferioridad (diferencia estimada: 8,4%; IC95%: 1,9-14,8; p<0,001), sino también la superioridad de DRV/r sobre LPV/r (ITT; p=0,012). El 4% de los pacientes de la rama de DRV/r y el 9% de los la rama de LPV/r abandonaron el tratamiento asignado303. A las 192semanas el 68,8% de los pacientes con DRV/r y el 57,2% de los tratados con LPV/r mostraron una CVP<50copias/ml, corroborando la no-inferioridad y la superioridad del DRV/r. Se confirma así el buen perfil de tolerabilidad del DRV tras 4años de tratamiento304.
GEMINIEl estudio GEMINI comparó SQV/r (1.000/100mg BID) frente a LPV/r (300/100mg BID en cápsulas) en 337pacientes, todos los cuales recibieron, además, una combinación coformulada de TDF/FTC. A las 48semanas la proporción con CVP<50copias/ml mediante un análisis por ITT fue del 64,7% en el grupo de SQV/r y del 63,5% en el grupo de LPV/r (IC95%: −9,6 a −11,9). El estudio demostró no-inferioridad de SQV/r frente a LPV/r. Las elevaciones lipídicas fueron similares, con un incremento mayor de triglicéridos en el grupo de LPV/r y de colesterolLDL en el de SQV/r305.
BMS-089El estudio BMS 089 comparó ATV (400mg QD) frente a ATV/r (300/100mg QD). Se incluyeron 200pacientes que recibieron además d4T de liberación retardada (100mg QD) y 3TC (300mg QD). A las 48semanas, la proporción de pacientes con CVP<50copias/ml (ITT) fue del 75% en el grupo de ATV/r y del 70% en el de ATV. En este estudio se demostró la no-inferioridad de ATV/r frente a ATV sin potenciar. Las causas de fracaso terapéutico fueron diferentes en ambos brazos. Hubo más fracasos virológicos en la rama de ATV no potenciado (10% frente al 3%), pero tal diferencia no alcanzó la significación estadística. Los pacientes de la rama de ATV/r que experimentaron fracaso virológico no tenían mutaciones en el gen de la proteasa, mientras que estas se detectaron en 3 de los 10pacientes que fracasaron con ATV sin potenciar. También hubo más mutaciones de resistencia a 3TC en el grupo no potenciado (7 de 10 frente a 1 de 3). Las suspensiones de tratamiento por hiperbilirrubinemia fueron más frecuentes con ATV/r que con ATV. El estudio puso de manifiesto la mayor eficacia virológica y barrera genética de ATV/r306.
Estudio 1182.33El estudio 1182.33 intentó comparar 2dosis de TPV/r (500/100mg BID y 500/200mg BID) frente a LPV/r (400/100mg BID). El comité de vigilancia decidió parar el estudio por una mayor tasa de elevación asintomática de enzimas hepáticas en la rama de TPV/r 500/200mg BID y por no alcanzar el criterio de no-inferioridad a la semana60 en la rama de TPV/r 500/100mg BID. Debido a estos resultados no se recomienda el uso de TPV/r en terapias de inicio (número de identificación en ClinicalTrials.gov NCT00144105).
Estudio 730La combinación TDF/FTC+LPV/r (comprimidos BID) se ha comparado con la combinación TDF/FTC+LPV/r (comprimidos QD) en 664pacientes que recibían el primer tratamiento. La proporción de pacientes con cargas virales <50copias/ml (ITT) fue del 77% en el grupo QD y del 76% en el grupo BID. Este ensayo clínico demuestra, pues, la no-inferioridad de LPV/r administrado una vez al día frente a LPV/r administrado 2veces al día. Durante las primeras 8semanas del estudio los pacientes en cada grupo fueron aleatorizados además a recibir las cápsulas o los comprimidos de LPV/r, sin que se encontrasen diferencias entre ambas presentaciones respecto a la incidencia de efectos adversos o a las discontinuaciones por toxicidad298.
Estudio CASTLEEste ensayo comparó la combinación TDF/FTC+ATV/r QD con la combinación TDF/FTC+LPV/r (cápsulas) BID en 883pacientes sin TAR previo, habiendo demostrado la no-inferioridad de ATV/r frente a LPV/r en su presentación de cápsulas. La proporción de pacientes con cargas virales <50copias/ml (ITT) fue del 78% en el grupo de ATV/r y del 76% en el grupo de LPV/r. ATV/r mostró mejor perfil lipídico (colesterol total, triglicéridos y colesterol no-HDL). La ictericia y la hiperbilirrubinemia fueron más frecuentes en el grupo de ATV/r, mientras que la diarrea y las náuseas lo fueron en el grupo de LPV/r307. A las 96semanas, el 74% frente al 68% de los pacientes en las ramas de ATV/r y de LPV/r, respectivamente, tenían una CVP<50copias/ml (ITT; p<0,05), con lo que se confirmó la no-inferioridad de ATV/r con respecto a LPV/r. Los abandonos en ambas ramas fueron del 7%308.
Estudio GS-US-236-0103La combinación de ATV/r+FTC/TDF como TAR de inicio también ha sido evaluada en el estudio 0103, un ensayo clínico aleatorizado a doble ciego que comparó EVG/COBI/FTC/TDF (coformulados en pastilla única) y ATV/r+FTC/TDF. La variable primaria de evaluación fue la proporción de pacientes con CV<50copias/ml en la semana 48 mediante un análisis por ITT, según el algoritmo Snapshot de la FDA. Se trataron 708pacientes y lograron el objetivo primario 316/353 (89,5%) de los tratados con EVG/COBI/FTC/TDF y 308/355 (86,8%) de los tratados con ATV/r+FTC/TDF (diferencia: 3,0%; IC95%: −1,9-7,8%), confirmándose, pues, la no-inferioridad de EVG/COBI/FTC/TDF frente a ATV/r+FTC/TDF. En 5 de los 12pacientes del grupo de EVG/COBI/FTC/TDF en los que se efectuó una prueba genotípica de resistencia se objetivaron mutaciones, las cuales conferían resistencia a los inhibidores de la integrasa en 4 de ellos (Q148R en 2, N155H en 2, T66I en uno y E92Q en uno). Ninguno de los 8pacientes del grupo de ATV/RTV+FTC/TDF en los que se investigó la existencia de resistencia habían desarrollado mutaciones. Ambos regímenes se toleraron bien y las interrupciones del tratamiento por efectos adversos fueron bajas en ambos brazos. El tratamiento con EVG/COBI/FTC/TDF indujo menos elevaciones de los triglicéridos hasta la semana4 que el tratamiento con ATV/r+FTC/TDF (mediana de 0,09 y 0,26mmol/l, respectivamente; p=0,006). En ambos brazos se documentaron pequeños incrementos en la concentración de creatinina sérica, con los consiguientes descensos en el filtrado glomerular estimado309.
Estudio GS-US-216-0114El estudio 0114 es un ensayo clínico de fase iii, aleatorizado, doble ciego, de no-inferioridad, en el que se comparó COBI frente a RTV como potenciador de ATV en 692pacientes sin TAR previo y con un filtrado glomerular estimado ≥70ml/min. Todos los pacientes recibieron FTC/TDF como pareja de ITIAN. La aleatorización se estratificó según que la CVP basal fuese mayor o igual/menor de 100.000copias/ml. La variable primaria de eficacia fue la proporción de pacientes con CVP<50copias/ml en la semana48, según el algoritmo Snapshot de la FDA, con un margen inferior de no-inferioridad del 12%. Se aleatorizaron 344pacientes al brazo de COBI y 348pacientes al brazo de RTV. Lograron el objetivo de eficacia el 85% de los tratados con COBI y el 87,2% de los tratados con RTV (diferencia: −2,2; IC95%: −7,4-3,0), demostrándose, por tanto, la no-inferioridad de COBI frente a RTV. El incremento en la cifra de linfocitos CD4+ fue de 213células/μl con COBI y de 219células/μl con RTV. En 24pacientes con fracaso virológico en los que se pudo hacer genotipificación del virus no se detectaron mutaciones primarias para IP ni para TDF; se detectó la mutación M184V/I en 2pacientes tratados con COBI y en ninguno de los tratados con RTV. Los pacientes con COBI presentaron más hiperbilirrubinemia (65% frente al 57%), mayor descenso del filtrado glomerular estimado (−13 frente a −9ml/min) y una tendencia sin significación estadística a presentar cifras menores de colesterol y triglicéridos310.
Resumen sobre ensayos de inhibidores de la proteasa en pacientes sin terapia previaLos estudios han demostrado que la administración de un IP potenciado con RTV (LPV/r, SQV/r, FPV/r, ATV/r, DRV/r) tiene ventajas de eficacia y barrera genética respecto a los IP no potenciados. El principal inconveniente de la potenciación es el aumento de efectos adversos.
Recomendaciones sobre los inhibidores de la proteasa- •
Como IP de primera elección se recomiendan ATV/r QD, DRV/r QD y LPV/r BID o QD. (A-I)
- •
FPV/r o SQV/r se pueden utilizar como pautas alternativas. (B-III)
Los inhibidores del correceptor CCR5 actúan bloqueando la entrada de VIH-1 en la célula diana. Estos fármacos son activos solamente si el virus tiene tropismo R5. MVC es el único inhibidor del correceptor CCR5 que ha sido aprobado para tratamientos en pacientes pretratados con tropismo R5 (tabla 8).
Antagonistas del correceptor CCR5 e inhibidores de la integrasa
| Grupo terapéutico | Antagonistas correceptor CCR5 | Inhibidores de la integrasa | |
| Nombre genérico | Maraviroc | Raltegravir | Elvitegravir |
| Nombre comercial | Celsentri® | Isentress® | Stribild®(no comercializado en España a enero 2013)Contiene la combinación QUAD (EVG/COB/TDF/FTC) |
| DosisRecomendación | 150, 300 o 600mg BID dependiendo de las interacciones con otros fármacos. 300mg BID en ausencia de inhibidores o inductores de CYP3A4Puede tomarse con o sin alimentos | 400mg BIDPuede tomarse con o sin alimentos | 1 comprimido/día(EVG 150mg/COB 150mg/TDF 300mg/FTC 200mg) |
| Presentación comercial | Comprimidos de 150 y de 300mg | Comprimidos de 400mg | Comprimidos |
| Biodisponibilidad oral | 23-33%a23% (dosis única de 100mg)a33% (estimada para 300mg)a | No se ha determinado su biodisponibilidad absoluta en humanosa | No se ha determinado su biodisponibilidad absoluta en humanosb |
| Efecto de los alimentos | Un desayuno rico en grasa redujo el AUC un 33%. Sin embargo, dado que no hubo restricciones alimentarias en los estudios de eficacia, puede administrarse con o sin alimentosb | Una comida con contenido moderado de grasa aumentó el 13% el AUC. Una comida con elevado contenido graso duplicó el AUC. En general, los alimentos aumentaron la variabilidad farmacocinética. Sin embargo, dado que no hubo restricciones alimentarias en los estudios de eficacia, puede administrarse con o sin alimentosb | QUAD debe ser administrado con alimentos. Tras la administración de una dosis única de QUAD, una comida ligera (∼373kcal, 20% grasa) aumentó el AUC de EVG un 34% y de TDF un 24%. Una comida grasa (∼800kcal, 50% grasa) aumentó el AUC de EVG un 87% y de TDF un 23%. Los cambios en el AUC de cobicistat y FTC no fueron clínicamente significativosb |
| Semivida plasmática | 13,2ha | 9ha | 12,9hb |
| Cmax | 0,618μg/ml (VIH-1+ asintomáticos con 300mg/12h)b | 4,5μM (IC90%: 2,0-10,2) (=2,17μg/ml) | 1,7 ± 0,4μg/mlb |
| Cmin | 0,034μg/ml (VIH-1+ asintomáticos con 300mg/12h)b | 0,14μMb (=0,068μg/ml) | 0,45 ± 0,26μg/mlb |
| AUC | AUC0-12h: 2.550μg·h/ml (VIH-1+ asintomáticos con 300mg/12h)b | AUC0-12h: 14,3μM·hb (IC90%: 7,6-26,6) (=6,89μg·h/ml) | 23,0 ± 7,5μg·h/mlb |
| CI50/90 frente a VIH-1 in vitro | CI50: 0,1 a 4,5nM (0,05-2,3ng/ml)b(1nM=0,5ng/ml)bCI90: 0,57 (0,06-10,7) ng/mla | CI95: 31±20nMa (=14,9ng/ml) | CI50: 0,02-1,7nMb (0,0089-0,7616ng/ml) |
| Actividad | VIH-1 tropismo R5 | VIH-1 y VIH-2, tropismo R5, X4 y dual | VIH-1 y VIH-2b |
| Penetración en LCR (LCR:plasma) | Grado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | Penetración insignificanteaLas concentraciones de raltegravir en LCR (LCR 14,5ng/ml; conc. plasmáticas 261ng/ml) fueron 4,5 veces superiores a la CI50para la cepa salvaje del virus, tomando como CI50 3,2ng/ml (Croteau D. AAC 2010;54:5156-5160)Grado 3 de penetración en LCR según la clasificación del estudio CHARTER (de 1 a 4, menor a mayor) (Letendre S Poster 430. Croi 2010) | No ha sido evaluado en humanos |
| Grupo terapéutico | Antagonistas correceptor CCR5 | Inhibidores de la integrasa | |
| Metabolización | CYP3A4 (no inductor, ni inhibidor) | Glucuronidación (UGT1A1)(No inductor, ni inhibidor de CYP3A4 ni de UGT1A1) | Elvitegravir: CYP3A (mayoritario) y UGT1A1/3bCobicistat: CYP3A>2D6bCobicistat inhibe: CYP3A y CYP2D6 y los transportadores: glucoproteína-P (P-gp), BCRP, OATP1B1 y OATP1B3. Elvitegravir es un inductor leve del CYP2C9) |
| Efectos adversos | Náuseas, vómitos, flatulencia, dolor abdominalParestesia, disgeusiaErupción cutáneaAstenia | Diarrea, náuseasCefalea | DiarreaCefaleaMareoNáuseasAcidosis lácticaAsteniaCobicistat reduce levemente el filtrado glomerular renal estimado de creatinina (aunque no altera el filtrado glomerular real), debido a la inhibición de la secreción tubular de creatininab |
| InteraccionesAsociaciones contraindicadasc | Hypericum perforatum (hierba de San Juan)Combinación de 2 inductores enzimáticos importantes (por ejemplo, rifampicina + efavirenz) | No se han descrito | Antirretrovirales: IP e ITINNbRitonavir (cobicistat tiene la misma acción: potenciador farmacocinético)bAlfuzosinaAnticonceptivos orales (valorar métodos alternativos): aumento de niveles del progestágeno (norgestimato) y reducción del estrógeno (etinilestradiol)CisapridaColchicina (si insuficiencia renal o hepática)Derivados de la ergotaminaFluticasonaHypericum (hierba de San Juan)LovastatinaMidazolam por vía oralPimozidaRifampicinaSalmeterolSildenafilo (en hipertensión pulmonar)SimvastatinaTriazolamEn general, evitar fármacos que puedan reducir su eficacia como los inductores (carbamacepina, oxcarbazepina, fenobarbital, fenitoína y rifabutina, entre otros) o los antiácidos (espaciando 2h el antiácido se evita esta interacción) |
Información procedente de la ficha técnica europea. EPARS: European Public Assessment Reports [consultado 14 Nov 2012]. Disponible en: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/medicines/medicines_landing_page.jspμrl=menus/medicines/medicines.jsp∣=
Información procedente de la ficha técnica americana (fuentes: FDA and First Data Bank, Inc) [consultado 14 Nov 2012]. Disponible en: http://www.rxlist.com/drugs/alpha_a.htm
No se han incluido todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en castellano) y www.hiv-druginteractions.org (en inglés). Debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
El estudio MERIT es un ensayo que comparó MVC (300mg BID) con MVC (600mg QD) y con EFV (600mg QD) en pacientes infectados por VIH-1 R5-trópico y sin TAR previo. Los pacientes recibieron además AZT/3TC. El grupo de MVC QD fue interrumpido a la semana16 por no alcanzar el criterio virológico definido en el protocolo. A las 48semanas, la proporción de pacientes con CVP<50 copias/ml (ITT) fue del 65,3 y del 69,3% en el grupo de MVC y de EFV (límite inferior del IC97,5%: −10,9%). La proporción con CVP<400copias/ml (análisis por ITT) fue del 70,6 y del 73,1% en el grupo de MVC y de EFV (límite inferior del IC97,5%: −9,5%). El límite inferior del IC97,5%, de no-inferioridad, que se estableció para este ensayo clínico fue de −10%. Por lo tanto, el estudio demostró la no-inferioridad de MVC para el criterio de 400copias/ml, pero no para el de 50copias/ml. La discontinuación del TAR por falta de eficacia fue más frecuente con MVC (11,9%) que con EFV (4,2%), pero por efectos adversos fue mayor con EFV (13,6%) que con MVC (4,2%). La recuperación inmunológica fue mayor con MVC (170 frente 144linfocitos CD4+/μl, respectivamente)311,312. En un reanálisis post-hoc en el que se excluyeron 107pacientes (15%) con virus X4 o D/M, gracias a un ensayo de tropismo más sensible, el límite inferior de no-inferioridad al 97,5% para la diferencia entre tratamientos se situó por encima de −10% para cada objetivo, consiguiendo, pues, la no-inferioridad de MVC frente a EFV312.
Recomendación- •
MVC solo debe emplearse como TAR de inicio en pacientes con virus con tropismo R5, cuando no sea posible un tratamiento con ITINN, un IP o un InInt (C-I). La recomendación se basa en resultados del estudio MERIT
Los inhibidores de la integrasa (InInt) actúan alterando la integrasa viral que no pueden unir los extremos reactivos del ADN viral al ADN celular (tabla 8).
Ensayos que avalan la recomendación de inhibidores de la integrasaEl estudio STARTMRK es un ensayo clínico, aleatorizado, doble ciego, multicéntrico e internacional que compara EFV con RAL, ambos combinados con TDF/FTC en 566pacientes sin TAR previo313. El objetivo primario de eficacia fue la consecución de una CVP<50copias/ml a la semana48. El margen de no-inferioridad fue del 12%. El 53% de los pacientes tenían carga viral basal >100.000copias/ml y el 47% una cifra de linfocitos CD4+<200células/μl. El objetivo primario del estudio se consiguió en el 86,1% de los pacientes del grupo de RAL y en el 81,9% de los del grupo de EFV (diferencia: 4,2%; IC95%: −1,9-10,3). El grupo de RAL tardó menos tiempo en alcanzar la indetectabilidad que el grupo de EFV (p<0,0001). Los efectos adversos relacionados con los fármacos fueron más frecuentes en el grupo de EFV que en el de RAL (p<0,0001). La comunicación de los resultados, aún ciegos, a 240semanas del estudio STARTMRK confirma la eficacia virológica e inmunológica tanto de RAL o de EFV combinados con TDF/FTC. Es de destacar que después de 5años de seguimiento la tasa de supresión virológica fue superior con RAL que con EFV (71% vs 61%). La recuperación inmunológica también fue superior con RAL (+374 frente a +312 linfocitos CD4+)313.
En el estudio QDMRK se comparó RAL 800mg QD frente a RAL 400mg BID, ambos en combinación con TDF/FTC, en 775pacientes sin TAR previo, con el objetivo de investigar la no-inferioridad de la pauta QD frente a la BID con un margen de no-inferioridad definido de antemano del −10%. En el análisis primario de eficacia a las 48semanas la proporción de pacientes con CVP<50copias/ml en los brazos QD y BID fue del 83,2 y del 88,9%, respectivamente (diferencia: −5,7; IC95%: −10,7-0,83). La respuesta virológica fue menor con RAL QD que con RAL BID, con independencia de la CVP basal. Se documentaron mutaciones de resistencia a RAL tras el fracaso virológico en 9/27 (33%) y 2/12 (17%) pacientes evaluables de las pautas QD y BID, respectivamente. La concentración media de RAL en el valle resultó inferior en el grupo QD (83nM) que en el grupo BID (380nM). El fracaso virológico fue más frecuente en los pacientes que recibían RAL QD y que tenían concentraciones bajas de RAL. No hubo diferencias en seguridad entre ambos brazos de tratamiento. Los resultados de este estudio muestran que la eficacia virológica de RAL 800mg QD es inferior a RAL 400mg BID cuando se administra con TDF/FTC314.
El ensayo clínico SPRING-2 es un estudio de fase iii, aleatorizado y doble ciego, que comparó la no-inferioridad de DTG, 50mg QD, frente a RAL, 400mg BID, en 822pacientes infectados por el VIH sin TAR previo (411 por brazo). Los investigadores pudieron elegir entre TDF/FTC o ABC/3TC como pareja de ITIAN. La variable primaria de eficacia fue la proporción de pacientes con CVP<50copias/ml en la semana 48, según el algoritmo Snapshot de la FDA, con un margen inferior de no-inferioridad del 10%. Se utilizó TDF/FTC en el 59% de los pacientes del brazo de DTG y en el 60% de los del brazo de RAL y se eligió ABC/3TC en el 41% de los pacientes del brazo de DTG y en el 40% de los del brazo de RAL. La proporción de pacientes que lograron el objetivo primario de eficacia fue del 88% con DTG y del 85% con RAL (diferencia entre ambos brazos: 2,5% [IC95%: −2,2-7,1], demostrándose, pues, la no-inferioridad de DTG frente a RAL. El incremento en la cifra de linfocitos CD4+ fue de 230células/μl en ambos brazos. Se documentó fallo virológico en 20 (5%) de pacientes tratados con DTG y en 28 (7%) de los tratados con RAL. No se detectaron mutaciones de resistencias a DTG ni a ITIAN en los tratados con DTG y se detectaron mutaciones de resistencia a inhibidores de la integrasa y a ITIAN en uno y 4 pacientes tratados con RAL, respectivamente. No hubo diferencias en lo relativo a efectos adversos o alteraciones en las pruebas de laboratorio de grado 3 o 4 entre ambos brazos. Se detectó un pequeño incremento en la cifra de creatinina en el brazo de DTG por bloqueo de la secreción renal del fármaco, sin que se afectara el filtrado glomerular real. Las retiradas del tratamiento por efectos adversos fueron poco frecuentes y no hubo diferencias entre ambos brazos315.
Recomendación- •
RAL puede emplearse como tratamiento de inicio en dosificación BID combinado con TDF/FTC o ABC/3TC. (A-I)
La idea de utilizar pautas de TAR libres de ITIAN surgió en un momento en que los fármacos de esta familia tenían una toxicidad significativa, como por ejemplo lipoatrofia (d4T, ddI, AZT), neuropatía periférica (d4T y ddI), pancreatitis aguda (ddI y d4T) y mielosupresión (AZT). Además, las pautas libres de ITIAN resultaban atractivas porque llevaban asociada una reducción en el número de pastillas que, al menos teóricamente, podía mejorar el cumplimiento terapéutico y la calidad de vida. La necesidad de pautas libres de ITIAN es hoy en día menos acuciante, puesto que las pautas de ITIAN actualmente recomendadas (FTC/TDF y 3TC/ABC) presentan ventajas indudables con respecto a otras que actualmente se consideran alternativas. Tanto FTC/TDF como 3TC/ABC resultan muy eficaces, pueden administrarse coformuladas en regímenes QD y apenas tienen toxicidad aguda (en ausencia del HLA-B*5701). La toxicidad a medio y a largo plazo de FTC/TDF como 3TC/ABC también es menor que otras combinaciones de ITIAN consideradas actualmente como alternativas, aunque conviene mencionar la toxicidad renal (poco frecuente) y ósea (de significado clínico todavía incierto) de TDF y la asociación entre el uso de ABC y el infarto agudo de miocardio, cada vez más puesta en duda.
Monoterapia con inhibidores de la proteasaEn el estudio MONARK se compararon la eficacia y la seguridad de LPV/r en monoterapia frente a LPV/r+AZT/3TC como TAR de inicio en pacientes con CVP basal <100.000copias/ml. En la semana 48, por ITT, lograron CVP<50copias/ml 53 de 83pacientes (64%) en el brazo de monoterapia y 40 de 53 (75%) en el brazo de triple terapia (p=0,19). En un análisis OT la proporción de pacientes con CVP<50copias/ml en la semana 48 fue del 80 y del 95% para los pacientes con monoterapia y triple terapia, respectivamente (p=0,02)316.
Combinaciones de inhibidores de la proteasa potenciados con ritonavir con inhibidores de la transcriptasa inversa no nucleósidosEn el estudio ACTG5142 se compararon 3pautas como TAR de inicio: EFV+2ITIAN, LPV/r+2ITIAN y una pauta libre de ITIAN (EFV+LPV/r). Se aleatorizaron en total 757 pacientes con una mediana de linfocitos CD4+ de 191células/μl y una mediana de CVP de 4,8log10 copias/ml. El tiempo hasta el fracaso virológico resultó más largo para la pauta de EFV+2ITIAN que para LPV/r+2ITIAN (p=0,006) pero no fue diferente para EFV+LPV/r en comparación con las otras 2pautas con ITIAN. En la semana 96, la proporción de pacientes con CVP<50copias/ml fue del 89% en el grupo de EFV, del 77% en el grupo de LVP/r y del 83% en el grupo de EFV+LPV/r (p=0,003 para la comparación entre EFV y LPV/r). No se observaron diferencias en el tiempo hasta la discontinuación del tratamiento por efectos tóxicos. En los pacientes con fracaso virológico la aparición de cepas con mutaciones de resistencia resultó más frecuente en el brazo libre de ITIAN que en los brazos con ITIAN. De este estudio se puede concluir que el fracaso virológico es menor con EFV que con LPV/r cuando se combinan con ITIAN, y que la pauta libre de ITIAN formada por EFV+LPV/r tiene una eficacia similar a la pauta de EFV+2ITIAN, pero se asocia con más resistencia farmacológica en caso de fracaso282.
Combinaciones de inhibidores de la proteasa potenciados con ritonavir con raltegravirEn el estudio SPARTAN se incluyeron pacientes sin TAR previo que fueron aleatorizados en proporción 2:1 a recibir ATV sin potenciar, 300mg BID+RAL 400mg BID (n=63) o bien una combinación estándar de ATV/r, 300/100mg+FTC/TDF (n=31). El estudio fue interrumpido en la semana24 por problemas de eficacia virológica y de seguridad. Durante las primeras 24 semanas se documentó fracaso virológico en 11pacientes del brazo ATV+RAL y en 8 del brazo ATV/r+FTC/TDF. Resultaron aptos para el estudio genotípico de resistencias (CVP>4.000copias/ml) 6aislados del brazo ATV+RAL, en los que se detectaron mutaciones de resistencia a RAL en 4 (en el quinto se documentó resistencia fenotípica a RAL). Solo un aislado del brazo ATV/r+FTC/TDF cumplió criterios para la genotipificación, que no desveló ninguna mutación de resistencia. Hubo discontinuaciones por efectos adversos en 4/63 (6,3%) de los pacientes del brazo ATV+RAL y en ninguno de los pacientes del brazo ATV/r+FTC/TDF. Se documentó elevación de bilirrubina total de grado 4 en 13 (20,6%) de los pacientes con ATV+RAL y en ninguno de los pacientes del brazo ATV/r+FTC/TDF317.
En el estudio PROGRESS se comparó LPV/r+RAL con LPV/r+TDF/FTC en 206pacientes sin TAR previo. El objetivo primario de eficacia fue el logro de una CVP<50copias/ml en la semana 48 por ITT-TLOVR. El objetivo primario del estudio se consiguió en el 83,2% de los pacientes del grupo de LPV/r+RAL y en el 84,8% del grupo de LPV/r (diferencia: −1,6%; IC95%: −12,0-8,8). El grupo con RAL tardó menos tiempo en alcanzar la indetectabilidad que el grupo con TDF/FTC (p<0,001). La proporción de pacientes que interrumpió el estudio como consecuencia de los efectos adversos relacionados con el TAR fue del 2,0% en la rama de LPV/r+RAL y del 1,9% en la de LPV/r+TDF/FTC318.
El estudio ACTG A5262 fue un estudio de brazo único donde se trataron 112pacientes sin TAR previo con la combinación de DRV/r+RAL. Se consideró que existía fracaso virológico ante cualquiera de las 3siguientes circunstancias: a)CVP confirmada ≥1.000copias/ml en la semana12; b)incremento de la CVP>0,5log10 copias/ml desde la semana 4 a la semana 12, o c)CVP>50copias/ml en o a partir de la semana24. Según un análisis por ITT, se documentó fracaso virológico a 24 semanas en el 16% de los pacientes (IC95%: 10-24%) y a 48 semanas en el 26% (IC95%: 19-36%). En un análisis ajustado por sexo y edad, el fracaso virológico se asoció con CVP>100.000copias/ml (HR: 3,76; IC95%: 1,52-9,31; p=0,004) y con cifras bajas de linfocitos CD4+ (0,77 por cada incremento de 100células/μl; IC95%: 0,61-0,98; p=0,037). Las mutaciones de resistencia a RAL resultaron frecuentes en los pacientes con fracaso virológico y CVP>100.000copias/ml319.
Combinaciones de inhibidores de la proteasa potenciados con ritonavir con maravirocEn el estudio A4001078 se aleatorizó a 121pacientes sin TAR previo infectados por cepas de VIH-1 R5 trópicas a ATV/r+MVC 150mg al día QD o a ATV/r+FTC/TDF. Todos los pacientes tratados con MVC tuvieron concentraciones plasmáticas superiores a la IC50 del virus a lo largo de las 24h del intervalo de dosis. En la semana 24 tenían CVP<50copias/ml 54 de 61 (89%) pacientes aleatorizados al brazo de FTC/TDF y 48 de 60 (80%) de los pacientes aleatorizados al brazo de MVC. Se documentó hiperbilirrubinemia de grado 3-4 en 35 (59,3%) de los pacientes tratados con MVC y en 30 (49,2%) de los tratados con FTC/TDF. En ninguno de los 5pacientes con fracaso virológico y muestras disponibles para genotipificación (3 con MVC y 2 con TDF/FTC) se documentó la existencia de mutaciones de resistencia a los fármacos ni tampoco hubo cambio de tropismo en los tratados con MVC320.
Se han comunicado los resultados de un pequeño estudio piloto en pacientes sin TAR previo en el cual se ha estudiado la combinación de LPV/r+MVC QD en 17pacientes frente a LPV/r+TDF/FTC en 15pacientes, sin que se observaran diferencias en eficacia y seguridad entre ambos brazos en la semana 24321.
Recomendaciones- •
La monoterapia con un IP/r no se recomienda como tratamiento de inicio.
- •
No se deben usar pautas libres de ITIAN para el TAR de inicio (A-III). Esta recomendación se basa en los siguientes puntos:
La necesidad de TAR de inicio sin ITIAN es ahora menos acuciante que en el pasado dado que los ITIAN de elección actuales (FTC/TDF, 3TC/ABC) son más eficaces y seguros que los usados previamente.
De los estudios publicados se puede concluir que algunos FAR (por ejemplo, RAL) parecen mejor protegidos en combinación con 2ITIAN que en biterapia con un IP/r.
Ninguna pauta sin ITIAN ha demostrado hasta la fecha claras ventajas frente a la triple terapia desde el punto de vista de la eficacia y de la seguridad.
Varios ensayos clínicos han estudiado el momento idóneo de iniciar el TAR en pacientes con infecciones oportunistas diagnósticas de sida. El ACTG A5164322 incluyó a pacientes con infecciones oportunistas diferentes de la tuberculosis y demostró que el TAR precoz (administrado antes de las 2semanas del inicio del tratamiento del episodio oportunista) redujo de forma significativa (49%) el riesgo de progresión clínica (nueva infección oportunista o muerte) con respecto a diferir el TAR (entre 30 y 270días). Varios estudios han mostrado excelentes resultados del TAR precoz en pacientes con tuberculosis (véase más adelante), que no se han podido confirmar en la meningitis tuberculosa ni en la criptocócica. Dos ensayos clínicos efectuados respectivamente en Vietnam y en Uganda demostraron que el TAR precoz no redujo la mortalidad de la meningitis tuberculosa323 y la aumentó en la meningitis criptocócica324. Se desconocen las causas de tales resultados, pero probablemente el peor manejo clínico de estas 2infecciones oportunistas en países con escasos recursos y el SIRI en un compartimento cerrado como el SNC podrían justificarlo.
Con respecto a las neoplasias asociadas al sida (sarcoma de Kaposi, linfomas y carcinoma de cérvix), el TAR debe iniciarse de forma precoz evitando en lo posible en los pacientes que reciban quimioterapia la utilización de pautas con inhibidores de la proteasa debido a que aumentan su toxicidad.
Recomendaciones- •
En pacientes que se diagnostican simultáneamente de la infección VIH-1 y de una infección oportunista definitoria de sida, el TAR debe administrarse precozmente (en el primer mes e idealmente en las primeras 2semanas). (A-I)
- •
Los pacientes con meningitis criptocócica (y tuberculosa) deben seguir controles estrictos durante el tratamiento, ya que son un grupo difícil de tratar por la gravedad intrínseca de la infección y las consecuencias nocivas de la recuperación inmunológica (SIRI). (B-II)
Se entiende por simplificación del TAR el cambio de un esquema terapéutico que ha conseguido la supresión de la replicación viral por otro más sencillo que sigue manteniendo la supresión. Sus objetivos son mejorar la calidad de vida, facilitar la adherencia y prevenir o revertir algunos efectos adversos.
Con la simplificación se consigue reducir el número de comprimidos o la frecuencia de tomas, aprovechar la comodidad de las coformulaciones, eliminar las restricciones alimentarias, mejorar los efectos secundarios y reducir o eliminar las interacciones.
Esta estrategia empezó a utilizarse con la aparición de los ITINN, que permitían simplificar las pautas de tratamiento con IP de primera generación, que eran complejas y en general tóxicas.
Si bien en la actualidad la mayoría de pacientes inician tratamiento con pautas de TAR más simples, con los nuevos fármacos y las coformulaciones existen cada vez más opciones para simplificar a pautas más cómodas y mejor toleradas.
Se puede simplificar el TAR reduciendo el número de fármacos, el número de comprimidos o el número de tomas.
Reducción del número de fármacosLos primeros estudios de simplificación del TAR tuvieron como objetivo la reducción del número de FAR en lo que se denominó estrategia de inducción-mantenimiento, consistente en una primera fase de inducción con 3 o 4FAR seguida del mantenimiento con menos de 3fármacos.
Los resultados de los primeros ensayos en los que se evaluó esta estrategia a partir de pautas con IP de primera generación fueron desalentadores. Los estudios más recientes han explorado la simplificación a monoterapia con IP/r, tras un período de inducción con triple terapia con pautas que incluyen ITIAN, y el mantenimiento con triple terapia con 2ITIAN y un IP no potenciado (ATV) después de un período de inducción con IP/r (ATV/r).
Terapia de mantenimiento con un inhibidor de la proteasa potenciado con ritonavirSe ha explorado la estrategia de simplificar a monoterapia con LPV/r, tras un tiempo de inducción con triple terapia que incluye a este fármaco. El estudio OK04 incluyó a 205pacientes con CVP indetectable durante al menos 6meses (mediana: 28meses) que estaban tomando un TAR que incluía LPV/r asociado a 2ITIAN. Es un estudio aleatorizado, abierto, de no-inferioridad, que comparaba la estrategia de continuación del tratamiento triple frente a la monoterapia con LPV/r, considerando válida la reinducción con 2ITIAN si aparecía un rebrote viral. A las 48semanas el porcentaje de pacientes sin fracaso virológico fue del 90 y del 94%, respectivamente (diferencia: −4%; límite superior del IC95% para la diferencia: 3,4%, cumpliendo el criterio de no-inferioridad de la monoterapia, con o sin reintroducción de los ITIAN, frente al tratamiento triple). El porcentaje de pacientes con CVP<50copias/ml a las 48semanas (ITT), considerando las reinducciones como fallos, fue del 85% en el grupo de monoterapia y del 90% en el de continuación (p=0,31). Los episodios de viremia de bajo nivel, entre 50 y 500copias/ml, fueron más frecuentes en los pacientes tratados con monoterapia (4 frente a ninguno)325.
La terapia de mantenimiento con LPV/r fue evaluada en un ensayo clínico en el que se incluyeron 155pacientes sin TAR previo, a los que se aleatorizó en una proporción 2:1 a iniciar tratamiento con ZDV/3TC junto con LPV/r (n=104) o con EFV (n=51). Entre las 24 y 48 semanas de tratamiento y tras al menos 3 controles con CVP<50copias/ml, los pacientes que tomaban LPV/r pasaron a mantenimiento con LPV/r en monoterapia. Considerando fracaso a cualquier viremia detectable, a las 96 semanas de seguimiento el 48% de los pacientes en tratamiento con LPV/r y el 61% con EFV presentaban CVP<50copias/ml (IC95% de la diferencia: −29-4%; p=0,17). En un nuevo análisis en el que se incluyeron como respondedores a los pacientes que tras reintroducir los mismos ITIAN consiguieron de nuevo CVP<50copias/ml, el 60% de pacientes en tratamiento con LPV/r y el 63% con EFV respondieron al tratamiento (IC95%: −19-13%; p=0,73). Se objetivaron viremias de bajo nivel en los pacientes en monoterapia. Las alteraciones lipídicas de grado 3-4 fueron más frecuentes en el grupo del LPV/r326.
En estos 2estudios se pone de manifiesto la importancia del periodo durante el cual la CVP permanece indetectable antes del paso a monoterapia. Otros estudios, como el KalMo327, confirman estos resultados, pero en el caso del MOST328 hubo que suspenderlo precozmente. Se trataba de un ensayo clínico en el que se incluyeron pacientes con carga viral indetectable que se aleatorizaron a continuar con el mismo tratamiento o a pasar a monoterapia con LPV/r. El objetivo primario fue el fallo virológico en el SNC y/o en el tracto genital. El fallo virológico a nivel sanguíneo se definió como la presencia de 2cargas virales consecutivas >400células/μl. Se incluyeron 60 pacientes; 6 pacientes de la rama de monoterapia presentaron fallo virológico, todos ellos con una cifra nadir de linfocitos CD4+<200células/μl y en las primeras 24semanas de tratamiento. Además, 5 de ellos tenían una CVP elevada en LCR y 4, sintomatología neurológica. La reintroducción de los ITIAN bloqueó de nuevo la replicación viral. Los autores concluyeron que esta es una estrategia que no puede recomendarse de forma general y que hay que seleccionar a los pacientes.
La estrategia de mantenimiento con un IP/r ha sido también explorada con ATV/r329,330 y DRV/r331–340.
Se dispone de resultados preliminares de 2estudios piloto, abiertos, de un solo brazo de simplificación a ATV/r: el ACTG 5201329 y OREY330. En el estudio OREY se incluyeron 61pacientes que no habían tenido ningún fracaso previo (CVP<50copias/ml, al menos 24 semanas). En el análisis a las 48 semanas, el 79% de los pacientes tenía una CVP<400copias/ml. La reintroducción de la triple terapia fue generalmente satisfactoria (7 de 9pacientes). El desarrollo de mutaciones primarias a IP fue raro330. No se han realizado estudios aleatorizados con ATV/r en monoterapia.
Con DRV/r, se han realizado 2ensayos clínicos aleatorizados importantes, el MONET y el MONOI. En el estudio MONET331 se incluyeron 256pacientes en TAR con 2ITIAN y un ITINN o un IP/r sin experiencia previa a DRV/r ni historia de fallo virológico, con carga viral indetectable (<50copias/ml) durante al menos 6meses. Se les aleatorizó a DRV/r (800/100 QD) (n=129) en monoterapia o en combinación con 2ITIAN optimizados (n=127). Se trata de un estudio de no-inferioridad en el que el objetivo primario fue el tiempo hasta la pérdida de la respuesta virológica (TLOVR). Se definió el fracaso terapéutico por la presencia de 2determinaciones consecutivas de CVP>50copias/ml antes de la semana 48 o por la suspensión del tratamiento en estudio. En el análisis a la semana 48 se confirmó la no-inferioridad de la rama de DRV/r en monoterapia. En el análisis por ITT, considerando el cambio de tratamiento como un fracaso, el 85,3% de los pacientes que tomaban DRV/r frente al 84,3% de los que tomaban además 2ITIAN presentaban CVP indetectable (diferencia: −1; límite inferior del IC95%: −9,9). En cuanto a la aparición de resistencias, se detectó un paciente por rama con resistencia genotípica, pero no fenotípica, a DRV. En la semana 96 no pudo demostrarse la no-inferioridad de la rama de monoterapia en el análisis primario (CVP<50copias/ml, TLOVR, cambio=fracaso): 78% frente a 82% en los brazos de monoterapia y triple terapia, respectivamente (IC95%: −14,3-5,8%)332. Las diferencias en favor de la triple terapia se mantenían en un análisis efectuado en la semana 144: 69% frente al 65% (diferencia: −5,9%; IC95%: −16,9-5,1%)333. En general, la tasa de blips y discontinuaciones fue superior en la rama de monoterapia, si bien en la mayoría de los casos se observó una resupresión posterior con el mismo tratamiento o tras intensificación335. De hecho, a las 144 semanas, en el análisis por ITT sin considerar como fracaso la reintroducción de nucleósidos, la monoterapia cumplió el criterio de no-inferioridad respecto a la terapia triple (84% frente al 83,5%; IC95%: −8,7-9,7%)333.
En el estudio MONOI337 se incluyeron pacientes en TAR que presentaban CVP<400copias/ml durante los 18 meses previos y CVP<50copias/ml en el momento de la inclusión, sin historia de fallo virológico a IP y que no habían recibido nunca DRV/r. Constaba de 2fases: en la primera se introducía en el tratamiento DRV/r en la semana −8 y en la segunda se les aleatorizaba en proporción 1:1 a DRV/r (600/100mg BID) o a DRV/r (600/100 BID)+2ITIAN. El objetivo primario fue la proporción de pacientes que mantenían la respuesta virológica en la semana 48 (CVP<400copias/ml), aunque el seguimiento fue más prolongado. En la fase inicial se incluyeron 242 pacientes y se aleatorizaron 226. En el análisis por protocolo, DRV/r en monoterapia logró demostrar la no-inferioridad (delta 10%) frente a la triple terapia (94,1% frente al 99,0%; IC95%: −9,1-0,8%), pero no en el análisis por ITT (87,5% frente al 92%; IC95%: −11,2-2,1%). Se produjeron 3fracasos virológicos (>400copias/ml) en pacientes con DRV/r en monoterapia, sin detectarse mutaciones de resistencia para DRV, y con posterior resupresión viral tras la reintroducción de los ITIAN. Los factores asociados con fracaso virológico en los pacientes en monoterapia con DRV/r fueron la presencia de un blip inicial, un menor tiempo en TAR antes de la monoterapia y una adherencia menor del 100% durante la monoterapia338. En la semana 96, en el análisis por ITT, 91 de los 103 pacientes del brazo de la monoterapia (88%; IC95%: 81-94) y 87 de 104 en el brazo de la triple terapia (84%; IC95%: 75-90) presentaron CVP indetectable. Por protocolo fueron 95 y 90%, respectivamente. Entre la semana 48 y la 96 se produjeron 6fracasos virológicos: 2 en el brazo de la monoterapia y 4 en el de la triple terapia. Las causas principales fueron baja adherencia e interrupción del tratamiento. En los casos de fracaso en la monoterapia la reintroducción de los ITIAN hizo recuperar la CVP indetectable. No aparecieron mutaciones de resistencia a DRV ni acumulación de mutaciones a ITIAN en los pacientes que fracasaron339. Si bien 2 pacientes con CVP indetectable presentaron síntomas neurológicos (cefalea y crisis convulsiva en un paciente con epilepsia no tratada) con carga viral detectable en el LCR, el análisis citobioquímico del LCR en ambos casos fue normal. Este hallazgo cuestionaría la capacidad de la monoterapia para mantener la supresión del VIH-1 en el SNC. Recientemente se han presentado los datos de redistribución de grasa corporal, observándose menores cambios en la rama que no llevaba ITIAN, al igual que en otros estudios previos340.
Recientemente se han publicado los resultados de una revisión sistemática de los ensayos clínicos aleatorizados en los que se comparó la terapia de mantenimiento con IP/r solos frente a la triterapia en pacientes suprimidos341. Se analizaron los datos de 10ensayos clínicos, que incluyeron un total de 1.189pacientes. Con el criterio de valoración más conservador (CVP<50copias/ml en 2determinaciones consecutivas), el cociente de riesgos de supresión virológica a las 48 semanas de la monoterapia en comparación con la triterapia en el análisis por ITT fue de 0,94 (IC95%: 0,89-1,00) y en el análisis por protocolo de 0,93 (IC95%: 0,90-0,97). La reintroducción de los ITIAN en 44pacientes con fracaso virológico logró la resupresión viral en el 93% de los casos. Los autores concluyen que los pacientes virológicamente bien suprimidos con una adherencia excelente podrían optar por la monoterapia de mantenimiento con IP/r si se considera de gran importancia evitar las complicaciones a largo plazo de los nucleósidos que están recibiendo. El incremento absoluto del riesgo de fracaso virológico al año con la monoterapia se sitúa aproximadamente en el 10-13% en el peor de los casos, con una alta probabilidad de recuperar el control virológico cuando se reintroducen los ITIAN. La monoterapia no sería una opción para médicos y pacientes que no estén dispuestos a aceptar ese riesgo. La ausencia de resultados de seguimiento a largo plazo y la escasez de datos en relación con la replicación del VIH-1 en el SNC son también obstáculos para recomendar un uso más extendido de esta estrategia.
Terapia de mantenimiento con un inhibidor de la proteasa potenciado con ritonavir en combinación con lamivudinaUna nueva estrategia terapéutica, actualmente en evaluación, es la simplificación a una pauta de biterapia con 3TC y un IP/r en pacientes con supresión de la replicación viral bajo un régimen de triple terapia. Esta estrategia se ha explorado en un ensayo clínico piloto realizado recientemente en Italia342, en el cual se incluyeron 40pacientes tratados con un régimen de ATV/r+2ITIAN que tenían la CVP<50copias/ml durante al menos un periodo de 3meses. Alcanzaron la semana 48 del estudio 38 pacientes. La simplificación del tratamiento a ATV/r+3TC fue segura y eficaz. Dos pacientes presentaron fracaso virológico, en ambos casos sin mutaciones de resistencia y con resupresión de la replicación viral tras la reintroducción de los ITIAN. En ambos pacientes las concentraciones plasmáticas de ATV fueron indetectables. Más recientemente se ha comunicado que a las 96 semanas tras el inicio del estudio el 85% de los pacientes continuaba presentando CVP indetectable en el análisis por ITT)343. Esta estrategia se está evaluando actualmente en un ensayo clínico multicéntrico aleatorizado (Estudio SALT-GeSIDA 7011; clinicaltrials. gov: NCT01307488).
Terapia de mantenimiento con atazanavir no potenciado con ritonavirSe trata de una estrategia de inducción-mantenimiento que consiste en iniciar un tratamiento con ATV/r y posteriormente suspender RTV con el fin de evitar los efectos secundarios que produce este, incluso a dosis bajas. Los estudios más significativos son el INDUMA y el ARIES. El INDUMA344 es un estudio aleatorizado, abierto, multicéntrico de no-inferioridad, en el que se incluyeron 252pacientes sin TAR previo, que iniciaron un tratamiento de inducción con 2AN+ATV/r. Entre las semanas 26 y 30, los que tenían CVP<50 copias/ml y seguían con el tratamiento (n=172) se aleatorizaban 1:1 a continuar con el mismo tratamiento o a recibir 2ITIAN+ATV 400mg QD, con un seguimiento de 48 semanas. La variable primaria fue la proporción de pacientes que mantenían CVP<50copias/ml a las 48 semanas. La mitad de los pacientes llevaban ABC/3TC como pareja de ITIAN. En la semana 48 la rama de ATV demostró no-inferioridad con respecto a la de ATV/r (la proporción de pacientes con CVP<50copias/ml fue 75% (n=85) en la rama de ATV/r y 78% (n=87) en la rama de ATV (diferencia: 2,9; IC95%: −9,8-15,5). En cuanto al perfil de seguridad, se produjeron menos casos de hiperbilirrubinemia y de dislipidemia en la rama de ATV. Los autores concluyeron que esta es una opción para pacientes que no toman tenofovir. No se detectaron resistencias frente a IP en los pacientes que presentaron fracaso virológico de ninguno de ambos brazos.
De forma similar, el estudio ARIES345,346 es un ensayo clínico abierto, multicéntrico, de no-inferioridad, en el que se incluyó a pacientes sin TAR previo a los que se pautó ABC/3TC+ATV/r y posteriormente, en la semana 36, se aleatorizaron en razón 1:1 a seguir con el mismo régimen o a suspender RTV durante 48 semanas, si la CVP era <50copias/ml y previamente no habían presentado fallo virológico. Se aleatorizaron 419pacientes, incluyéndose en el análisis a los 379 (90%) que completaron las 48 semanas de seguimiento. El objetivo primario fue la proporción de pacientes con CVP<50copias/ml en la semana 48 (TLOVR). Los autores observaron que la eficacia de ambos tratamientos es similar, independientemente de la CVP basal (86% frente al 81% en los brazos de ATV y ATV/r, respectivamente (IC95%: −1,75-12,48), siendo infrecuente el fallo virológico (2%). La eficacia se mantuvo en el análisis efectuado a las 144 semanas, y tanto el perfil lipídico como los valores de bilirrubina fueron más favorables en el grupo de simplificación a ATV347.
La simplificación a una pauta con ATV no potenciado en combinación con ABC/3TC en pacientes HLA-B*5701-negativos con supresión virológica (<75copias/ml) durante al menos 6meses con un régimen de ATV/r+TDF/FTC se ha evaluado recientemente en el estudio ASSURE, un ensayo clínico abierto en el que 296pacientes fueron aleatorizados en proporción 2:1 a recibir ATV+ABC/3TC (n=199) o a continuar con la pauta previa de ATV/r+TDF/FTC (n=97). El objetivo principal de este estudio fue demostrar la no-inferioridad (delta: 12%) de ATV+ABC/3TC, y su variable principal la proporción de pacientes con CVP indetectable (<50copias/ml) en la semana 24 mediante un análisis TLOVR. En una comunicación preliminar de los resultados se ha informado que en la semana 24 presentaban CVP indetectable el 86,9% de los pacientes de la rama de ATV+ABC/3TC frente al 86,6% de los que continuaron recibiendo ATV/r+TDF/FTC (IC95%: −7,97 a −8,64%). Así pues, la pauta de ATV+ABC/3TC cumplió el criterio predefinido de no-inferioridad348. Además, en el grupo de ATV+ABC/3TC se observó una mejoría significativa de los biomarcadores del metabolismo óseo (fosfatasa alcalina, PTH, telopéptido C y osteocalcina)348.
Reducción del número de comprimidos y/o de dosisLa reducción del número de comprimidos y/o de dosis puede conseguirse sustituyendo el IP por un FAR de otro grupo o utilizando fármacos que se presentan coformulados. Se ha evaluado la sustitución del IP/r por EFV, NVP o ABC y también por otro IP/r que se pueda administrar en QD.
Simplificación con efavirenzSe han realizado múltiples estudios evaluando esta estrategia, muchos de ellos no comparativos. La mayoría de los ensayos comparativos incluyeron un escaso número de pacientes y casi todos se realizaron a partir de pautas con IP de primera generación. En general, esta estrategia fue comparable o mejoró los resultados virológicos en relación con la pauta comparadora con IP no potenciados de primera generación349–353.
En un estudio más reciente, que incluyó 262pacientes que realizaban TAR basado en IP/r y con CVP<50copias/ml, se cambió el mismo a EFV en una pauta QD (EFV+ddI+3TC) o BID (EFV con los ITIAN de base). El estudio fue abierto, aleatorizado y de no-inferioridad y su objetivo primario fue el mantenimiento de supresión virológica en la semana 48. La pauta QD no fue inferior a la BID. En general, ambas pautas se asociaron con una baja tasa de fallo virológico y con una mejora importante de la satisfacción de los pacientes, de la adherencia y de la calidad de vida. Los autores concluyeron que el cambio de un IP/r a EFV es seguro y bien tolerado354.
Simplificación con nevirapinaComo en el caso de EFV, la mayoría de los ensayos en los que se ha evaluado esta estrategia han incluido un escaso número de pacientes y casi todos se realizaron a partir de pautas con IP de primera generación En general, la eficacia terapéutica a las 24-48semanas fue similar a la pauta de continuación con el IP no potenciado, y en la mayoría se observó una mejoría del perfil lipídico355–357. En uno de los estudios la eficacia virológica fue mayor en el grupo de simplificación358. En un estudio español reciente (MULTINEKA) se aleatorizó a 67 pacientes en TAR estable y con CVP<50copias durante al menos 6 meses a recibir LPV/r con NVP o con 2ITIAN. A las 48 semanas de tratamiento no se detectó fracaso virológico en ninguno de los pacientes. Se describió un posible beneficio en la toxicidad mitocondrial en los pacientes aleatorizados a NVP359.
NVP está contraindicada en mujeres con >250linfocitos CD4+/μl y en varones con >400linfocitos CD4+/μl por riesgo de hepatotoxicidad grave. Sin embargo, varios estudios independientes360–365 han coincidido en señalar que cuando se introduce NVP como una estrategia de simplificación en pacientes pretratados con cifras de linfocitos CD4+ por encima de esos límites, el riesgo de toxicidad es mucho menor que en los pacientes que previamente no han realizado TAR. Estos resultados son suficientemente consistentes y han llevado a modificar la ficha técnica del producto (véase ficha técnica).
Simplificación con abacavir y otros análogos de nucleósidoLos resultados de los ensayos clínicos que han evaluado esta estrategia no han sido concluyentes y han puesto de manifiesto el riesgo de fracaso virológico que la misma lleva asociado.
En el estudio TRIZAL y en otros la eficacia fue similar, pero se observó una mayor incidencia de fracasos en pacientes que previamente habían recibido tratamientos subóptimos366–368. En el estudio TRIZEFAL 209pacientes fueron aleatorizados a recibir ZDV/3TC/ABC en combinación fija con EFV o LPV/r, durante 24-36 semanas. Los pacientes que alcanzaron una CVP<50copias/ml en ambos brazos continuaron con ZDV/3TC/ABC. A las 72 semanas, en el análisis por ITT, solo el 31 y el 43%, respectivamente, mantuvieron la CVP indetectable; el 34 y el 25% de ellos cambiaron la pauta por toxicidad369.
En distintos estudios se ha puesto de manifiesto la existencia de un riesgo elevado de fracaso terapéutico y desarrollo de mutaciones a ITIAN cuando se utilizan pautas de simplificación con 3TC/ABC+TDF370 y 3TC+ddI+TDF, por lo que, pese a su sencillez, ambos regímenes se desaconsejan como estrategia de simplificación.
Comparación directa de efavirenz, nevirapina y abacavir en simplificaciónEn el ensayo NEFA se aleatorizaron 460pacientes en tratamiento con 2ITIAN+un IP, y con CVP<200copias/ml durante al menos 6 meses371, a recibir NVP (n=155), EFV (n=156) o ABC (n=149). El 50, el 58 y el 46% de los pacientes, respectivamente, habían recibido previamente regímenes de TAR subóptimos con uno o 2ITIAN. La eficacia terapéutica (CVP<200copias/ml) por ITT a las 48 semanas fue similar en los 3grupos (77, 72 y 77%; p=NS). Se produjeron más fracasos virológicos con ABC (6% con NVP, 4% con EFV y 12% con ABC; p<0,05), sobre todo en los pacientes que habían recibido previamente tratamientos subóptimos. Estos resultados se han confirmado a los 3años372. El análisis genotípico de las cepas de los pacientes con fracaso virológico evidenció un mayor número de mutaciones de resistencia asociadas a ITIAN entre los que recibieron ABC368. El número de pacientes que suspendió el tratamiento por efectos adversos fue menor en el grupo de ABC (17% con NVP, 17% con EFV y 6% con ABC; p<0,01). La simplificación a cualquiera de los ITINN, sobre todo a NVP, produjo beneficios en el perfil lipídico, con reducción del colesterol no-HDL con ABC. Los niveles de triglicéridos se redujeron en los 3brazos. Los marcadores de resistencia a la insulina mostraron una tendencia a la mejoría. Sin embargo, no mejoraron las alteraciones en la distribución de la grasa373,374.
Simplificación con atazanavirEl ATV es un IP de dosificación QD con buen perfil metabólico que ha permitido una nueva estrategia de simplificación en la que un IP sustituye a otro.
El SWAN es un estudio abierto en fase IIIb, en el que 419pacientes en tratamiento estable con IP (potenciado o no) y con CVP indetectable se aleatorizaron 2:1 a ATV 400mg QD (en caso de tomar TDF se pautó ATV/r 300/100mg) (n=278) o a continuar con el IP (n=141). A la semana 48 el fracaso virológico fue menor en los que se simplificó el TAR a pautas con ATV (7% frente a 16%; p<0,01)375. En cuanto a la seguridad, las suspensiones del tratamiento fueron más frecuentes en los pacientes del grupo control que en los que simplificaron a ATV (21% frente al 34%; p<0,01) y el perfil lipídico fue mejor en el grupo que simplificó a ATV. Los datos de los estudios ATAZIP y SIMPATAZ confirman la seguridad y la eficacia de esta estrategia de simplificación, en este caso cambiando LPV/r por ATV/r376,377.
El estudio REAL es un ensayo clínico en el que se incluyó a 201pacientes en TAR estable durante al menos 12semanas con pautas que contenían un IP administrado BID, con CVP indetectable y lipohipertrofia, a los que se aleatorizaba a continuar con el mismo tratamiento o cambiar el IP a ATV/r (300/100mg). El control inmuno-virológico se mantuvo, se observó una mejoría del perfil lipídico en la rama de ATV pero no se objetivaron diferencias en cuanto a la composición corporal tras 96 semanas de seguimiento378.
Otra estrategia es la evaluada en el estudio AI424-067379, un ensayo abierto en el que se incluyeron 246pacientes en tratamiento con un IP/r y con hiperlipidemia y CVP<50copias/ml. Se aleatorizaron los pacientes a cambiar a ATV (400mg) en el día 1 (cambio inmediato) o mantener su tratamiento y cambiar a ATV (400mg) a las 24semanas (cambio retrasado). A las 12semanas, los pacientes que cambiaron a ATV mostraron una mejoría significativa de las cifras de colesterolLDL (−15 y +1%, respectivamente; p<0,0001). Los 2grupos tuvieron una eficacia virológica similar a las 48semanas, aunque la frecuencia de fracaso terapéutico (CVP>50copias o discontinuación del tratamiento) superó el 20% en ambos. Los autores concluyeron que el cambio inmediato o diferido de un IP potenciado a ATV no potenciado en pacientes con hiperlipidemia se asocia con mejoría en los parámetros lipídicos sin pérdida de la supresión virológica.
Simplificación a pautas de administración una vez al díaEl cambio a un régimen QD es una de las formas de simplificación más atractivas en pacientes virológicamente suprimidos con pautas más complejas. Los resultados de un metaanálisis sugieren que con las pautas QD la adherencia es mejor que con los regímenes BID380. El cambio a una pauta QD en pacientes que han alcanzado la supresión virológica con pautas más complejas se ha evaluado en varios ensayos clínicos.
En un ensayo clínico realizado en Francia se aleatorizaron 355pacientes virológicamente suprimidos con pautas que contenían IP (la mayoría no potenciados) a seguir con su TAR o cambiar a una terapia QD (ddI+FTC+EFV). En la semana 48 seguían con CVP indetectable el 87% de los pacientes de la rama QD y el 79% de los que no habían cambiado el tratamiento (p<0,05)381. En un estudio español no aleatorizado se analizó a 169pacientes virológicamente suprimidos con TAR, de los que 84 siguieron con el mismo régimen de TAR y 85 cambiaron a una pauta con ddI+TDF+NVP QD; la eficacia virológica de la pauta QD fue buena (86% frente al 76% por ITT), pero el número de linfocitos CD4+ disminuyó en la rama QD (decremento medio de 95células/μl)382.
Otros ensayos clínicos han confirmado que la combinación ddI+TDF es eficaz virológicamente pero que con ella la recuperación de linfocitos CD4+ es menor o incluso se produce una caída del número de CD4+ aunque la CVP esté suprimida. Este descenso era más patente cuando se administraban las dosis estándar (plenas) de ddI383. Los regímenes de administración QD que contengan la combinación ddI+TDF deben evitarse. De hacerlo, se debe reducir la dosis de ddI a 250mg/día en pacientes con más de 60kg de peso y a 200mg/día en pacientes por debajo de dicho peso.
Con la aparición de las combinaciones a dosis fijas de ITIAN administrados QD las pautas de TAR se han simplificado aún más. Estas pautas se emplean cada vez más en el TAR de inicio, y diversos ensayos clínicos han demostrado que son eficaces y seguras en simplificación. El SWEET es un ensayo clínico en el que 234pacientes en tratamiento durante al menos 6meses con ZDV/3TC (coformulados)+EFV y con CVP<50copias/ml se aleatorizaron a TDF/FTC (coformulado)+EFV o a seguir con el mismo tratamiento. A las 24 semanas se observó una mejoría en las cifras de hemoglobina y en el perfil lipídico en la rama de TDF/FTC, manteniéndose la respuesta al tratamiento (CVP<50copias/ml; 93% frente al 88%; p=0,26). A las 48 semanas384 el 5% de los pacientes que continuaron con ZDV/3TC y el 3% de los que cambiaron a TDF/FTC discontinuaron el tratamiento por efectos adversos. No se objetivaron diferencias estadísticamente significativas de eficacia virológica entre las 2ramas (85% de los pacientes que continuaron el tratamiento frente al 88% de los que lo cambiaron tenían una CVP<50copias/ml en el análisis por ITT). En un subestudio de 100pacientes a los que se realizó DEXA del tejido graso se observó que la cuantía de grasa se mantuvo o aumentó en los pacientes que cambiaron a TDF/FTC, pero disminuyó en el grupo que continuó con ZDV/3TC (diferencia media de 448g; IC95%: 57-839g; p=0,025). No se observaron diferencias entre los 2grupos en cuanto a toxicidad renal. Los investigadores concluyeron que el cambio de ZDV/3TC por TDF/FTC en pacientes en tratamiento con EFV y respuesta virológica mantenida es seguro desde el punto de vista virológico y se asocia con un incremento de la cifra de hemoglobina y una mejoría de los parámetros lipídicos y de la distribución de la grasa corporal.
Los beneficios del cambio de ZDV/3TC a TDF/FTC fueron confirmados en un estudio español, el ensayo RECOMB. En este ensayo abierto se incluyó a 80pacientes con CVP<50copias/ml en tratamiento con ZDV/3TC (+un IP o un ITINN), a los que se aleatorizó a seguir con ZDV/3TC o cambiar a TDF/FTC. En la semana 24, el 85% de los pacientes en tratamiento con TDF/FTC presentaba una CVP<50copias/ml frente al 80% de los que recibían ZDV/3TC (p=0,77)385. Se observó además un incremento significativo de grasa en las extremidades en los pacientes cuya masa grasa basal era inferior a 7,2kg. Por último mejoraron las cifras de colesterolLDL en los pacientes de la rama de TDF/FTC. A las 72 semanas el 90% de los pacientes que recibieron TDF/FTC frente al 83% de los tratados con AZT/3TC presentaban una CVP<50copias/ml (p=0,52); la mediana de incremento de la cifra de linfocitos CD4+ fue similar. Además, en el grupo de TDF/FTC se observó un incremento progresivo de grasa en las extremidades, sobre todo si el índice de masa corporal basal era superior a 25kg/m2 y llevaban más de 5años con AZT/3TC. Otro ensayo similar es el TOTEM, en el que se incluyó a 91pacientes con CVP<400copias/ml y dislipidemia, a los que se aleatorizó a seguir con los mismos ITIAN (más del 50% de ellos con ZDV/3TC) o cambiar a TDF/FTC. En los pacientes que cambiaron se observó una mejoría significativa en el perfil lipídico (triglicéridos y colesterolLDL) a las 12semanas385.
En el estudio BICOMBO se analizaron la eficacia y la seguridad de la simplificación con ABC/3TC frente a TDF/FTC. Se trataba de un estudio aleatorizado y abierto que incluyó a 335pacientes que recibían tratamiento con un régimen que incluía 3TC, con supresión virológica durante al menos 6meses y que fueron aleatorizados a sustituir los ITIAN por las combinaciones coformuladas de ABC/3TC (n=167) o TDF/FTC (n=168)386. El estudio se diseñó para evaluar la no-inferioridad de ambas combinaciones respecto a fracaso terapéutico o virológico. En el grupo de TDF+FTC, la proporción de fracaso terapéutico fue del 13,3%, frente al 19,2% en el de ABC+3TC, no demostrándose la no-inferioridad de ABC/3TC frente a TDF/FTC (IC95%: −2-14%). Sin embargo, ABC/3TC demostró la no-inferioridad frente a TDF/FTC en el objetivo de fracaso virológico (2,4% frente al 0%; IC95%: 0,05-6%). Las suspensiones por efectos adversos fueron del 10% en el grupo de ABC/3TC frente al 5% en el grupo de TDF+FTC (p=0,004). En cuanto al perfil lipídico, las reducciones en las cifras de colesterol total, colesterolHDL, colesterolLDL y triglicéridos fueron mayores en la rama de TDF/FTC, y en un subestudio metabólico los pacientes que recibieron ABC/3TC presentaron un perfil lipídico más aterogénico387. No se hallaron diferencias significativas entre las 2ramas en lo que respecta a las concentraciones de los biomarcadores de riesgo cardiovascular388.
El incremento en la cantidad de grasa periférica y las alteraciones en la función renal o la densidad mineral ósea fueron similares. La toxicidad hepática fue muy baja en ambos grupos. La determinación previa del HLA-B*5701 podría haber modificado estos resultados.
El diseño del estudio STEAL fue similar al del BICOMBO, pero se determinó previamente el HLA-B*5701. En este estudio abierto, programado para 96 semanas, se incluyeron 357pacientes HLA-B*5701 negativos con CVP<50copias/ml, a los que se aleatorizó a sustituir la pareja de ITIAN por los combos ABC/3TC o TDF/FTC. El objetivo primario fue el fallo virológico (2determinaciones consecutivas >400copias/ml en ITT). Los objetivos secundarios incluían sida, muerte, efectos adversos, episodios graves no relacionados con sida, alteraciones metabólicas y composición corporal. El fallo virológico fue muy poco frecuente (5,6% en pacientes con ABC/3TC y 3,9% en los que tomaban TDF/FTC; IC95%: −2,8-6,1%; p=0,62). La combinación TDF/FTC se asoció con menos episodios no-sida que la formada por ABC/3TC (1,2 frente a 4,8 episodios por 100pacientes-año; HR: 0,24; IC95%: 0,08-0,73; p=0,012), debido sobre todo a una menor incidencia de episodios cardiovasculares. En el grupo de TDF/FTC se produjo un descenso significativo en la densidad mineral ósea (media de la diferencia en la puntuación t de la cadera: 0,16; IC95%: 0,08-0,23; p<0,001), aunque no hubo una mayor incidencia de fracturas389. De forma similar al estudio BICOMBO, se realizó también un estudio exhaustivo de biomarcadores de riesgo cardiovascular y no se encontraron diferencias entre los 2grupos comparativos390.
La simplificación a una pauta QD con TDF/FTC/EFV coformulados fue evaluada en un ensayo clínico abierto, aleatorizado, en pacientes con diferentes pautas de TAR virológicamente controlados391. Se incluyeron en él pacientes que presentaban una CVP<200copias/ml durante al menos 3meses. Se estratificaron en función de que estuvieran tomando ITINN o IP y se aleatorizaron 2:1 a simplificar el tratamiento a TDF/FTC/EFV coformulados (una tableta única diaria) o a seguir con el mismo tratamiento. Se evaluaron la eficacia y la seguridad en el momento basal y a las semanas 4, 12, 24, 36 y 48. Además, se valoró la calidad de vida y las preferencias de los pacientes en cuanto a los FAR. De los 300pacientes incluidos, 97 continuaron con el mismo tratamiento. A las 48 semanas, el 89% de los pacientes con TDF/FTC/EFV frente al 88% de los que continuaron con el mismo tratamiento presentaban una CVP<200copias/ml (análisis TLOVR; IC95%: −6,7-8,8%), indicando no-inferioridad de la rama de TDF/FTC/EFV. El 87% de los pacientes que cambiaron a TDF/FTC/EFV frente al 85% de los que no cambiaron el tratamiento presentaban una CVP<50copias/ml (IC95%: −5,9-11,1%). Las tasas de discontinuación fueron similares, aunque la suspensión por efectos adversos fue más frecuente en la rama de TDF/FTC/EFV (5% frente al 1%), sobre todo por sintomatología relacionada con el SNC. No se objetivaron diferencias en la tasa de filtrado glomerular, ni en la adherencia, pero sí una mejoría en la cifra de triglicéridos en la rama de TDF/FTC/EFV (−20 frente a −3 mg/dl; p=0,035). En cuanto a los datos de calidad de vida392, los pacientes en los que se simplificó el TAR referían una mejoría de muchos de los síntomas relacionados con el VIH, encontraban el nuevo tratamiento más fácil de tomar y de seguir, y lo preferían frente a los regímenes de TAR previos.
En el estudio NODY393 se evaluaron la eficacia y la seguridad de la simplificación de NVP desde una pauta convencional de 2veces al día a la de una vez al día. Se trataba de un estudio de 48 semanas, abierto, en el que se incluyeron 298pacientes estables que estaban tomando NVP 2veces al día durante al menos 12-18 semanas y presentaban una CVP<50copias/ml. Estos pacientes se aleatorizaron a continuar con el mismo tratamiento o a tomar nevirapina QD. El objetivo primario fue la seguridad hepática del tratamiento QD, para lo cual se analizó la proporción de pacientes con ALT/AST de grado 3-4, y los objetivos secundarios fueron el desarrollo de hepatitis clínica y la eficacia inmuno-virológica y clínica. Se definió la no-inferioridad con un delta del 10% para el desarrollo de hepatotoxicidad. El estudio demostró no-inferioridad por protocolo de NVP QD frente a la pauta inicial de 2veces al día.
La simplificación a una pauta QD con una coformulación a dosis fijas de RPV/TDF/FTC ha sido evaluada en el estudio SPIRIT, un ensayo clínico abierto, aleatorizado, en pacientes con supresión de la replicación viral (CVP<50copias/ml) durante al menos 6meses con un régimen de un IP/r+2ITIAN394. No se incluyeron en él pacientes que hubiesen recibido previamente ITINN o tuviesen resistencia genotípica conocida frente a estos fármacos, ni que hubiesen sido tratados previamente con más de 2pautas distintas de TAR. Los 476pacientes incluidos fueron aleatorizados a razón 2:1 a recibir RPV/TDF/FTC o a continuar con la pauta previa. El 37% de los pacientes estaban recibiendo ATV/r; el 33%, LPV/r, y el 20%, DRV/r. El objetivo principal del estudio fue demostrar la no-inferioridad (delta: 12%) de la simplificación a RPV/TDF/FTC, y su variable primaria la proporción de pacientes con CVP<50copias/ml en la semana 24 (análisis Snapshot de la FDA). En el análisis a las 24 semanas presentaban CVP indetectable el 93,7% de los pacientes del grupo tratado con RPV/TDF/FTC frente al 89,9% de los que continuaron con el régimen del IP/r (diferencia: 3,8%; IC95%: −1,6-9,1%), cumpliendo así la pauta QD de RPV/TDF/FTC el criterio de no-inferioridad predefinido. En el grupo de RPV/TDF/FTC se registraron menos acontecimientos adversos gastrointestinales y el perfil lipídico fue más favorable que en los que continuaron con IP/r, observándose diferencias estadísticamente significativas (p<0,001) a favor de RPV/TDF/FTC en todos los parámetros, incluyendo el cociente colesterol total/colesterolHDL394.
Otros tipos de simplificaciónCon el desarrollo de fármacos de nuevas familias, en estos últimos años se han realizado ensayos de simplificación en pacientes multitratados, en los que se han evaluado la eficacia y la seguridad de la sustitución de ENF (parenteral) por otros FAR de administración oral. En la mayoría de los estudios se ha sustituido ENF por RAL, manteniéndose la eficacia del tratamiento395–399. Aunque la mayoría de ellos fueron estudios observacionales, se han comunicado los resultados de un ensayo clínico400 en el que se incluyó a 170pacientes con VIH-1 resistente a las 3familias clásicas de FAR, que tenían una CVP<400copias/ml durante al menos 3meses bajo tratamiento con ENF. Se les aleatorizó a razón 1:1 a seguir con ENF o a cambiar a RAL. El objetivo primario fue la proporción acumulada de pacientes con fallo virológico definido como CVP≥400 copias/ml hasta la semana 24. Se observó fallo virológico en un paciente por rama. La conclusión de los autores fue que el cambio a RAL es eficaz y bien tolerado a las 24semanas y ofrece la ventaja de la simplicidad y la tolerabilidad en relación a ENF. También se ha explorado con éxito la sustitución de ENF por otros fármacos, como ETR401.
La simplificación de pautas con IP/r a RAL fue evaluada en los estudios SWITCHMRK1 y 2402 y en el SPIRAL403. En los estudios SWITCHMRK, 2ensayos clínicos paralelos, multicéntricos, aleatorizados a doble ciego, se incluyó a pacientes virológicamente controlados y estables con 2ITIAN y LPV/r. No se excluyó a los pacientes que habían fracasado con otros regímenes terapéuticos, siempre que en el momento de la inclusión en el estudio presentaran una CVP<50copias/ml desde al menos 3meses antes. Los pacientes se aleatorizaron 1:1 a mantener LPV/r o cambiar a RAL, con la misma base de ITIAN. Los objetivos primarios fueron: porcentaje de cambio en las cifras de lípidos en la semana 12, proporción de pacientes con CVP<50copias/ml en la semana 24 (margen prefijado de no-inferioridad: −12%) y frecuencia de acontecimientos adversos a las 24semanas. Se aleatorizaron 707pacientes y se trató a 702 (350 con RAL y 352 con LPV/r). El cambio a RAL fue bien tolerado y se asoció con una mejoría significativa de los parámetros lipídicos, pero no demostró la no-inferioridad desde el punto de vista virológico en la semana 24, pues 293 de 347pacientes (84,4%) frente a 319 de 352 (90,6%) tuvieron una CVP<50copias/ml en los grupos de RAL y LPV/r, respectivamente; la diferencia observada entre los 2tratamientos en el análisis por ITT fue de −6,2% (IC95%: −11,2 a −1,3). Probablemente estos resultados fueron debidos a los criterios de inclusión de los pacientes, y más en concreto al hecho de permitir la entrada en el estudio de pacientes con fracaso virológico previo cuando se plantea sustituir LPV/r por RAL, que tienen distinta barrera genética. La enseñanza de este estudio es que se debe elegir muy bien a los pacientes cuyo tratamiento se simplifica y la estrategia de simplificación a seguir.
En el estudio SPIRAL403 los resultados fueron muy distintos. En este caso se trataba de un ensayo clínico abierto, multicéntrico, de no-inferioridad (con un límite inferior del IC95% de −12,5%), de 48 semanas de seguimiento, en el que se incluyó a 273pacientes en tratamiento con un régimen basado en IP/r, con CVP<50copias/ml durante al menos 6meses, a los que se aleatorizó 1:1 a seguir con el mismo tratamiento o cambiar el IP/r a RAL. El objetivo primario fue la proporción de pacientes sin fallo virológico a las 48 semanas. En este caso sí que se cumplió el criterio de no-inferioridad, pues a las 48 semanas el 89,2% de pacientes con RAL y el 86,6% de pacientes con IP/r no habían experimentado fracaso virológico (diferencia: 2,6%; IC95%: −5,2-10,6). Además, el cambio a RAL se asoció con una mejoría significativa del perfil lipídico y de diferentes biomarcadores asociados con arteriosclerosis (inflamación, resistencia insulínica e hipercoagulabilidad)404–406. La diferencia entre los resultados de ambos estudios se ha explicado por el diseño y especialmente por la duración de la supresión virológica previa al cambio de tratamiento, que fue mayor en el SPIRAL.
También se ha explorado la posibilidad de utilizar RAL una vez al día como simplificación. Esta estrategia se ha evaluado en el estudio ODIS407, un ensayo clínico en el que se incluyó a 222pacientes en tratamiento con un IP/r+TDF/FTC o ABC/3TC y una CVP<50copias/ml durante al menos las 24semanas precedentes, a los que se aleatorizó 1:2 a sustituir el IP/r por RAL 400mg BID (n=73) o por RAL 800mg QD (n=149). Además, a la semana 12, si no existían diferencias entre las 2ramas en cuanto a eficacia, los pacientes incluidos en la rama BID podían ser de nuevo aleatorizados a seguir BID (n=35) o a pasar a QD (n=38). Los datos publicados corresponden a las 24 semanas. Un total de 13 pacientes (5,9%) desarrollaron fracaso virológico, 12 (6,4%) en el grupo QD y uno (2,9%) en BID. La frecuencia de fracaso virológico en pacientes con resistencia a los ITIAN previos fue del 16,2% (12/74), mientras que fue inferior al 1% (1/148) en el resto (p<0,001). La frecuencia de fracaso virológico fue particularmente elevada en pacientes con mutaciones de resistencia a los ITIAN. Estos resultados ponen de nuevo de manifiesto la importancia de elegir adecuadamente los pacientes a los que se va a simplificar. Este mismo análisis es discutido en un editorial que acompaña la publicación del estudio SWITCHMRK408.
Recomendaciones- •
Es importante seleccionar muy bien los pacientes a los que se desea simplificar el TAR y la estrategia a seguir. La simplificación no se puede realizar a costa de la pérdida de eficacia virológica. Solo se puede plantear una simplificación si no ha existido fracaso previo y si se utilizan fármacos plenamente activos para mantener el éxito virológico. (A-I)
- •
Los pacientes con supresión vírica prolongada (≥6meses) y buena adherencia (>90%) son los mejores candidatos a simplificación. (B-II)
- •
No se recomienda simplificar de un IP/r a ABC si el paciente ha recibido previamente tratamientos subóptimos con ITIAN. (A-I)
- •
Está contraindicada la simplificación a ABC asociado a TDF y 3TC o a TDF y ddI. (A-II)
- •
En pacientes con riesgo cardiovascular elevado la simplificación a NVP o RAL puede añadir ventajas metabólicas. (B-I)
- •
En pacientes en su primera pauta terapéutica con IP/r y con CVP indetectable se puede sustituir el IP/r por EFV, RPV, NVP o ATV (A-I) y simplificar a pautas QD como EFV+TDF+3TC (o FTC), RPV+TDF+FTC, EFV+ddI+3TC (o FTC), ATV/r+TDF/FTC o ATV+ABC/3TC. (A-I)
- •
El cambio a un régimen de un único comprimido añade ventajas adicionales de adherencia. (A-I)
- •
La sustitución de ENF por RAL en pacientes suprimidos virológicamente se ha demostrado eficaz y segura. (A-I)
- •
Las monoterapias con DRV/r (QD) o LPV/r (BID) deben usarse de forma excepcional y siempre y cuando el paciente presente toxicidad o intolerancia a los ITIAN. Además, esta estrategia debe restringirse a pacientes que cumplan todas las siguientes condiciones: a)ausencia de fracaso previo con IP; b)CVP<50copias/ml durante al menos 6meses, y c)haber demostrado una excelente adherencia (C-I). Los pacientes con una cifra nadir de linfocitos CD4+<100células/μl pueden tener un mayor riesgo de fracaso con esta pauta terapéutica, por lo que su indicación en este contexto debe valorarse con especial cuidado.
- •
Otras posibles simplificaciones deben realizarse en el seno de ensayos clínicos, no en la práctica clínica. (A-III)
El fracaso del TAR se puede definir desde 2puntos de vista: virológico e inmunológico. Se entiende por fracaso inmunológico la incapacidad de conseguir y mantener una cifra adecuada de linfocitos CD4+ a pesar de haber conseguido la supresión virológica duradera en plasma (CVP<50copias/ml de VIH-1). Esta situación se conoce asimismo como respuesta inmunológica discordante. En estos casos no existe evidencia que demuestre que un cambio de régimen de TAR consiga una mayor recuperación de linfocitos CD4+, por lo que no se recomienda cambiar el TAR, salvo cuando incluya combinaciones que se asocian específicamente a un descenso de linfocitos CD4+ (por ejemplo, ZDV, ZDV/3TC, o TDF+ddI).
En este capítulo se analiza solo el fracaso virológico. Se define la supresión virológica como el mantenimiento de la CVP<50copias/ml, aunque algunas técnicas tienen establecido el umbral de detección en 20copias/ml. Se define fracaso virológico, por tanto, como 2CVP confirmadas >50copias/ml (a partir de las 24semanas del inicio del TAR).
Existe cierta incertidumbre sobre el significado de valores de CVP comprendidos entre 50 y 200copias/ml. Por debajo de 200copias/ml la tasa de éxito de la amplificación de los estudios de resistencia o de tropismo es sensiblemente menor. La concentración de las muestras mediante 3ciclos de ultracentrifugación y la utilización de un mayor volumen de partida (3ml) han demostrado que se puede conseguir una elevada tasa de resultados válidos en los genotipos, por lo cual se recomienda hacer ambas cosas en estos casos409. Por otra parte, con considerable frecuencia se obtienen resultados detectables de CVP cerca del límite de detección de la técnica y por debajo de 200copias/ml en sujetos que posteriormente mantienen nuevamente la supresión virológica sin cambio alguno en su TAR410. Por todo ello, algunos autores han propuesto definir fracaso virológico como 2valores consecutivos de CVP>200copias/ml4. De hecho, un análisis retrospectivo de 2estudios de los ACTG realizados en 1.479sujetos sin TAR previo concluyó que situar el umbral de fracaso virológico en más de 200 en lugar de en más de 50copias/ml era más útil en la práctica, puesto que la mayoría de sujetos con CVP comprendida entre 50 y 200copias/ml quedaron nuevamente indetectables sin cambio alguno en su TAR y no se consiguieron datos válidos de sus genotipos en la mayoría de ellos411.
Por el contrario, un estudio reciente ha demostrado una mayor probabilidad de fracaso en los pacientes con CVP muy baja pero que no consiguen la indetectabilidad total412. Así, tras 12meses de seguimiento, el 34,2 y el 13,0% de los pacientes con CVP entre 40 y 49copias/ml en el momento basal, frente al 11,3 y el 3,8% con CVP entre 0 y 40copias/ml, y el 7,0 y el 1,2% con CVP indetectable, presentaban fracaso virológico confirmado con más de 50 o más de 400copias/ml, respectivamente (ambos: p<0,0001; log-rank test). En el análisis multivariante, la CVP en T0 entre 40-49copias/ml (HR: 4,67; IC95%: 2,91-7; p<0,0001) y la CVP entre 0-40copias/ml (HR: 1,97; IC95%: 1,25-3,11; p<0,0001) fueron predictores independientes de tener CVP detectable (>50copias/ml) a los 12meses de seguimiento.
Los valores de CVP entre 200 y 1.000copias/ml se asocian universalmente en todos los estudios a selección de mutaciones de resistencia y deben considerarse siempre como fracasos virológicos413. En todos los estudios de registro de TAR se genotipan sistemáticamente todas las muestras con CVP>400copias/ml. En la práctica clínica es deseable genotipar todas las muestras con CVP confirmada >50copias/ml, especialmente si es >200copias/ml.
La incidencia de fracaso virológico, sus causas y el perfil de mutaciones de resistencia seleccionadas han cambiado radicalmente desde el inicio del TAR de forma paralela a la mejoría paulatina en la eficacia de los regímenes utilizados. Tras la introducción de los ITINN y los IP/r, y la sustitución de los ITIAN timidínicos por TDF o ABC coformulados, se ha reducido significativamente no solo la incidencia del fracaso virológico al primer TAR sino también la frecuencia y el tipo de mutaciones emergentes. Se ha observado una reducción de las TAM frente a ITIAN y de las mutaciones en el gen de la proteasa87. La toxicidad fue inicialmente la causa más frecuente de fracaso terapéutico por retirada del tratamiento. Con los regímenes usados actualmente se ha reducido drásticamente la frecuencia de fracaso terapéutico (endpoint compuesto). Las tasas de fracaso virológico (definido como CVP confirmada >50copias/ml hasta la semana 48) en primeras líneas de TAR son inferiores al 15% (6-14,5%) con las pautas actualmente consideradas como preferentes (ITIAN no timidínicos, coformulados, e ITINN, IP/r o RAL), aunque al menos en una cuarta parte de ellos no se consigue amplificar el estudio genotípico de resistencias por tratarse de CVP bajas (<500copias/ml)171,251,277,302,307,414.
Los sucesivos fracasos terapéuticos con acumulación de mutaciones de resistencia frente a las diferentes familias de FAR limitan las posibilidades de éxito del TAR, obligan al uso de regímenes más caros y complejos, y se asocian con mayor incidencia de progresión a sida y muerte.
Factores que influyen en el fracaso terapéuticoEstos factores pueden clasificarse en 3grupos, según dependan del paciente, de los fármacos o del virus.
Entre los primeros, el más importante es la adherencia al tratamiento, que es el mejor predictor de respuesta virológica. Cuando se detecta un fracaso virológico sin mutaciones de resistencia debe valorarse la falta de adherencia como la causa más probable. La ausencia de mutaciones mayoritarias en el genotipo poblacional estándar (genotipo Sanger) en este escenario no excluye que puedan existir en poblaciones virales minoritarias, no detectables por las pruebas de resistencia genotípicas convencionales, especialmente frente a FAR de baja barrera genética (3TC, FTC, ITINN de primera generación e inhibidores de la integrasa). Asimismo, la aparición súbita de una CVP elevada (con valores similares a los del setpoint previo al inicio del TAR) en un sujeto tratado sugiere el abandono del tratamiento por parte del paciente.
Entre los factores que dependen del fármaco se considera en primer lugar la potencia del régimen terapéutico, la falta de concentraciones adecuadas en sangre, por malabsorción o interacciones medicamentosas, y los errores de dosificación.
Entre los factores que dependen del VIH-1, el más importante es la resistencia a los FAR, que resulta de la interacción de la capacidad replicativa y diversidad del virus y la presión farmacológica. La resistencia a los FAR puede ser transmitida a otras personas y es variable según sea el área o colectivo de pacientes estudiados y el método empleado. Las cifras de infección reciente por virus resistentes varían entre el 7,7 y el 19,2%. Se estima que en nuestro medio el 9-12% las infecciones recientes por VIH-1 tienen mutaciones de resistencia415. De hecho, la monitorización de las mutaciones M184V, K65R y K103N, que se asocian a 3TC/FTC, TDF y EFV, han demostrado una reducción significativa en años recientes, a pesar del incremento en el uso de estos fármacos416, la cual puede deberse a la administración de los mismos en forma de comprimido único, como sugiere la mayor prevalencia de tales mutaciones en pacientes que los toman por separado que en los que los toman coformulados417.
Las mutaciones que confieren resistencia a ITINN, algunas de las que confieren resistencia a los inhibidores de la integrasa (muy infrecuentes en la actualidad) o determinadas mutaciones frente a ITIAN comprometen la eficacia de los tratamientos en sujetos que no han recibido TAR previo. El uso de técnicas ultrasensibles que permiten detectar mutaciones de resistencia en variantes virales minoritarias no detectables con las técnicas convencionales (véase más adelante) ha demostrado asociarse a una mayor tasa de fracaso virológico solo en los sujetos con mutaciones minoritarias frente a ITINN que inician pautas basadas en ITINN de primera generación (NVP o EFV [OR: 3,0])100, aunque probablemente se trate de un efecto de familia.
Dos estudios de cohortes han coincidido al describir que si se logra una CVP<50copias/ml en el primer TAR, el rebrote de la CVP suele asociarse a mal cumplimiento o toxicidad y solo en muy escasas ocasiones puede atribuirse a falta de potencia, interacciones medicamentosas o problemas de absorción.
El fenómeno de la inmigración obliga a valorar asimismo el origen de los pacientes con infección por VIH-1 en situación de fracaso virológico. El uso de NVP en dosis única como profilaxis de transmisión maternoinfantil, el TAR poco potente, la escasa monitorización o problemas de distribución y almacenaje de los FAR, etc., explican el aumento de incidencia de fracaso virológico en determinados países con escasos recursos económicos. Una elevada proporción de estos pacientes son portadores de VIH-1 de subtipos no-B y, por tanto, con patrones de mutaciones de resistencia potencialmente distintos. Existe una mayor predisposición a seleccionar las mutaciones K65R, K70E, K103N, V106M, y Y181C en el subtipo C, y en general hay menos experiencia con FAR en subtipos no-B418. La prevalencia de subtipos no-B se está incrementando en España, alcanzando el 15% entre los seroconversores recientes (a partir de 2005)415. La introducción de subtipos no-B en España se ha asociado claramente con la inmigración87. Por otra parte, algunos pacientes de origen subsahariano padecen infección por VIH-2 o infección dual por VIH-1 y VIH-2. El VIH-2 desarrolla patrones diferentes de mutaciones tras la exposición a ITIAN y es intrínsecamente resistente a ITINN y ENF.
Criterios de cambio de tratamiento antirretroviral por fracaso virológicoAnte un fracaso virológico es recomendable repetir la CVP para confirmarlo y cambiar el TAR lo antes posible para evitar la acumulación de mutaciones y la elevación de la CVP, facilitando de este modo la respuesta al nuevo tratamiento.
Los blips son valores aislados de CVP cerca del umbral de detección (50copias/ml, y especialmente 20copias/ml) en pacientes en TAR y con CVP suprimida, que vuelve a reportarse como suprimido en el control posterior sin cambio del TAR. Habitualmente son valores <200copias/ml. En la mayoría de los estudios los blips aislados no se asocian con un mayor riesgo de fracaso, pero sí con una mayor frecuencia posterior de blips. Sin embargo, si se presentan en más de una ocasión sí se relacionan con un incremento en el riesgo de fracaso virológico, e incluso se ha observado evolución genética y selección de resistencias durante los blips frecuentes. En un paciente con blips frecuentes debe evaluarse la potencia del TAR, y especialmente la adherencia al mismo, así como asegurarse de que el régimen de TAR administrado tenga una elevada barrera genética frente al desarrollo de resistencia.
Objetivo del tratamiento tras un fracaso virológicoEl objetivo terapéutico es conseguir de nuevo la supresión viral mantenida (<50copias/ml). Para ello se debe instaurar un nuevo régimen con 3FAR plenamente activos siempre que esto sea posible, o sus equivalentes en caso de FAR con actividad intermedia3. No hay ningún dato respecto a pautas de solo 2FAR activos en TAR de rescate, a pesar de que potencialmente podrían ser suficientes en algunos escenarios de rescate precoz con escasa resistencia, baja CVP y cifra elevada de linfocitos CD4+, y siempre que incluyan un IP/r activo419. Cuando no pueda disponerse de 3FAR activos, se contará con otros que conserven la mayor actividad residual (estudio de resistencias) y tengan la mejor tolerabilidad posible.
No se debe retrasar el cambio de TAR. A partir de datos de los estudios TORO ya se establecieron 4factores asociados a la eficacia virológica del TAR de rescate: a)recuento de linfocitos CD4≥100células/μl (OR: 2,1; IC95%: 1,5-3,1); b)CVP basal <5log10 (OR: 1,8; IC95%: 1,2-2,6); c)haber recibido ≤10FAR los individuos multitratados (OR: 2,4; IC95%: 1,6-3,4), y d)disponer de 2 o más FAR activos en el nuevo TAR (OR: 2,3; IC95%: 1,6-3,3). En todos los estudios de rescate se han identificado como factores de mala respuesta una CVP elevada (habitualmente definida como >100.000copias/ml) y cifras bajas de linfocitos CD4+420. Estos sujetos, debido a su mayor riesgo de fracaso virológico, deben tratarse con pautas especialmente validadas y con la mayor potencia antirretroviral posible.
Para conseguir el objetivo de CVP indetectable pueden ser útiles algunas estrategias, tales como:
- •
Facilitar la adherencia al TAR. La mala adherencia suele ser la causa de la mayoría de los fracasos virológicos. Antes de iniciar el TAR de rescate hay que identificar las causas de mala adherencia y se deben tomar medidas para corregirlas, pues en caso contrario se reproducirán tras el nuevo TAR de rescate. El nuevo régimen de TAR debe ser lo más cómodo de tomar y lo mejor tolerado posible. En algunos grupos de pacientes con mala adherencia al TAR se debe procurar que el tratamiento sea directamente observado, o al menos supervisado con frecuencia. No hay datos que confirmen que la adherencia sea mejor con pautas de rescate QD que con pautas BID, por lo que nunca debe escogerse una pauta QD solo para facilitar la adherencia, si no tiene la eficacia óptima para esa situación de rescate.
- •
Pruebas de resistencia. La realización de una prueba de resistencia genotípica o fenotípica en cada fracaso optimiza el nuevo tratamiento, aumenta su eficacia y mejora el pronóstico de los pacientes421,422. Los resultados más útiles se obtienen si esta prueba se realiza mientras el paciente está tomando el régimen de TAR que ha fracasado. En España se usa habitualmente el genotipo. En su interpretación debe tenerse en cuenta que las mutaciones detectadas están presentes con seguridad. Sin embargo, las no detectadas pueden encontrarse en poblaciones minoritarias (menores al 15-20% de la población viral total) y pueden no detectarse en el genotipo poblacional. Cuanto más tiempo transcurra entre la suspensión del TAR y la realización del genotipo, más fácil es que desaparezcan las mutaciones, especialmente las que causan mayor deterioro en la fitness o capacidad replicativa viral, que desaparecen con mayor rapidez al ceder la presión farmacológica que las mantenía. En los casos en que se disponga de estudios genotípicos previos es muy importante valorar la suma de todos los genotipos (genotipo acumulado), pues ello ha demostrado que mejora la eficacia en la elección del nuevo régimen antirretroviral respecto a la valoración aislada del genotipo del último fracaso87,423. Existen ciertas discordancias menores, según sea el sistema con el que se realiza la interpretación de las resistencias. La concordancia mayor se observa en los ITINN (93%) y en los IP (84%); en cambio, para los ITIAN solo es del 76%. Aunque es preferible disponer de una muestra con una CVP>1.000copias/ml para realizar el genotipado de VIH-1, la rentabilidad de su realización por debajo de este valor sigue siendo elevada, por lo que en ningún caso debe dejar de realizarse el estudio de resistencias solo por este motivo, debido a su mayor riesgo de fracaso virológico424.
- •
Tropismo. Debe determinarse sistemáticamente el tropismo del VIH-1 en cada fracaso virológico, exceptuando los casos en que previamente ya se haya documentado la existencia de tropismo no-R5. La determinación debe realizarse simultáneamente con el genotipo estándar, y su resultado debe ser evaluado conjuntamente con el del genotipo de la transcriptasa inversa, la proteasa y la integrasa, si procede.
- •
Revisar el historial de tratamiento del paciente. Identificar FAR que no fueron tolerados en el pasado, así como identificar fracasos virológicos previos durante el tratamiento con pautas de baja barrera genética. Especialmente en aquellos episodios en que no se realizara estudio genotípico de resistencias próximo a la fecha de fracaso, debe sospecharse la presencia de mutaciones que confieran resistencia frente a 3TC/FTC o ITINN de primera generación, que en genotipos posteriores (años) pueden haber desaparecido.
- •
Índice ponderado de resistencia genotípica para cada fármaco. La interpretación de las pruebas de resistencia genotípicas depende del número, del tipo y del patrón de mutaciones seleccionadas. En la actualidad se han desarrollado índices ponderados (scores) que definen la sensibilidad a determinados FAR (ATV/r, SQV/r, LPV/r, TPV/r, y ETR) basados en datos extraídos de estudios realizados en la vida real. Algunos algoritmos de interpretación, como el español de la RIS (véase más adelante) o el de Stanford (disponible en http://hivdb.stanford.edu/), ponderan el peso de las mutaciones para todos los FAR, incluidos los inhibidores de la integrasa. Estos scores marcan el «peso» o valor de cada una de las mutaciones según el grado de resistencia (valores negativos) o de hipersusceptibilidad (valores positivos), siendo de gran utilidad en la valoración de la actividad de los FAR que se plantean incluir en el TAR de rescate425. Los scores puntúan cada una de las mutaciones y finalmente se obtiene un resultado de mayor o menor susceptibilidad al fármaco.
- •
Cociente inhibitorio genotípico. El cociente inhibitorio genotípico es la razón entre las concentraciones plasmáticas del fármaco y el número de mutaciones relevantes en el gen de la proteasa. Se considera un marcador predictivo de respuesta a un IP, a pesar de que en él todas las mutaciones cuentan por igual. Otra aproximación es el cociente inhibitorio normalizado, que calcula la relación entre las concentraciones mínimas plasmáticas del fármaco y el número de veces por encima de la IC50 obtenido en el fenotipo viral. A pesar de su interesante valor teórico, no se ha establecido su uso en la práctica clínica.
- •
Pruebas de resistencia genotípicas con mayor sensibilidad. La PCR alelo específica y diferentes plataformas de ultradeep sequencing —454 Roche, myseq Illumina— se encuentran en un grado avanzado de desarrollo y son metodologías que permiten encontrar variantes virales minoritarias (o en baja cantidad, en porcentajes de la población viral del 0,1-15%) con mutaciones de resistencia, que no son detectadas mediante el genotipo estándar o poblacional100. Las técnicas basadas en PCR tienen la mayor sensibilidad teórica para detectar variantes minoritarias puntuales, pero presentan bastantes limitaciones técnicas. Las técnicas ultrasensibles basadas en la ultrasecuenciación (tecnología 454) aumentan la sensibilidad de detección de mutaciones minoritarias y, al disponer de secuencias completas independientes, permiten establecer nítidamente su asociación, realizar estudios filogenéticos más completos y estudiar la evolución en el tiempo de las diferentes poblaciones. Está bien establecido actualmente que la existencia de mutaciones minoritarias condiciona la respuesta a un TAR basado en ITINN de primera generación (NVP o EFV). Esto es debido a que la presencia de una sola mutación produce resistencia de alto nivel al fármaco. Es la única situación en que hasta ahora se ha podido demostrar el impacto clínico de las mutaciones minoritarias94,95. La presencia de variantes con K103N en un nivel >2.000copias/ml o >2% de la población viral se ha correlacionado con mayores tasas (OR: 47,4) de fracaso virológico a EFV426. Es de esperar que acabe confirmándose asimismo el impacto de las mutaciones minoritarias frente a otros FAR de baja barrera genética, especialmente los inhibidores de la integrasa (RAL) y 3TC o FTC. Asimismo, es posible que sean determinantes respecto a FAR con mayor barrera genética, pero en las situaciones en que, existiendo ya mutaciones que comporten una resistencia de nivel bajo/intermedio, la detección de otras mutaciones (aunque por debajo del 15-20%) pueda comportar un incremento significativo de la resistencia, por ejemplo frente a ETR. Contrariamente, la presencia de mutaciones minoritarias no tiene impacto en la respuesta a pautas triples basadas en IP/r en pacientes sin tratamiento previo427,428.
- •
Monitorización plasmática de las concentraciones de fármacos. Es poco útil en la actualidad en la optimización de un TAR de rescate debido a la existencia de datos clínicos muy sólidos con las pautas actuales de rescate y a la variabilidad interindividual de los niveles plasmáticos de los FAR. En casos seleccionados, puede ayudar a optimizar el TAR, mejorando así su eficacia429 (véase el apartado de farmacocinética).
Los ensayos clínicos aleatorizados que han evaluado la eficacia de diferentes combinaciones de FAR en tratamientos de segunda línea son escasos. El objetivo terapéutico es la resupresión de la CVP (<50copias/ml). Los cambios precoces evitan la acumulación de mutaciones y permiten secuenciar FAR, incluso dentro de cada familia. Con los FAR actualmente disponibles resulta sencillo diseñar un TAR de rescate precoz, aunque siempre se debe contar con una prueba de resistencias y tropismo del VIH-1.
En el caso de pacientes que iniciaran el tratamiento con 3ITIAN, las mutaciones más frecuentes son: la M184V/I con TAM, si el esquema incluía ITIAN timidínicos, o con la K65R (TDF) o L74V y Y115F (ABC), si el TAR incluía ITIAN no timidínicos.
Si el fracaso es con una pauta con 2ITIAN y un ITINN, una única mutación (por ejemplo: K103N, L100I, Y181C) es capaz de generar resistencia de alto nivel a EFV y NVP. El patrón de mutaciones es ligeramente distinto según que la pauta incluya NVP (Y181C, K103N, G190A, K101E, A98G) o EFV (K103N, L100I, Y188L, G190A, K101E). El fracaso virológico se acompaña habitualmente de otras mutaciones a ITIAN (M184V, L74V o K65R) con una incidencia superior a lo que ocurre cuando el primer régimen está compuesto por 2ITIAN y un IP/r, que protege mejor la actividad de los ITIAN. Un cambio precoz evita la acumulación de mutaciones de resistencia que comprometería la eficacia de los ITINN de nueva generación. En este escenario la utilización de ETR puede estar comprometida según sea el patrón de mutaciones seleccionado (véase más adelante). Aunque ha existido debate acerca de si los fracasos a NVP seleccionan mayor resistencia frente a ETR que los fracasos a EFV, debido a que el patrón de mutaciones que finalmente se selecciona es complejo, en el análisis con mayor poder estadístico realizado sobre un estudio aleatorizado (estudios DUET, con 599pacientes tratados) los fracasos previos a NVP no se asociaron con una peor respuesta a ETR que los fracasos a EFV, aunque su impacto pudo quedar diluido por la actividad de DRV/r420.
El uso de IP/r en el primer TAR ha reducido significativamente el número de fracasos virológicos y la selección de mutaciones frente a los IP y a los FAR acompañantes. Todos los IP/r tienen una elevada barrera genética y, de hecho, solo excepcionalmente aparecen mutaciones primarias o secundarias frente al IP en un primer fracaso virológico, aunque nunca condicionan resistencia fenotípica al IP. El desarrollo de resistencias en los IP es un proceso gradual que requiere la acumulación de varias mutaciones en el gen de la proteasa. Existen mutaciones seleccionadas específicamente por un IP (no potenciado) que no presentan resistencia cruzada con otros IP. Se consideran mutaciones específicas o signature mutations: D30N (NFV), I47A y L76V (LPV), G48V (SQV), I50L (ATV) o I50V (FPV y DRV), y alguna de ellas puede, por el contrario, producir hipersusceptibilidad a otros IP: la I47A confiere elevada resistencia fenotípica a LPV (>100veces), resistencia cruzada con FPV e hipersusceptibilidad a SQV; la I50L causa hipersusceptibilidad a todos los IP excepto a ATV, y la I50V causa resistencia a LPV, FPV y DRV e hipersusceptibilidad a TPV. Otras mutaciones, en cambio, causan resistencia cruzada a la mayoría de IP (I54L, I84V). La continua aparición de nuevas mutaciones obliga a consultar las bases de datos específicas con información actualizada sobre patrones de resistencia y su significado clínico (Los Álamos, Universidad de Stanford, o la Plataforma de Resistencias de la RIS). El rescate de un tratamiento a 2ITIAN más un IP/r debe realizarse con 3FAR activos, que pueden incluir ITINN, ITIAN, IP/r y otros de las nuevas familias.
Con frecuencia, el fracaso virológico del primer TAR puede seleccionar mutaciones de resistencia (M184V, K65R, L74V, etc.) que comprometen la actividad de ABC y parcialmente también la de TDF. Ello obligaba utilizar ITIAN timidínicos (AZT especialmente, o d4T) en los tratamientos de segunda línea, que se han asociado con toxicidad crónica y a menudo irreparable (toxicidad mitocondrial, lipoatrofia, resistencia insulínica y otras alteraciones metabólicas). Actualmente se puede evitar el uso de estos análogos timidínicos gracias a su sustitución por FAR de las nuevas familias.
Si se pretende utilizar un IP/r, DRV/r ha demostrado ser más eficaz que LPV/r en determinadas situaciones en este entorno de rescate «precoz» o con fracaso limitado a pautas previas (estudio TITAN).
TITANEs un ensayo en fase iii, aleatorizado, que comparó la eficacia de DRV/r frente a LPV/r, ambos con un tratamiento optimizado, en pacientes en fracaso virológico pero con menor número de tratamientos previos que en los estudios POWER. Los criterios de inclusión fueron: CVP>1.000copias/ml, duración del TAR≥12semanas, y que nunca hubieran recibido LPV/r. Los datos deberían analizarse con criterios de no-inferioridad. No obstante, con la finalidad de realizar una comparación entre ambas opciones terapéuticas, el diseño del estudio recogía a priori que se realizaría un estudio de superioridad por ITT en caso de cumplirse la no-inferioridad. Se incluyeron 595pacientes. A las 48semanas se observó (ITT) que el 77% de los pacientes tratados con DRV/r y el 67% del grupo LPV/r alcanzaron la variable principal del estudio: CVP<400copias/ml (diferencia media estimada: 10%; IC95%: 2-17; p<0,001). DRV/r cumplió, por tanto, los criterios de superioridad frente a LPV/r. También se observaron diferencias con criterios de superioridad de DRV/r cuando se analizó la proporción con CVP<50copias/ml (71% frente al 60%; diferencia media estimada: 11%; IC95%: 3-19; p=0,005). En el análisis de subgrupos respecto a CVP<50copias/ml, DRV/r resultó también superior a LPV/r si: la cifra basal de linfocitos CD4+ era baja, la CVP era >100.000copias/ml, existía ≥1mutación primaria a IP, fold-change de LPV/r >10 o fold-change a DRV <10. El fracaso virológico fue del 10% en el grupo de DRV/r y del 22% en el de LPV/r. En el análisis de las mutaciones de resistencia solo el 21% (6/28) de los pacientes que fracasaron con DRV/r desarrollaron mutaciones adicionales en el gen de la proteasa, mientras que lo hizo el 36% (20/56) del grupo LPV/r. Las mutaciones frente a ITIAN fueron menos frecuentes en el grupo de DRV/r (14% frente al 27%). La seguridad y la tolerancia de DRV/r fueron comparables a las de LPV/r, con menos diarrea de grado 2-4 y mejor perfil lipídico.
ODINEs un estudio en fase IIIb, que ha comparado las dosis de DRV/r 800/100 QD frente a 600/100 BID junto a un régimen de rescate que solo incluía ITIAN en pacientes con fracaso a un TAR y que no presentaran ninguna mutación frente a DRV (score de la IAS-USA). Incluyó 590pacientes con CVP>1.000copias/ml y un recuento de CD4>50células/μl. El objetivo primario fue la demostración de la no-inferioridad de la dosis QD de DRV/r en el porcentaje de pacientes con CVP<50copias/ml (ITT-TLOVR, delta predefinido del 12%). A las 48 semanas el porcentaje fue del 72,1% para la dosis QD y del 70,9% para la dosis BID (diferencia: 1,2%; IC95%: −6,1-8,5%), cumpliendo, pues, el criterio de no-inferioridad. La evolución del recuento de linfocitos CD4 fue similar entre ambas ramas. Solo uno de 294 pacientes de la rama QD desarrolló una mutación de resistencia a DRV durante el fracaso, frente a ninguno en la rama BID. La tasa de efectos adversos de grados 3/4 se redujo a la mitad en el grupo QD (7,8% frente al 15,2%) y las concentraciones plasmáticas de colesterol total, de colesterolLDL y de triglicéridos fueron significativamente menores en la rama QD. Debe destacarse que en realidad el 46% de los pacientes incluidos no habían recibido ningún IP y que la mediana de mutaciones primarias para IP fue de 0, por lo que la aplicación de los resultados de este estudio en pacientes con varias mutaciones primarias en la proteasa es limitada105. El estudio establece la no-inferioridad de la pauta de DRV/r QD en pacientes que no han recibido previamente IP y en los que no presentan mutaciones primarias en la proteasa.
Lopinavir potenciado con ritonavir en monoterapiaEn el estudio HIV STAR 195sujetos que habían presentado fracaso virológico a su primer TAR triple basado en ITINN de primera generación (86% NVP, 14% EFV) fueron aleatorizados a recibir un régimen de rescate con LPV/r en monoterapia (400/100mg BID) frente a LPV/r asociado a TDF+3TC430. Ninguno había sido previamente tratado con IP. El 82% habían seleccionado la mutación M184V frente a 3TC, el 6,7% la K65R, y el 29% ≥3TAM. A las 48 semanas las tasas de sujetos con CVP<50copias/ml fueron del 61,2 y del 82,5%, respectivamente (OR de fracaso virológico para LPV/r monoterapia: 2,5; IC95%: 1,3-4,9; p=0,01), por análisis ITT en el que las pérdidas o la adición de ITIAN se consideraron fracasos. Asimismo, el estudio ACTGA5230, a pesar de no disponer de una rama de control, ha confirmado estos resultados, puesto que en 122sujetos con fracaso a un TAR de primera línea formado por 2ITIAN y un ITINAN, su rescate con LPV/r en monoterapia se asoció a tasas bajas de indetectabilidad en la semana 24 (63% con CVP<50copias/ml)431.
Así pues, LPV/r en monoterapia no es un fármaco de elección en TAR de rescate en pacientes que fracasan a una primera línea de TAR triple basado en ITINN, a pesar de que no hayan recibido nunca IP.
Inhibidores de la transcriptasa inversa no nucleósidos de segunda generaciónEtravirinaEtravirina es un ITINN de segunda generación activo frente a determinadas cepas de VIH-1 con mutaciones de resistencia frente a EFV y NVP.
Estudio TMC125-C227Fue un estudio aleatorizado, abierto, en fase ii, que evaluó la utilidad de ETR en TAR de rescate precoz. Se incluyeron 116pacientes que nunca habían estado expuestos a IP pero que tenían mutaciones de resistencia a ITINN por fracaso de un régimen de TAR previo con ITINN, interrupción de un ITINN o profilaxis de transmisión vertical con ITINN. Se utilizó una dosis de ETR de 800mg BID, en una formulación anterior a la actual, que ofrecía una farmacocinética similar a la obtenida con los 200mg BID actuales. Los pacientes fueron aleatorizados a recibir ETR o el IP seleccionado por el investigador (potenciado con RTV en el 96,5%, mayoritariamente [63%] con LPV/r), siempre junto a 2ITIAN. Más del 90% de sujetos tenía alguna mutación frente a ITINN (K103N la más frecuente) y el 70% tenían ≥2 de ellas. Asimismo presentaban una media de 2mutaciones frente a ITIAN y el 37% de los pacientes reciclaron al menos un ITIAN. El estudio se suspendió prematuramente al confirmarse una peor respuesta en la rama de ETR que en la que recibió IP. Este estudio es un ejemplo de la necesidad de utilizar más de 2FAR activos y evitar la monoterapia funcional en pacientes pretratados.
ETR no es una opción óptima en el rescate de pacientes que no han sido tratados con IP, que han fracasado a regímenes de TAR triple basados en ITINN de primera generación y han seleccionado resistencia frente a ITIAN e ITINN. Su utilidad en el rescate de primeras líneas de TAR triple basado en ITINN en que no se haya seleccionado resistencia significativa en el fracaso no ha sido evaluada.
Recomendaciones de cambio de tratamiento antirretroviral por fracaso virológico precoz- •
El cambio del TAR por fracaso virológico debe efectuarse de modo precoz para evitar la acumulación de mutaciones y facilitar la respuesta al nuevo tratamiento. (A-II)
- •
El TAR nuevo debe contener 3fármacos totalmente activos según los estudios de resistencia, o su equivalente si se incluyen fármacos parcialmente activos. (A-I)
- •
Se debe conseguir CVP indetectable (<50copias/ml) en cualquier tratamiento de rescate. (A-I)
- •
Se debe realizar un estudio de resistencias y una prueba de tropismo para confeccionar el mejor régimen alternativo (A-II). La prueba de resistencias debe realizarse mientras el paciente está recibiendo el tratamiento fallido o lo más precozmente posible tras la suspensión. Si se dispone de pruebas genotípicas previas, debe tenerse siempre en cuenta el conjunto total de mutaciones detectadas (genotipo acumulado). (A-II)
- •
En la elección del nuevo TAR se deben analizar las causas que motivaron el fracaso (adherencia o interacciones medicamentosas), la historia farmacológica, las toxicidades que haya presentado y las mutaciones de resistencia previas. (B-II)
- •
En fracasos a una primera línea de TAR triple basada en ITINN de primera generación, el TAR de rescate basado en ETR es inferior al basado en un IP/r, por lo que ETR no constituye una elección preferente en este contexto. (A-I)
- •
DRV/r (600/100mg BID) ha demostrado superioridad frente a LPV/r BID como IP/r de elección en tratamiento de rescate precoz, siendo el fármaco de elección. Los estudios tienen suficiente poder estadístico para confirmar esta superioridad si existe al menos una mutación primaria en la proteasa, pero no si no existe ninguna (A-I). Puede utilizarse la dosificación 800/100mg QD en sujetos que no hayan recibido previamente IP, y/o que habiéndolos recibido no presenten mutaciones para DRV ni mutaciones primarias en la proteasa. (A-I)
- •
Deben evitarse los análogos de timidina en el tratamiento de rescate si existen otras alternativas. (A-I)
El tratamiento tras el fracaso virológico de al menos 2líneas de TAR se ha denominado terapia de rescate avanzado. En esta situación la mayoría de los enfermos han experimentado fracaso con las 3familias de FAR más utilizadas: ITIAN, ITINN e IP. Sin embargo, en los estudios genotípicos todavía existen algunos FAR que conservan actividad moderada o elevada frente al VIH-1.
Existen numerosos ensayos clínicos que han comparado diferentes tratamientos de rescate. En todos ellos se han utilizado un IP/r, una pareja de ITIAN y el fármaco nuevo en evaluación. Son estudios difícilmente comparables entre sí por la heterogeneidad de la población, de los tratamientos previos, de los criterios de eficacia, del tiempo de seguimiento y del tipo de terapia optimizada utilizada dependiendo de los fármacos disponibles comercialmente en la época de realización del estudio.
Tanto ENF como TPV/r, DRV/r, ETR, MVC y RAL han demostrado superioridad frente a placebo en todos los parámetros de eficacia en sus respectivos estudios principales, siempre en combinación con el mejor tratamiento optimizado disponible en el momento de realización del estudio432–438.No existen estudios comparativos entre ellos en TAR de rescate.
Inhibidores de la proteasa potenciadosLopinavir/rLa experiencia de LPV/r en terapia de rescate se ha obtenido de los ensayos clínicos realizados con el resto de IP/r que utilizaron LPV/r como IP comparador (véase más adelante). Tanto TPV/r como DRV/r han demostrado superioridad respecto a LPV/r en este escenario. LPV/r no es un fármaco de elección en TAR de rescate avanzado.
FosamprenavirCONTEXTCONTEXT es un ensayo clínico aleatorizado y abierto de fase iii que comparó la eficacia de FPV/r frente a LPV/r, ambos con 2ITIAN en pacientes tratados previamente con uno o 2IP. Se incluyeron 300pacientes, que fueron aleatorizados a recibir FPV/r (1.400/200mg QD), FPV/r (700/100mg BID) o LPV/r (400/100mg BID). Los resultados mostraron que FPV/r QD era inferior a LPV/r y no se pudo demostrar la no-inferioridad de FPV/r BID frente a LPV/r. La proporción de pacientes con CVP<400 y a 50copias/ml a las 48 semanas fue del 50 y del 37%, respectivamente, para FPV/r QD, del 58 y del 46% para FPV/r BID, y del 61 y del 50% para LPV/r. FPV/r no es un fármaco de elección en TAR de rescate.
SaquinavirMaxCmin2Evaluó la eficacia de SQV/r (1.000/100mg BID) frente a LPV/r (400/100mg BID) en un estudio abierto y aleatorizado que incluyó 339pacientes. A las 48 semanas (ITT, interrupción=fracaso) el 25% de los tratados con LPV/r y el 39% de los tratados con SQV/r presentaron fracaso terapéutico definido como CVP≥200copias/ml en cualquier momento del estudio (p=0,005). El tiempo hasta el fracaso fue similar con ambas pautas (p=0,27). En este estudio se utilizaron cápsulas duras con 200mg de SQV, y la adherencia, los efectos adversos y el diseño abierto probablemente resultaron negativos para el brazo con SQV/r. SQV/r no es un fármaco de elección en TAR de rescate.
AtazanavirAI424 045El estudio AI424 045 comparó ATV/r frente a ATV más SQV y frente a LPV/r en pacientes en fracaso virológico. Se requería una CVP>1.000copias/ml, que hubieran realizado al menos 2regímenes previos y experiencia con algún FAR de las 3clases (ITIAN, ITINN e IP). Todos los pacientes recibieron además TDF y otro ITIAN. A la semana 24 se demostró que la eficacia del brazo que combinaba ATV y SQV era inferior a LPV/r, por lo que se dio opción de cambiar el tratamiento. Tanto a las 24 como a las 48 semanas ATV/r resultó no inferior a LPV/r respecto al criterio de valoración primario (reducción de CVP) y la consecución de CVP<50 o 400copias/ml. Sin embargo, la proporción de pacientes con CVP<50copias fue del 38% en el grupo de ATV/r frente al 45% en el de LPV/r. A las 96 semanas el criterio de valoración primario demostró una eficacia similar de ATV/r (−2,29log10) y LPV/r (−2,08log10). En los pacientes en tratamiento la proporción con CVP indetectable fue similar en ambos brazos, pero el estudio carece de poder estadístico para demostrar diferencias para este objetivo secundario. En un subestudio que analizó la respuesta virológica a las 48 semanas según la presencia de mutaciones en las posiciones D30, V32, M36, M46, I47, G48, I50, F53, I54, A71, G73, V77, V82, I84, N88 y L90, la respuesta fue similar si había ≤4mutaciones. Sin embargo, cuando el número de ellas era ≥5, ningún paciente (0/9) del grupo de ATV/r y 5/18 (28%) de los de LPV/r lograron la indetectabilidad. Por otra parte, se identificaron mutaciones asociadas a resistencia a ambos fármacos. Las mutaciones en las posiciones M46, I54, I84, o L90 reducen la eficacia de ATV/r a menos de un 30%, al igual que las posiciones M46, I54 o I84 en LPV/r. ATV/r no es un fármaco de elección en TAR de rescate avanzado.
TipranavirRESIST 1 y 2Los RESIST son estudios en fase iii en los que se comparó TPV/r con otro IP/r elegido por cada investigador. Los criterios de inclusión exigían recibir una pauta con IP, estar en situación de fracaso virológico (CVP>1.000copias/ml) y haber en el estudio genotípico ≥1mutación primaria en los codones D30, M46, G48, I50, V82, I84 o L90, y ≤2 en los codones L33, V82, I84 o L90. Los pacientes fueron aleatorizados a TPV/r o un IP/r comparador. A todos los pacientes se les administró un tratamiento optimizado que solo podía incluir ENF de entre los nuevos fármacos. Los pacientes partían de CVP elevadas (mediana de 4,8log10copias/ml) y CD4 bajos (mediana <200células/μl). En el RESIST-1 la mayoría de los pacientes del IP/r comparador recibieron LPV/r (61%), mientras que en el RESIST-2 los más usados fueron APV/r (40%) y LPV/r (38%). ENF se indicó en el 36% de los pacientes del RESIST-1 y en el 12% de los del RESIST-2 (había pacientes tratados previamente con este fármaco).
Los resultados conjuntos de ambos estudios a las 48 semanas mostraron una proporción de pacientes con CVP<50copias/ml del 22,8 y del 10,2% con TPV/r y con el IP/r comparador, respectivamente, demostrándose la superioridad de TPV/r. El uso de ENF mejoró los resultados en ambos brazos, alcanzando el 52% (<400copias/ml) y el 35,8% (<50copias/ml) en el brazo del TPV/r.
Mutaciones de resistencia a tipranavirSe han identificado 19mutaciones en 14posiciones de aminoácidos (L10V, L24I, M36I, K43T, M46L, I47V, I50L/V, I54A/L/M/V, I54L, Q58E, T74P, L76V, V82L/T, N83D y I84V) asociadas con resistencia a TPV. Según el «peso» que tengan en la respuesta al tratamiento con TPV (favorezcan la respuesta, tengan un impacto reducido o un gran impacto en la resistencia) se les ha dado un valor en el score actualizado (L10V: 1; L24I: −2; M36I: 2; K43T: 2; M46L: 1; I47V: 6; I50L/V: −4; I54A/M/V: 3; I54L: −7; Q58E: 5; T74P: 6; L76V: −2; V82L/T: 5; N83D: 4, y I84V: 2). En la elaboración de este score se ha tenido en cuenta la eficacia de la medicación que acompañaba a TPV. Cuando el score era ≤3 la respuesta en las semanas 8 y 48 es máxima y mínima si el score es >10. La proporción de pacientes que respondían aumentaba cuanto mayor era la eficacia del régimen acompañante439. Su perfil de mutaciones que le confieren resistencia difiere ligeramente del de DRV, por lo que puede plantearse su uso en el rescate de determinados fracasos a DRV. No obstante, ambos comparten las mutaciones L33F, I47V, I54M, T74P y I84V, por lo que es realmente infrecuente encontrarse con cepas en las que la diferencia de actividad estimada entre ambos sea clínicamente significativa.
Debido a que requiere una dosis mayor de RTV (200mg BID), tiene un peor perfil de interacciones farmacocinéticas, posee una incidencia de trastornos lipídicos y digestivos menos favorable que DRV (a pesar de que no se han comparado directamente), y a que no hay experiencia de su utilización con MVC, ETR o RAL, su uso ha quedado restringido a aquellos casos en que su actividad residual estimada sea claramente superior a la de DRV/r BID, y no se requiera el uso de ETR (contraindicada con TPV/r).
DarunavirPresenta una alta afinidad por la proteasa y es muy potente in vitro e in vivo frente a la cepa salvaje y las mutantes con resistencias a múltiples FAR, incluyendo IP/r. Tiene un patrón relativamente específico de mutaciones en la proteasa, diferenciado en gran parte respecto al resto de IP, lo que le permite mantener actividad frente a gran parte de las mutaciones habitualmente seleccionadas por el resto de IP.
Es el IP/r de elección en un TAR de rescate avanzado, a dosis BID (600/100mg BID), y debe incluirse siempre en el régimen de rescate, exceptuando casos de toxicidad intratable o resistencia adquirida de alto nivel al fármaco (muy infrecuente).
POWER 1 y 2Los ensayos POWER compararon la eficacia y la tolerancia de diferentes dosis de DRV/r (fase ii) frente a un IP/r comparador, asociados ambos a una selección optimizada de otros FAR. Los pacientes incluidos tenía CVP>1.000copias/ml, habían sido tratados previamente con FAR de las 3 familias y tenían ≥1mutación primaria a IP. La aleatorización se estratificó por el número de mutaciones, la CVP y el uso de ENF. A las 24 semanas, la dosis de DRV/r se unificó a 600/100mg BID. En el análisis combinado de ambos estudios, a las 48 semanas, se incluyeron solamente los pacientes que recibieron la dosis de DRV/r de 600/100mg BID desde el principio (n=131) frente al grupo control (n=120). La variable principal de eficacia fue la respuesta virológica confirmada (reducción de la CVP≥1log10copias/ml y tiempo hasta la pérdida de respuesta virológica, TLOVR). En cuanto a eficacia, la reducción de la CVP se alcanzó en el 61% de los pacientes del grupo de DRV/r y en el 15% del grupo comparador (diferencia: 46%; IC95%: 35-57; p<0,0001). La proporción con CVP<50copias/ml (ITT-TLOVR) fue del 45% en los tratados con DRV/r y del 10% en el grupo control. La eficacia superior de DRV/r frente al IP/r comparador se mantuvo independientemente del uso de ENF, de la CVP basal, de las mutaciones primarias frente a IP o del número de FAR activos en la terapia optimizada. En la semana 96 el 39% de los tratados con DRV/r y el 9% del grupo comparador persistían con CVP<50copias/ml (p<0,001, ITT-TLOVR).
El estudio POWER 3 incrementó los datos de eficacia, seguridad y tolerancia de DRV/r (dosis de 600/100mg BID), ratificando los resultados obtenidos en los estudios POWER 1 y 2, y no dispone de rama control. En la semana 144, el 37 y el 9% de los pacientes, respectivamente, persistían con CVP< 50copias/ml.
TITANHa sido analizado anteriormente.
GRACEEl estudio GRACE (Gender, Race and Clinical Experience) es un estudio en fase IIIb, multicéntrico y abierto, que incluyó pacientes con experiencia previa a FAR y con una CVP≥1.000copias/ml. Todos los pacientes recibieron DRV/r 600/100mg BID con un tratamiento optimizado que incluía ITIAN y ITINN, incluida ETR. El objetivo principal fue evaluar si había diferencias en función del sexo o la raza de los pacientes respecto a la eficacia (CVP<50copias/ml) del tratamiento a las 48 semanas. Se incluyeron 429pacientes, de los cuales el 67% eran mujeres y el 84% eran de raza negra. El 32,8% de las mujeres suspendieron el TAR frente al 23,2% de los hombres (p<0,05). En la semana 48, en el análisis en ITT-TLOVR, el 50,9% de las mujeres y el 58,5% de los hombres alcanzaron una CVP<50copias/ml. Las diferencias en la respuesta virológica ajustada por la CVP y recuento de linfocitos CD4+ basales fue −9,6 (IC95%: −19,85-0,68). En el análisis en ITT-TLOVR, en el que se censuraron los pacientes que fracasaron por causas diferentes al fracaso virológico, el 73% de las mujeres y el 73,5% de los hombres alcanzaron una CVP<50copias/ml (diferencia: −3,9; IC95%: −13,89-6,02). Los autores resaltaron la eficacia y la seguridad de DRV/r en el TAR de rescate, tanto en hombres como en mujeres, aunque una elevada proporción de mujeres abandonan el TAR por razones distintas al fracaso virológico.
ODINHa sido analizado anteriormente.
Mutaciones de resistencia a darunavirSe han identificado 11mutaciones en el gen de la proteasa (V11I, V32I, L33F, I47V, I50V, I54L/M, T74P, L76V, I84V, L89V) relacionadas con pérdida de sensibilidad a DRV. La respuesta virológica a DRV/r en TAR de rescate se va reduciendo paralelamente al número de estas mutaciones, al igual que sucede con todos los IP/r y ETR. Los porcentajes de respuesta (CVP<50copias/ml a 48 semanas) con 0, 1, 2 y 3 mutaciones fue del 72, del 53, del 37 y del 29%, respectivamente, en los estudios POWER (7% con ≥4mutaciones).
Este dato debe ser muy tenido en cuenta para valorar el número de FAR acompañantes en el régimen de rescate según la actividad residual de DRV cuando esta esté comprometida.
Inhibidores de la transcriptasa inversa no nucleósidos de segunda generaciónEtravirinaEtravirina es un ITINN de segunda generación activo frente a determinadas cepas de VIH-1 con mutaciones de resistencia frente a EFV y NVP.
DUET 1 y 2Los ensayos clínicos en fase iii DUET se diseñaron para analizar la eficacia y la seguridad de ETR en pacientes con experiencia previa a múltiples FAR. Con un diseño similar, DUET1 y 2 son ensayos multinacionales, paralelos, aleatorizados a doble ciego, de ETR frente a placebo. Los criterios de inclusión fueron: CVP>5.000copias/ml, TAR estable durante al menos 8 semanas, presencia de ≥1mutación frente a ITINN y ≥3mutaciones frente a IP. Todos los pacientes recibieron DRV/r e ITIAN según terapia optimizada. El uso de ENF fue opcional. La variable principal del estudio era una CVP<50copias/ml en la semana 24 (análisis por ITT y TLOVR). Se incluyeron 612pacientes en el DUET1 y 591 en el DUET2. Los resultados fueron: CVP<50copias/ml en tratados con ETR: 56% en el DUET1 y 62% en el DUET2, frente al 39 y el 44% en los grupos placebo, respectivamente (p<0,01 y p<0,001). La CVP se redujo en 2,4 y 2,3log10 en los grupos con ETR y 2,3 y 1,7log10en los de placebo (diferencia sin significación estadística). Entre los pacientes que recibieron ETR y ENF en el tratamiento optimizado, el 60 y el 73% alcanzaron una CVP<50 copias/ml, frente a56 y el 68% en los de los grupos placebo, respectivamente. Los resultados conjuntos de ambos estudios a las 48 semanas confirman los datos previos, y el 61% de los pacientes con ETR alcanzaron una CVP<50copias/ml frente al 40% de los pacientes asignados al grupo placebo (p<0,0001); el descenso de la CVP fue de −2,25log10 y −1,49log10, respectivamente420. En la semana 96, el análisis combinado de ambos estudios indica que la eficacia de ETR más terapia optimizada fue superior a la del tratamiento comparador: 57% frente al 36%, respectivamente (p<0,001). El 91% de los pacientes asignados a ETR que alcanzaron una CVP<50copias en la semana 48 persistían con la misma eficacia virológica en la semana 96. Por otra parte, el número de eventos clínicos asociados a sida o muerte fue menor en el grupo que recibió ETR (p=0,06) y alcanzó diferencias estadísticamente significativas cuando se compararon exclusivamente los pacientes de ambos grupos que además recibían ENF habiéndola utilizado previamente (5,9% en el grupo de ETR frente al 10,1% en el grupo placebo; p=0,02).
La tolerancia a la ETR fue buena, y el efecto secundario más frecuente fue un exantema leve o moderado. Este lo manifestaron el 19% de los pacientes tratados con ETR frente al 12% de los del grupo control (p<0,0001); apareció en las primeras semanas, la mayoría fue de grado leve-moderado (solo el 1% se consideró de grado3) y solo en el 2% se retiró el tratamiento por su causa. Hubo una incidencia más elevada de exantema entre las mujeres. Un tercio (32%) de las mujeres que recibieron DRV/r+ETR desarrollaron exantema, frente al 19% de las que recibieron ETR y placebo. Asimismo, una proporción más elevada de mujeres con DRV/r debió suspender ETR por exantema (5% en el grupo DRV/r+ETR frente al 2% en el grupo ETR+placebo). La historia de exantema previo con NVP o EFV no se asoció con la aparición de exantema frente a ETR420.
Mutaciones de resistencia a etravirinaSe han identificado 17mutaciones en su score ponderado que disminuyen la tasa de respuesta a ETR en los estudios DUET: V90I, A98G, L100I, K101E/H/P, V106I, E138A, V179D/F/T, Y181C/I/V, G190A/S y M230L440. Entre estas mutaciones, Y181I/V/C, seguida de L100I, K101P y M230L son las que generan una mayor resistencia a ETR. Estas mutaciones pueden seleccionarse tras el fracaso virológico a ITINN de primera generación, y siempre que vaya a usarse ETR debe evaluarse su actividad si existen mutaciones para ITINN. La puntuación obtenida con la suma de cada mutación se ha correlacionado con respuesta virológica observada en los estudios DUET.
Por otra parte, la compañía Monogram ha desarrollado un score que asigna a cada mutación un valor en función de su peso (valor 4: L100I, K101P, Y181C/I/V; valor 3: E138A/G, V179E, G190Q, M230L, K238N; valor 2: K101E, V106A/I, E138K, V179L, Y188L, G190S; valor 1: V90I, A98G, K101H, K103R, V106M, E138Q, V179D/F/I/M/T, Y181F, Y189I, G190A/E/T, H221Y, P225H, K238T). En este score la puntuación obtenida con la suma de los puntos de cada mutación se correlaciona con el fenotipo. Si el resultado es <4, ETR tiene un 90% de probabilidades de ser eficaz (fold change<2,9). La plataforma de resistencias de la Red de Investigación en Sida (RIS) adjudica 2puntos a las mutaciones Y181C/I/V, L100I, K101P, V179F, G190E, M230L y un punto al resto, considerando resistencia un score ≥3.
Los pacientes que han seleccionado K103N como única mutación en su genotipo poblacional tras fracaso a EFV o NVP contienen frecuentemente poblaciones minoritarias con otras mutaciones frente a ITINN. Hasta en el 45% de los casos se encuentran mutaciones específicas frente a ETR441. No se ha confirmado aún el impacto en la respuesta a una pauta de rescate con ETR de este fenómeno. Sin embargo, en aquellos casos en que tras un fracaso a ITINN de primera generación se aísle alguna mutación frente a ITINN en el genotipo convencional debería confirmarse siempre que sea posible la presencia de otras mutaciones minoritarias, con el fin de seleccionar la mejor pauta de rescate para cada paciente.
Inhibidores de la uniónEnfuvirtidaENF inhibe la fusión del VIH-1, evitando su penetración y su replicación. Se administra por vía subcutánea 2veces al día y su principal efecto adverso es la reacción en el punto de inyección (tabla 9).
Inhibidores de la fusión
| Nombre genérico | Enfuvirtida (T-20) |
| Nombre comercial | Fuzeon® |
| Dosis recomendada | 90mg c/12h s.c. |
| Presentaciones comerciales | Vial de 90mg |
| Semivida plasmática | 3,8 ± 0,6h |
| Biodisponibilidad | 80% (vía s.c.) |
| Semivida plasmática | 3,8 ± 0,6h |
| Cmax | 4,59 ± 1,5μg/ml (VIH-1+) |
| Cmin | 2,6-3,4μg/ml (VIH-1+) |
| AUC | 55,8 ± 12,1μg·h/ml (VIH-1+) |
| CI50 | CI50: 0,259μg/ml (media geométrica) en un ensayo de entrada de recombinación de genotipos VIH-1 |
| Actividad | VIH-1 |
| MetabolizaciónExcreción | Catabolismo en sus aminoácidos constituyentesNo hay datos |
| Efectos adversos | Reacciones locales leve-moderadas en el punto de inyección, dolor de cabeza y fiebre |
| Interacciones | Escaso riesgo de interacción metabólica. Estudios in vitro e in vivo con una amplia variedad de isoenzimas hepáticas no han mostrado efecto inhibitorio de T-20 sobre ellas |
Nota: debido a que la información científica relacionada con los fármacos antirretrovirales se renueva constantemente, se recomienda consultar la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias.
Los estudios TORO son 2 ensayos abiertos en fase iii en los que se comparó la actividad antiviral de ENF en combinación con una pauta optimizada frente a una pauta optimizada en pacientes en fracaso viral con múltiples tratamientos previos. Se incluyeron alrededor de 1.000pacientes, con una mediana de CVP basal >100.000copias/ml y de linfocitos CD4+ <100 células/μl. A las 24 semanas el descenso de la CVP fue mayor en los tratados con ENF. ENF produjo un descenso de CVP de −0,93log10 (TORO I) y −0,78log10 (TORO II) (p<0,0001 frente a la rama comparadora). A la semana 48, en el análisis combinado de los 2 estudios, el descenso de la CVP fue de −1,48log10 copias/ml en el grupo de ENF y −0,63log10 copias/ml en el de tratamiento optimizado (p<0,0001). La probabilidad de alcanzar una respuesta virológica fue más del doble en el grupo de ENF (descenso CVP>1log10 del 37% frente al 17%; CVP<400copias/ml, del 30% frente al 12%, y CVP<50copias/ml, del 18% frente al 8%; p<0,0001). El tiempo hasta el fracaso del grupo con ENF triplicó al del grupo control (32 y 11semanas, respectivamente; p<0,0001). Es decir, tanto el análisis primario de eficacia como el resto de análisis secundarios predefinidos demostraron que el tratamiento de rescate era más eficaz cuando se utilizaba ENF.
Se han identificado mutaciones en la región HR1 de la gp41 del virus que reducen la sensibilidad al ENF (G36D/S, I37V, V38A/M/E, Q39R, Q40H, N42T, N43D). Otras mutaciones o polimorfismos en otras regiones de la envoltura, como por ejemplo la región HR2, podrían disminuir la sensibilidad a ENF442. Por ello, secuenciar solo las mutaciones de la región HR1 podría ser inadecuado cuando se sospecha resistencia a ENF. La barrera genética de ENF es típicamente baja y desarrolla resistencia al fármaco con solo una mutación.
Por otra parte, la inclusión de ENF en el régimen de rescate ha demostrado sistemáticamente incrementar las tasas de respuesta en otros estudios en que se evaluaban otros FAR pero se permitía el uso de ENF434-438,443.
La inconveniencia de su administración por vía subcutánea 2veces al día limita su uso. Un documento de consenso español recomienda su uso en pacientes en los que no se pueda confeccionar un tratamiento óptimo con 3FAR activos.
Inhibidores del correceptor CCR5MaravirocMVC es el único antagonista del correceptor CCR5 actualmente comercializado. Impide la entrada del virus con tropismo R5. Presenta una actividad potente frente a cepas con tropismo R5 tanto silvestres como con mutaciones para ITIAN, ITINN o IP, con los que no comparte resistencia cruzada.
MOTIVATE 1 y 2Los estudios MOTIVATE 1 y 2 son 2 ensayos clínicos en faseiii y doble ciego en los que se aleatorizó a los pacientes incluidos a recibir MVC (QD o BID) frente a placebo, añadiendo a cada uno de los brazos un tratamiento optimizado. Los criterios de inclusión fueron: CVP>5.000copias/ml, tropismo R5 y resistencia a ≥1FAR o ≥2 IP. Los pacientes se estratificaron según el uso de ENF y la CVP. Se incluyó a más de 1.000 pacientes, de los que 209 recibieron placebo, 414 MVC QD (150 o 300mg) y 426 MVC BID (150 o 300mg). La variable principal del estudio fue la reducción de la CVP en la semana 48, y la proporción de pacientes con CVP<400 y a 50copias/ml fue una variable secundaria.
Los resultados combinados de ambos estudios a las 48 semanas muestran que el descenso de CVP fue: MVC QD: −1,68log; MVC BID: −1,84log, y grupo placebo: −0,78log; la proporción de pacientes con CVP<400copias/ml: 51,7, 56,1 y 22,5%, respectivamente (valor de p frente a placebo <0,0001); y CVP<50copias/ml: 43,2, 45,5 y 16,7%, respectivamente (valor p frente a placebo <0,001). El incremento del número de linfocitos CD4+ fue también significativamente mayor y más precoz en los grupos que recibieron MVC. Esta ventaja en la recuperación inmunológica ha sido también demostrada con independencia de la eficacia virológica. La eficacia (CVP<50copias/ml) fue también superior cuando los pacientes recibieron ENF por primera vez: el 64 y el 61% en los grupos que recibieron MVC y el 27% en los grupos que recibieron placebo con ENF. En la semana 96, el 41,3% de los pacientes que recibieron MVC en pauta BID persistían con una CVP<50copias frente al 7,2% de los pacientes del grupo placebo. En cuanto a variables de endpoints clínicos «duros», el tratamiento con MVC se asoció de manera significativa con un mayor tiempo hasta la aparición de episodios diagnósticos de sida en comparación con el placebo (p=0,042), siendo hasta la fecha el único estudio de rescate en que ha podido demostrarse este beneficio clínico incluyendo solo uno de los nuevos fármacos activos.
Los efectos adversos producidos por MVC no fueron superiores a los del grupo placebo. Los temores iniciales acerca de un posible mayor riesgo de hepatotoxicidad o de una mayor incidencia de tumores asociada al bloqueo del correceptor humano CCR5 no solo no se han confirmado sino que la incidencia de determinados episodios clínicos, y específicamente de algunos tumores, ha sido significativamente menor en la rama tratada con MVC.
Se analizó retrospectivamente la relación del fracaso virológico en los pacientes de los MOTIVATE con el tropismo viral. Dos tercios de los pacientes con MVC que fracasaron tenían un tropismo dual (R5/X4) o bien el tropismo varió en el periodo de tiempo comprendido entre la selección y el inicio del tratamiento. El cambio de tropismo de R5 a dual o X4 se observó en el 7,5% de los pacientes que fracasaron con MVC y solo en el 1,9% del grupo placebo. No se observó cambio de tropismo en el 4% de los fracasos a MVC. Entre los pacientes que fracasaron en los estudios MOTIVATE, los que recibieron MVC asociado a ≥2FAR activos presentaron una tasa de cambio de tropismo R5 a D/M o X4 similar a los tratados con placebo. Tras la retirada de MVC las cepas revertían rápidamente a R5. Todos los pacientes que recibían MVC presentaron un incremento significativo del número de linfocitos CD4+.
Resistencia a maravirocLa eficacia de MVC exige la presencia de tropismo viral R5 y no es activo cuando las poblaciones virales presentan tropismo viral X4 o dual (R5/X4). Algunos casos de fracaso virológico durante el tratamiento con MVC corresponden a un sobrecrecimiento de poblaciones virales con tropismo X4 prexistentes y no detectadas por la baja sensibilidad de la prueba basal (Trofile® inicial detectaba virus X4 con una certeza del 100% solo cuando la proporción era ≥10% de la población viral y la CVP>1.000copias/ml). La técnica de Trofile® ES detecta poblaciones minoritarias con tropismo X4 hasta en una proporción del 0,1%. Asimismo, el tropismo puede detectarse por métodos genotípicos. Recientemente se ha comunicado una predicción del tropismo y de respuesta a MVC similar con el genotipo tanto en pacientes sin tratamiento previo como en rescate. Recientemente se han actualizado las guías españolas para la determinación del tropismo, las cuales recomiendan la realización de pruebas genotípicas444. Esta evaluación debe realizarse en todos los episodios de fracaso virológico (exceptuando aquellos en los que ya conste un tropismo previo no R5) simultáneamente al genotipo poblacional estándar de la transcriptasa inversa y proteasa.
Además, se han identificado mutaciones en la molécula gp120 que permiten al virus unirse al receptor R5 en presencia de MVC sin cambiar de tropismo durante el tratamiento442. El perfil de mutaciones seleccionadas por MVC en los casos en que se mantiene el tropismo R5 es complejo y todavía no se conoce con exactitud su patrón.
MVC es un sustrato de la glucoproteína-P y su metabolismo se realiza a través del CYP3A4, por lo que presenta un potencial importante de interacciones con fármacos que utilizan la misma vía metabólica (véase el apartado de interacciones).
Inhibidores de la integrasaRaltegravirLa integrasa es una de las 3 enzimas fundamentales del ciclo de replicación del VIH-1. Su función es catalizar la inserción del ADN proviral en el genoma de la célula huésped. RAL es un fármaco capaz de inhibir la integrasa, y es activo frente a cepas de VIH-1 con mutaciones de resistencia a las 3 familias clásicas (ITIAN, ITINN e IP) y frente a cepa silvestre.
BENCHMRK 1 y 2Los ensayos clínicos en fase iii BENCHMRK 1 y 2 son estudios paralelos, aleatorizados y doble ciego, diseñados para analizar la eficacia de RAL en rescate de pacientes expuestos a múltiples FAR y en fracaso virológico. Los criterios de inclusión fueron CVP>1.000copias/ml y resistencia genotípica o fenotípica al menos a un FAR de cada una de las 3 clases (ITIAN, ITINN e IP). Se incluyeron 350pacientes (BENCHMRK-1) y 349 (BENCHMRK-2) en situación clínica muy avanzada (82% con criterios de sida) y que habían recibido una mediana de 12FAR durante 9,9años. Los pacientes fueron aleatorizados (2:1) a recibir RAL o placebo, ambos con terapia optimizada. En los resultados combinados de ambos estudios a las 48 semanas, la proporción de CVP<400copias/ml fue del 72,1% en el grupo de RAL y del 37,1% en el de placebo (p<0,001). La CVP<50copias/ml fue del 62,1% en el grupo de RAL y del 32,9% en el de placebo (p <0,001). La eficacia de RAL fue superior a la del placebo, independiente de la CVP, de la cifra de linfocitos CD4+ o del índice GSS/PSS ≥2.
Cuando en la terapia optimizada se asoció DRV/r y ENF la proporción de CVP<400copias/ml fue del 98% (RAL) y del 87% (placebo); si solamente recibían ENF, el 90% (rama de RAL) y el 63% (rama placebo) tenían <400copias/ml, mientras que si solo recibían DRV/r las proporciones fueron del 90 y del 55%, respectivamente. La seguridad y la tolerabilidad de RAL resultaron comparables a placebo. Se observó una mayor incidencia de neoplasias en el grupo asignado a RAL (3,5% frente al 1,7%), aunque no fue estadísticamente significativa ni se ha comunicado ninguna relación directa con el fármaco. Estudios posteriores que analizan la incidencia de neoplasias en ensayos clínicos y programas de acceso expandido no han observado una mayor incidencia de neoplasias entre los pacientes que reciben RAL.
El análisis combinado en las semanas 96, 156 y 192 de los estudios BENCHMRK demostró la eficacia duradera de RAL445. El 58% de los pacientes a 96 semanas (45% a 192 semanas) que recibieron RAL persistían con CVP<50copias/ml frente al 26% de los pacientes (16% a 192 semanas) de los del grupo placebo. Este porcentaje se incrementó al 79% cuando los pacientes recibían además otros fármacos activos.
RAL se metaboliza por glucuronización hepática y no es inductor o inhibidor de las isoenzimas del citocromo P450, por lo que su potencial de interacciones farmacológicas es bajo (véase el apartado de interacciones).
Mutaciones de resistencia a raltegravirEn estudios in vitro se han identificado hasta 41mutaciones del gen de la integrasa asociadas a resistencia a RAL. Un total de 105pacientes de 462 que recibieron RAL en los estudios BENCHMRK desarrollaron fracaso virológico en la semana 48, aunque solo en 94 de ellos se pudo realizar un estudio de resistencias. En 64/94 (68%) se detectaron mutaciones de resistencia a RAL con 3patrones de resistencia: 1)N155H+L74M, E92Q, T97A, V151I, G163R; 2)Q148K/R/H+G140S/A, E138K, y 3)Y143R/C+L74A/I, T97A, I203M, S230R. RAL es un fármaco de barrera genética baja, siendo necesarias solo 2mutaciones para la resistencia completa. Sin embargo, alguna mutación (Q148K/H e incluso N155H) confiere ya un nivel de resistencia elevado a RAL por sí sola.
Las resistencias cruzadas con otros fármacos de la familia de los InInt son muy frecuentes con elvitegravir, por lo que probablemente su uso secuencial no será posible. Elvitegravir ha demostrado una eficacia no inferior a RAL en rescate en estudios en faseiii446. Con otros InInt en desarrollo (dolutegravir) el grado de resistencia cruzada es mucho menor, permitiendo su uso en rescates a fracasos con RAL447.
Uso combinado de varios de los nuevos fármacos en un tratamiento retroviral de rescateEstudio TRIOEs un estudio abierto, no comparativo, que tuvo como objetivo evaluar la eficacia y la seguridad de un TAR de rescate que contenía RAL+DRV/r+ETR. Incluyó 103 pacientes con edad ≥18años, CVP>1.000copias/ml, sin TAR previo con fármacos en investigación, con historia de fracaso virológico a ITINN, e infección por VIH-1 multirresistente definida como: ≥3mutaciones primarias de resistencia a IP; ≥3mutaciones a ITIAN; ≤3mutaciones a DRV, y ≤3mutaciones a ITINN. Todos los sujetos recibieron RAL+DRV/r+ETR, junto con ITIAN (83%) y ENF (12%). En la semana 24, 93 pacientes (90%; IC95%: 85-96%) y en la semana 48, 89 pacientes (86%; IC95%: 80-93%) lograron una CVP<50copias/ml. La mediana de descenso de la CVP en la semana 48 respecto al valor basal fue de −2,4log10 (IQR: −2,9 a −1,9). La mediana de ascenso de los linfocitos CD4+ respecto al valor basal fue de 108/μl (IQR: 58-169). Durante el periodo del estudio, solo un paciente tuvo que suspender el tratamiento por toxicidad.
Otros estudios retrospectivos no aleatorizados han confirmado asimismo la elevada tasa de respuesta virológica que se obtiene combinando DRV/r+ETR+RAL en un TAR de rescate avanzado448.
Opciones de tratamiento en pacientes con múltiples fracasos virológicos y sin opciones terapéuticas. Pautas de tratamiento antirretroviral no supresorasCon los FAR actualmente disponibles puede conseguirse la supresión virológica completa y duradera (<50copias/ml) en la gran mayoría de pacientes con fracaso virológico, por avanzado que sea. El fracaso virológico en el paciente multitratado no siempre conduce de forma rápida al fracaso inmunológico y a la progresión clínica. De hecho, muchos pacientes continúan con recuentos de CD4 relativamente estables y aproximadamente solo un tercio experimenta un descenso. Este último hecho se observa más frecuentemente cuando la CVP es elevada, habitualmente >10.000-20.000copias/ml.
En un paciente en el que resulte imposible formar un TAR potencialmente eficaz con al menos 2FAR activos por problemas de resistencia, toxicidad, comorbilidad grave, adherencia o tolerabilidad, pueden plantearse otras opciones diferentes al TAR de rescate si la situación inmunitaria del paciente lo permite.
Diversos estudios han demostrado los efectos beneficiosos de mantener un TAR no supresor (frente a su interrupción completa) en caso de infección avanzada multirresistente sin opciones de iniciar una pauta supresora. En pacientes con un recuento de linfocitos CD4+<50/μl, el riesgo de desarrollar una enfermedad oportunista diagnóstica de sida era un 22% inferior si el paciente continuaba con el TAR a pesar de presentar fracaso virológico respecto a los que lo suspendieron. Esto se relaciona con la persistencia de una población viral de VIH-1 menor y con baja capacidad replicativa y, por tanto, probablemente menos lesiva.
Estos TAR no supresores deben ser regímenes cómodos de tomar, poco tóxicos, que disminuyan la capacidad replicativa viral y que no acumulen mutaciones que puedan comprometer futuros TAR de rescate. No se recomienda continuar con pautas no supresoras que contengan IP, ITINN o InInt, ya que la acumulación de resistencias frente a estos fármacos dificultaría la eficacia de futuros fármacos de estas familias. Pautas con 2 o 3 ITIAN, que incluyan 3TC o FTC, y simultáneamente AZT y TDF (por su antagonismo sobre la RT), podrían resultar parcialmente eficaces en algunos pacientes si su recuento de linfocitos CD4+ no es bajo. Pueden mantener transitoriamente (meses) una población viral con baja fitness y deben remplazarse por un régimen de rescate supresor con 3FAR activos en cuanto sea posible.
La mutación M184V compromete la capacidad replicativa del VIH-1. Un ensayo que incluía pacientes multirresistentes con esta mutación, aleatorizados a continuar solo con 3TC o a suspender el tratamiento, mostró, en la semana 48, que el 69% (IC95%: 51-83%) del grupo que interrumpió frente al 41% (IC95%: 26-59%) de los que continuaron con 3TC presentaron un evento clínico o fracaso inmunológico. En los que recibían 3TC el fracaso fue más tardío (p=0,01) y el descenso de CD4, el rebrote de CVP y el aumento de capacidad replicativa fueron menores. En función de estos estudios, se propone que el tratamiento de pacientes con múltiples fracasos contenga 3TC o FTC para mantener en la población viral la mutación M184V y reducir su capacidad replicativa. Sin embargo, este dato no se ha confirmado cuando se usan pautas de rescate supresoras que consiguen CVP indetectables.
Replicación viral baja en pacientes multitratadosDos determinaciones sucesivas de CVP>50copias/ml 6meses después de haber iniciado el TAR o tras haber conseguido previamente una CVP<50copias/ml se considera fracaso virológico. Con frecuencia, los pacientes con replicación viral baja y persistente del VIH-1, que podemos definir como una CVP entre 50-200copias/ml, mantienen estable el número de linfocitos CD4+. El tratamiento de elección es un régimen que consiga nuevamente la supresión mantenida por debajo de 50copias/ml. Sin embargo, la ausencia o la complejidad de otras alternativas terapéuticas, la conservación de la inmunidad y la dificultad de obtener resultados válidos con pruebas de resistencia genotípicas o determinación del tropismo viral por la baja viremia plasmática dificultan el manejo de estos pacientes.
En este escenario se han propuesto diferentes estrategias terapéuticas, aunque ninguna de ellas ha sido evaluada en estudios prospectivos longitudinales y comparativos. Si la pauta de TAR presenta una baja barrera genética y se confirma que la baja viremia no responde a un error de técnica de laboratorio, es prudente sustituirla bajo criterio clínico por otra con barrera genética elevada basada en un IP/r, independientemente de que no se consiga demostrar la presencia de mutaciones. No está bien establecida cuál es la mejor actitud en pacientes con replicación viral baja, aunque se conoce que estos pacientes pueden seleccionar nuevas mutaciones de resistencia, desarrollar fracasos y contribuir a la diseminación de la infección a otras personas. La posibilidad de obtener resultados en las pruebas de resistencia o tropismo viral, las alternativas disponibles y sobre todo la posibilidad de cambiar el TAR a un régimen que incluya al menos 2FAR plenamente activos condicionan la actitud del clínico en este escenario. Si el régimen de TAR presenta una barrera genética elevada y el riesgo de selección de resistencias es muy bajo, puede en determinados casos mantenerse al paciente con el mismo tratamiento.
Suspensión del tratamiento antirretroviralLa interrupción del TAR en pacientes multirresistentes se planteó ante la hipótesis de que la reaparición de la cepa silvestre permitiría una mejor respuesta tras la reintroducción del tratamiento. Los ensayos clínicos realizados para evaluar esta estrategia han evidenciado un descenso importante del número de linfocitos CD4+ durante la interrupción frente a los que continúan con TAR. No se recomienda la suspensión del TAR como opción electiva en ninguna situación en sujetos con resistencia múltiple.
Recomendaciones de cambio de tratamiento antirretroviral por fracaso virológico avanzado- •
El cambio del TAR por fracaso virológico debe efectuarse de modo precoz para evitar la acumulación de mutaciones y facilitar la respuesta al nuevo tratamiento. (B-II)
- •
El TAR nuevo debe contener 3FAR totalmente activos (A-I). Si no es posible diseñar un TAR de rescate con 3fármacos activos, la combinación de 2 plenamente activos y otros que conserven cierta actividad puede resultar eficaz en una elevada proporción de pacientes.
- •
Se debe realizar un estudio de resistencias y una prueba de tropismo para confeccionar el mejor régimen alternativo. (A-I)
- •
La prueba de resistencias debe realizarse mientras el paciente está recibiendo el tratamiento fallido o lo más precoz tras la suspensión. (A-II)
- •
Si se dispone de pruebas genotípicas previas, siempre se debe tener en cuenta el conjunto de mutaciones detectadas (genotipo acumulado). (B-II)
- •
Se debe conseguir CVP indetectable (<50copias/ml) en cualquier tratamiento de rescate. (A-I)
- •
En la elección del nuevo TAR se deben analizar las causas que motivaron el fracaso (adherencia o interacciones medicamentosas), la historia farmacológica, las toxicidades que haya presentado y las mutaciones de resistencia previas. (C-II)
- •
El nuevo TAR debe ser cómodo, bien tolerado por el paciente y lo menos tóxico posible. (C-II)
- •
DRV/r (600/100mg BID) ha demostrado superioridad frente a LPV/r BID como IP/r en el tratamiento de rescate, siendo el fármaco de elección en este contexto. Los estudios tienen suficiente poder estadístico para confirmar esta superioridad si existen al menos una mutación primaria en la proteasa, pero no si no existe ninguna (A-I). La dosis habitual debe ser 600/100mg BID, aunque en los casos sin mutaciones para DRV ni mutaciones primarias en la proteasa puede utilizarse la dosificación 800/100mg QD. (A-I)
- •
El uso de TPV/r queda restringido a los casos en que su actividad residual estimada sea claramente superior a la de DRV/r BID, y no se requiera el uso de etravirina (contraindicada con TPV/r). (A-II)
- •
Deben evitarse los análogos de timidina, especialmente d4T, si existen otras alternativas en cualquiera de las líneas del tratamiento de rescate. (C-I)
- •
No se deben realizar interrupciones estructuradas del TAR en situaciones de fracaso virológico con el objetivo de aumentar la eficacia del TAR de rescate. (A-I)
- •
No se recomienda suspender definitivamente el TAR en pacientes con fracaso virológico avanzado y sin opciones terapéuticas de rescate, incluso aunque reciban pautas con resistencia demostrada (B-II). En esta situación debe buscarse un tratamiento basado en fármacos que disminuyan la capacidad replicativa viral y no añadan más resistencia a la ya existente (por ejemplo, 3TC o FTC o TDF) y deben vigilarse estrechamente las cifras de linfocitos CD4 y la CVP. (B-II)
- •
El manejo de los pacientes con fracaso virológico avanzado es complejo. Es recomendable consultar con un clínico o virólogo con experiencia en resistencias y TAR de rescate que tenga acceso a fármacos de uso restringido a través de programas de acceso expandido si estos pueden tener interés en el caso para conseguir un TAR con las máximas posibilidades de éxito. (C-III)
Se entiende por adherencia al TAR la capacidad del paciente para implicarse correctamente en la elección, el inicio y el cumplimiento del mismo a fin de conseguir una adecuada supresión de la replicación viral.
El control virológico depende de múltiples factores, pero la adherencia incorrecta es la primera causa de fracaso terapéutico, relacionándose con mala respuesta virológica449, peor reconstitución inmune450 y mayor riesgo de mortalidad451,452. Por tanto, es muy importante que los pacientes sean conscientes de su enfermedad, entiendan claramente el objetivo del TAR, participen en la decisión de iniciarlo, se sientan capaces de cumplir dicho tratamiento y comprendan la enorme importancia que tiene una toma continuada y correcta de la medicación.
Durante los últimos años se ha intentado conocer los factores asociados a mala adherencia. Las diferencias entre los estudios, muchos de ellos carentes del adecuado rigor metodológico, hacen difícil realizar generalizaciones con alto grado de evidencia453. Se han identificado varios factores asociados con mala adherencia, entre los que destacan: mala relación médico-paciente, consumo activo de drogas, enfermedad mental, edad más joven, nivel educativo del paciente, idioma, falta de apoyo social, complejidad del tratamiento, efectos secundarios y, más recientemente, temor acerca de la aparición de alteraciones metabólicas y morfológicas7,454,455. Un tema controvertido es la relación de la adherencia con la edad, y su importancia va a ser creciente dado el envejecimiento de la población infectada por el VIH-1. Se ha encontrado peor cumplimiento en pacientes muy jóvenes y mejor cumplimiento en pacientes >65años455. El uso concomitante de otros fármacos, la mayor prevalencia de efectos adversos, de interacciones, la depresión y las alteraciones de memoria podrían causar menor adherencia en los mayores456. Lo que parece claro es que los trastornos neurocognitivos, más frecuentes en la población de más edad, suponen un factor negativo para la adherencia457 y que este aspecto deberá ser estudiado y atendido en los próximos años. Por el contrario, el apoyo emocional y vital, la capacidad para incluir la medicación en las actividades de la vida diaria sin ocultarla y la comprensión de la relación entre adherencia y desarrollo de resistencias son factores que predicen una adherencia correcta. Corregir los primeros e incrementar los segundos forma parte de la optimización del TAR y debe incorporarse a la rutina de seguimiento de los pacientes con infección por VIH-1 (tabla 10).
Causas de adherencia incorrecta y posibles estrategias de intervención
| Causas potenciales de incumplimiento | Posibles intervenciones | |
| Factores sociales, económicos, educativos | Falta de apoyo social y/o familiar. Escasos recursos. Bajo nivel educativo | Buscar alianza con familia y allegados. Conocer necesidades sociales. Reclutar organizaciones comunitarias. Educación intensiva, explicaciones claras y comprensibles y adaptadas |
| Factores del equipo asistencial | Falta de recursos. Atención masificada e impersonal. Ausencia de coordinación entre diferentes servicios de apoyo a la asistencia. Insuficiente formación en terapia antirretroviral. Falta de accesibilidad. Deficiente formación en la relación personal sanitario-paciente | Accesibilidad y continuidad de la asistencia. Equipo multidisciplinar. Recursos materiales y humanos suficientes y coordinadosFormación sólida en terapia antirretroviral y en atención al pacientePlantear terapia directamente observada en determinados ámbitos asistenciales |
| Factores relacionados con el tratamiento | Efectos adversos, tamaño y palatabilidad de las unidades galénicas, número de dosis diarias. Intrusión en la vida del paciente. Falta de adaptación a las preferencias y necesidades del paciente | Simplificar el régimen terapéutico. Pautas convenientes en número y dosificación de comprimidos, emplear fármacos co-formuladosIndividualizar tratamiento: resistencias, comorbilidad, preferencias, interaccionesTécnicas especiales para la toma de la medicación. Ayudar a desarrollar mecanismos de reacción (por ejemplo, anticipación y manejo de efectos adversos) |
| Factores relacionados con el paciente | No aceptación. Rechazo del diagnóstico. Rechazo del tratamiento (creencias y actitudes)Olvidos y barreras. Insuficiente comprensión de la enfermedad y su tratamiento. Insuficiente entendimiento de la relación riesgo/beneficioMotivos de dosificación y cumplimientoComorbilidad psiquiátricaUso y abuso de drogas | Negociar y consensuar el plan terapéutico. Fomentar la percepción de indicadores de la necesidad de tratamiento. Informar sobre riesgos y beneficios del tratamiento. Asociar cada toma con actividades cotidianas. Técnicas especiales y ayudas para el cumplimiento (diarios de medicación, alarmas, teléfonos, etc.). Mejorar la comunicación paciente-profesional sanitario. Información referente a la enfermedad y el tratamiento, motivo de la dosificación, riesgo del incumplimiento. Información oral y escrita. Verificar comprensión. Derivar para intervención psicológica en áreas disfuncionales o intervención psiquiátrica si se detecta patología psiquiátrica |
El inicio del TAR no suele ser urgente en los pacientes con infección crónica, exceptuando los que presentan enfermedades definitorias de sida322. En los pacientes con tuberculosis, empezar el TAR dentro de las 2-4semanas siguientes al inicio del tratamiento antituberculoso se asocia a una menor mortalidad, especialmente en los pacientes con <200linfocitos CD4/μl458–460.
Antes de iniciar la terapia conviene preparar al paciente, identificar las situaciones que puedan dificultar la adherencia correcta e intentar corregirlas461. Es importante conocer los factores dependientes del paciente (laborales, restricciones dietéticas, etc.) para diseñar un TAR a la medida. Cuando se inicie el TAR es imprescindible que se ofrezca una información detallada, soporte y accesibilidad en los aspectos relacionados con el tratamiento. En este sentido el uso de un teléfono directo puede facilitar el contacto entre el paciente y los profesionales.
Durante el TAR es fundamental evaluar periódicamente la adherencia, que debe tenerse en cuenta en las decisiones terapéuticas. Dado que no existe un método fiable de evaluación, se recomienda utilizar varias técnicas como la entrevista, pasar un cuestionario estructurado (existen cuestionarios disponibles validados en España462,463), el recuento de la medicación sobrante y el registro de recogida de la medicación en la farmacia, así como la evolución clínica y virológica. Para ello es indispensable que exista una buena coordinación entre todos los estamentos implicados y en particular entre clínicos y farmacéuticos.
La entrevista y los cuestionarios estructurados son de fácil acceso en cualquier ámbito asistencial; sin embargo, no son muy precisos, y en determinadas circunstancias pueden arrojar resultados no válidos. Los métodos más sofisticados y caros para evaluar la adherencia como determinación de los niveles plasmáticos de fármacos464 o dispositivos electrónicos que registran la toma de medicación (Medication Event Monitoring System [MEMS]) se circunscriben al campo de la investigación465. Con ninguno de ellos hay información suficiente como para recomendar su utilización rutinaria en clínica.
Las características virológicas del VIH-1 determinan que cuando existen niveles subterapéuticos de los FAR el virus puede replicarse y desarrollar resistencias. Los datos obtenidos durante los primeros tratamientos combinados, basados en IP sin potenciar, constataron que la máxima eficacia requería una adherencia prácticamente perfecta, clásicamente >95%466. Estudios recientes sugieren que con niveles menores se pueden alcanzar los objetivos terapéuticos en regímenes basados en ITINN o IP/r, especialmente en pacientes que consiguen viremias indetectables467–469. Debe destacarse que no solo es importante el porcentaje de dosis omitidas sino también los patrones de adhesión subóptima. Las interrupciones de tratamiento (más de 2días sin tomar ningún fármaco) presentan mayor repercusión en la respuesta virológica que la omisión ocasional de dosis470. En la terapia de inicio también se ha demostrado una relación lineal entre nivel de adherencia y efectividad: en tratamientos basados en ITINN, por cada 10% de incremento en el cumplimiento se observó un 10% más de pacientes que consiguen viremias indetectables sostenidas471. También pudieron evidenciarse diferencias entre distintos IP/r y su relación entre adherencia y efectividad, y la adherencia subóptima a DRV/r tuvo un impacto menor comparado con LPV/r en pacientes sin tratamiento previo472. Esta diferencia se mantuvo al realizar un análisis multivariante de regresión logística que ajustó múltiples factores de confusión (CVP y cifra de CD4 basales, edad y raza).
La relación entre adherencia y desarrollo de resistencias es más compleja que la idea establecida de que «la no adherencia aumenta el riesgo de resistencias». Se han encontrado diferencias en función de las familias de fármacos; así, en pautas basadas en IP no potenciados se comprobó la aparición de resistencias con niveles altos de cumplimiento, mientras que en pautas con ITINN las resistencias son inusuales en los pacientes muy cumplidores, dándose en los poco adherentes y especialmente en los pacientes con interrupciones prolongadas del tratamiento473. Por el contrario, en el caso de los IP potenciados la aparición de resistencias es mucho más difícil con cualquier nivel de adherencia, debido a su elevada barrera genética474.
En cualquier caso, ante un paciente con alta sospecha de presentar dificultades para el cumplimiento terapéutico es mejor evitar las pautas basadas en FAR de baja barrera genética (como ITINN de primera generación o raltegravir) e iniciar TAR con pautas basadas en IP/r, que evitan el riesgo de seleccionar resistencias relevantes en caso de incumplimiento y fracaso virológico.
Si se detecta falta de adherencia, debe intervenirse de forma activa para corregirla. La monitorización de la adherencia no debe utilizarse para explicar un fracaso o el desarrollo de resistencias, sino para prevenir que estos se produzcan mediante la detección precoz de los problemas e implementar rápidamente medidas correctoras475. A modo de ejemplo, cuando se analizó la adherencia fármaco por fármaco, en lugar de hacerlo de forma global, se detectó que el 30% de los pacientes tenían falta de adherencia diferencial —es decir, a alguno de los componentes del tratamiento—, y esta falta de adherencia se relacionó con fracaso virológico476. La coformulación de fármacos simplifica el TAR y podría prevenir y corregir este problema, mejorando la adherencia global477–479 e impidiendo la adherencia selectiva en pacientes que reciben tratamiento triple. De este modo se reducen las posibilidades de selección de resistencias417 por monoterapia encubierta.
Frecuentemente, en pacientes con fracaso virológico asociado a mala adherencia se intenta priorizar un tratamiento QD en el rescate para favorecer su cumplimiento. En este sentido, no existe evidencia suficiente para asegurar que en este escenario existirá mejor cumplimiento con un tratamiento QD que con otro administrado BID. Por este motivo, debe priorizarse siempre el mejor régimen de TAR según el estudio de resistencias disponible, la historia terapéutica del paciente, administrando los fármacos en las dosis en que han sido evaluados en sus respectivos ensayos clínicos.
Las estrategias son múltiples, algunas basadas en teorías psicológicas480 y la mayoría dirigidas a paliar los condicionantes sociales o individuales de los pacientes. Desde esta perspectiva cabe analizar las intervenciones sobre adherencia mediante las denominadas «entrevistas motivacionales»481, las visitas domiciliarias y la intervención específica sobre la pareja del paciente, todas ellas relativamente complejas, sin resultados definitivos y sin grandes diferencias frente al grupo control asesorado acorde a una buena práctica clínica. Las intervenciones dirigidas a ciertas poblaciones especiales (mujeres, latinos en Estados Unidos, y pacientes con historia previa de alcoholismo) no han logrado mejorar la adherencia al tratamiento453.
Las intervenciones sencillas de educación y soporte en aspectos prácticos del tratamiento han demostrado ser beneficiosas482. Probablemente la intervención que ha demostrado mayor eficacia ha sido el soporte interpersonal estructurado, en el que personal sanitario entrenado emplea estrategias individualizadas.
Respecto al tratamiento directamente observado (TDO), en un metaanálisis de 17estudios se ha demostrado que incrementa la probabilidad de conseguir CVP indetectable (HR: 1,24; IC95%: 1,08-1,41), mayor incremento de CD4 y adherencia >95%. Este beneficio es incluso mayor cuando se aplica a determinadas poblaciones con grandes dificultades para un cumplimiento óptimo, como usuarios de drogas o en prisiones, pero no parece mantenerse a largo plazo tras cesar la intervención482,483.
La adhesión al tratamiento puede decaer con el tiempo, y por tanto las estrategias diseñadas para optimizarla deben dirigirse no solo a incrementarla sino a mantenerla constante484,485.
GeSIDA y el PNS, conjuntamente con la Sociedad Española de Farmacia Hospitalaria, han revisado los factores que influyen en la adherencia, los métodos de evaluación y las posibles estrategias de intervención y actuación de un equipo multidisciplinar que debe ser integrado por médicos, farmacéuticos, enfermeras, psicólogos y personal de soporte. Remitimos a ese documento7 para profundizar en el tema de la adherencia al TAR.
Recomendaciones- •
Antes de iniciar el TAR se debe preparar al paciente, identificar y corregir las causas que pueden limitar su adherencia. Si el paciente no está preparado, en general es mejor retrasar el inicio del TAR. (A-III)
- •
Una vez iniciado el TAR, se recomienda efectuar un primer control a las 2-4semanas para corregir los factores inherentes al tratamiento o del propio paciente que puedan limitar la adherencia. (B-III)
- •
Si la adherencia es correcta, debe monitorizarse y reforzarse, coincidiendo con las visitas clínicas. (A-III)
- •
El control de la adherencia debe realizarse por un equipo multidisciplinar, y en él deben estar implicados no solo el médico sino también la enfermería, los profesionales de apoyo psicológico y la farmacia hospitalaria. (B-III)
- •
Cada unidad asistencial debería realizar un seguimiento periódico de la adherencia, no solo con vistas a detectar las faltas individuales de cumplimiento sino para conocer la magnitud del problema en su ámbito de trabajo; el análisis de los datos permitirá determinar las causas de los problemas detectados (abandonos, vacaciones terapéuticas, incumplimientos…) y elaborar estrategias concretas de actuación, tanto con los enfermos como en la estructura y en el funcionamiento del equipo asistencial. (B-III)
- •
En pacientes con cumplimiento irregular es preferible utilizar pautas basadas en IP/r frente a las basadas en ITINN para dificultar la selección de resistencias. (B-III)
- •
La combinación a dosis fijas de FAR simplifica el TAR y, por tanto, facilita la adhesión mantenida. El uso de regímenes completos en comprimido único constituye la estrategia más eficiente para prevenir la adherencia selectiva de fármacos. (A-III)
Los FAR pueden producir numerosos efectos secundarios que se presentan al inicio del tratamiento o a medio-largo plazo. En este último caso suelen asociarse a procesos fisiológicos o comorbilidades relacionados con el envejecimiento. Algunos de ellos son específicos de fármacos, y otros, de grupo. En las tablas 11–15 se muestra la toxicidad característica de cada familia y se resume la toxicidad por órganos y aparatos, la patogenia, la relación individual con cada fármaco, el diagnóstico y el tratamiento de los mismos. A continuación se comentan los que, por su frecuencia y/o potencial gravedad, tienen mayor relevancia clínica.
Toxicidad de cada familia de fármacos antirretrovirales
| Familia | Toxicidad | Prevalencia | Clínica |
| Inhibidores de la transcriptasa inversa nucleósidos o nucleótidosa | Toxicidad mitocondrial | 20-40% | Neuropatía periféricaMiopatíaCardiomiopatíaPancreatitisHepatomegaliaEsteatosis hepáticaHepatitisAcidosis lácticaMielotoxicidadAlteración tubular proximal renalLipoatrofiaHiperlipidemia y resistencia insulínica |
| Inhibidores de la transcriptasa inversa no nucleósidosb | Hipersensibilidad | 10-20% | Exantema (extensión y gravedad variable)Afectación multiorgánicaFiebre |
| Inhibidores de la proteasac | HiperlipidemiaResistencia a la insulina (IP clásicos; difícil de separar de los efectos de análogos de la timidina) y lipodistrofia | 25-50% | Hipertrigliceridemia (especialmente)HipercolesterolemiaDiabetes mellitusLipoacumulación intraabdominal |
| Inhibidores de la fusiónd | Inflamación dérmica local | 60-70% | DolorTumoración |
| Inhibidores de los receptores CCR5e | Mecanismo desconocido (¿bloqueo receptor CCR5?) | <2% | ↑ riesgo de infecciones respiratorias¿↑ riesgo de cardiopatía isquémica?e |
| Inhibidores de la integrasaf | Mecanismo desconocido | 5-10%f | Elevación de CPKf |
Las distintas manifestaciones de la toxicidad se suelen presentar de forma aislada. Aparecen generalmente en los 3 primeros meses de tratamiento, aunque algunas expresiones de la toxicidad mitocondrial (neuropatía, miopatía, acidosis láctica o lipoatrofia) suelen aparecer de forma tardía (meses o años).
ABC constituye una excepción, pues su toxicidad está mediada por hipersensibilidad. TDF puede inducir nefrotoxicidad.
Indinavir puede causar nefrolitiasis e insuficiencia renal como efectos adversos singulares. Atazanavir no comporta el riesgo de alteraciones metabólicas, como otros IP, pero puede producir hiperbilirrubinemia y nefrolitiasis como efectos adversos singulares.
La enfuvirtida (T-20), único representante de esta familia disponible en la actualidad, se administra por vía subcutánea.
Toxicidad de los antirretrovirales por órganos y aparatos
| Toxicidad | Fármaco/s | Diagnóstico | Patogenia | Actitud |
| Anemia | AZT (dosis-dependiente) | Síntomas clínicos de anemia | Inhibición de la proliferación de las células progenitoras eritroides | Suspensión de AZTTransfusión de hematíes (si hemoglobina <8g/dl o hay síntomas de anemia) |
| Miopatía | AZT (dependiente de la dosis)RAL | Después del primer semestre de tratamiento (AZT)Síntomas: mialgias o debilidad muscular proximal y elevación de enzimas musculares (CPK, LDH, aldolasa) | Toxicidad mitocondrial (AZT)Mecanismo desconocido (RAL) | Suspensión de AZT o RALSi clínica importante, prednisona 1-2mg/kg/día (AZT) |
| Neuropatía periférica | ddI (13-34%)d4T (15-20%)(dependiente de la dosis) | Hipoestesia, parestesia o dolor en zona distal de extremidades (especialmente pies)Diagnóstico diferencial con la neuropatía por el propio VIH-1 (aparece en pacientes sin tratamiento e inmunodepresión grave) | Interacción entre citoquinas y factores de crecimiento neuronalFavorecida por neuropatía previa y factores predisponentes (enolismo, desnutrición, diabetes, etc.) | Evitar asociaciones de fármacos neurotóxicosValorar mantener fármacos potencialmente implicados si la clínica no es grave y no hay alternativas razonablesSuspender los fármacos implicados si clínica progresiva o invalidante. La recuperación es lenta (meses o años)Si dolor leve: analgésicos habitualesSi dolor moderado-intenso: gabapentina, pregabalina o lamotrigina con o sin benzodiacepinas; si no eficacia, valorar carbamacepina y/o amitriptilina (producen efectos colinérgicos)Si dolor muy intenso o refractario: opiáceos |
| Toxicidad neuropsíquica | EFV (20-50%)(dependiente de la dosis; más prevalente si administración con alimentos) | Durante el primer mes de tratamiento, tras lo cual disminuye o desapareceEspectro clínico variado: mareo, ansiedad, somnolencia, trastornos del sueño, agravamiento de problemas psíquicos subyacentes, y alteraciones motoras | Desconocida | Evitar en pacientes con trastornos psiquiátricos mayoresValorar individualmente el estilo de vida y la actividad del paciente antes de prescribirloGeneralmente no es necesaria la suspensión de efavirenzSuspender en casos de manifestaciones graves o invalidantesAdministración por la noche, al menos 1-2h después de la cenaValorar benzodiacepinas o neurolépticos si alteraciones del sueño persistentesPuede intentarse ajuste de dosis si hay posibilidad de estudio farmacocinético, aunque no está definitivamente probado |
| Toxicidad | Fármaco/s | Diagnóstico | Patogenia | Actitud |
| Exantema y/o hipersensibilidad | No-nucleósidos (más frecuente NVP) (<20%)Inhibidores de la proteasa (más frecuente TPV (8-14%), DRV y FPV (3-5%)Nucleósidos (más frecuente ABC: 5% si no se realiza determinación de HLA B*5701; si se realiza, la frecuencia es 0%) | Durante los primeros 2 meses de tratamientoExantema maculopapular (casos leves)Fiebre, afectación mucosa, pulmonar, hepática o hematológica (casos graves)La hipersensibilidad se manifiesta por afectación multiórganica, a veces con escaso o nulo exantema, y puede haber eosinofilia | DesconocidaSe ha sugerido una reacción antígeno-anticuerpo similar a la enfermedad del suero o una toxicidad directa por metabolitos intermediariosIdentificación de pacientes con riesgo genético elevado de hipersensibilidad a abacavir (HLA-B*5701) | Comienzo escalonado de dosis de nevirapina. No dar corticoides profilácticos (no evitan el riesgo e incluso lo pueden aumentar) ni antihistamínicosSi exantema leve sin clínica de hipersensibilidad acompañante, se puede mantener el tratamiento y realizar una vigilancia estrechaEl exantema por ETR generalmente es leve-moderado y raramente obliga a retirar el fármacoSi exantema grave o clínica de hipersensibilidad, interrupción permanente del fármacoEn los casos donde pueda estar implicado ABC y la clínica no sea clara, puede ser razonable mantener el tratamiento durante 24h más con una vigilancia estrecha y valorar la evolución antes de retirar ABCTratamiento sintomático con antihistamínicos y/o corticoides una vez interrumpido el fármaco sospechosoTratamiento de soporte hemodinámico, o respiratorio en casos graves que lo requieranNO REINTRODUCIR NUNCA UN FÁRMACO RETIRADO POR SOSPECHA DE HIPERSENSIBILIDAD |
| Hepatitis | Nucleósidos (más frecuente AZT, ddI y d4T). No nucleósidos (más frecuente NVP)Inhibidores de la proteasa (más frecuente RTV a dosis plena) | El 50% de los casos aparece en el primer semestreAumento de transaminasas sin clínica (10-15% de pacientes)Hepatitis clínica (<1%)Factores de riesgo: infección por virus de la hepatitis B y C | Multifactorial: toxicidad mitocondrial (nucleósidos), efecto tóxico-inmunológico (no nucleósidos); si IP efecto mixto (toxicidad directa, recuperación inmune si VHB o VHC), rebrote de virus B tras suspender TAR con efecto anti-VHB | Considerar vigilancia estrecha y potencial suspensión si transaminasas >5 veces el límite superior de normalidadInterrumpir si transaminasas >10 veces límite superior de normalidad, manifestaciones clínicas de hipersensibilidad (fiebre o exantema), de fallo hepático (ictericia, encefalopatía o hemorragia) o acidosis láctica |
| Toxicidad gastrointestinal | Inhibidores de la proteasa sobre todo RTV (a dosis plenas), 40%; IDV, 25%; NFV, 25%; LPV/r, 25%; SQV, 5%Nucleósidos con menor frecuencia que IP (particularmente AZT y TDF) | Sabor desagradable (RTV suspensión)Molestias digestivas altas (IDV y APV)Diarrea (NFV y LPV/r) | MultifactorialInhibición de enzimas pancreáticas (inhibidores de la proteasa)Intolerancia a la lactosa que contienen como excipiente (todos los antirretrovirales) | Raramente grave, pero por su frecuencia e incomodidad puede limitar la adherencia al TARPara la diarrea, dietas rica en alimentos astringentes o fibra soluble o fármacos inhibidores de la motilidad intestinal (loperamida)Suspensión del fármaco si molestias persistentes o intensas |
| Pancreatitis | Nucleósidos (más frecuente ddI y d4T).Aumento de riesgo cuando se administra hidroxiurea, o TDF con ddI | Generalmente asintomáticaPuede haber manifestaciones clínicas de dolor abdominal y diarrea | Toxicidad mitocondrial | Retirar el fármaco potencialmente implicado |
| Insuficiencia renal y tubulopatía | TDFIDV | Elevación leve o moderada de la creatinina con/sin anomalías analíticas de disfunción tubular (hipofosfatemia)No suele acompañarse de clínica | Alteración tubular proximal (TDF)Nefritis intersticial por cristales (IDV) | Evitar en pacientes con insuficiencia renal. Evitar en lo posible la coadministración con otros fármacos potencialmente nefrotóxicosHidratación adecuada para prevenir o mejorar la elevación de creatininaSuspensión del fármaco si el filtrado glomerular es <50ml/min |
| Nefrolitiasis | IDVATV | Dolor cólico lumbar. Hematuria microscópica y ocasionalmente macroscópica. Ocasionalmente, fiebre (diagnóstico diferencial con pielonefritis)Más frecuente en ambientes calurosos | Precipitación de indinavir en orina concentrada (densidad >1.020) y pH básico (>5) | Prevención mediante ingesta adecuada de líquido (1.500ml de agua al día o más si ambiente caluroso o pérdidas extraordinarias de líquidos). Evitar bebidas carbónicasAntiinflamatorios no esteroideos para el dolorSuspensión transitoria del fármaco responsable si dolor intensoSuspensión definitiva del fármaco responsable si episodios repetidos sin desencadenante evidenteAjuste de dosis si posibilidad de estudio farmacocinético |
Evaluación y tratamiento de la dislipidemia
| Evaluación | Tratamiento |
| Realizar analítica en ayunasDescartar otras causas de hiperlipidemia secundariaTratamiento encaminado a prevenir la enfermedad aterosclerótica y a evitar las complicaciones inmediatas de la hipertrigliceridemia grave | Objetivo del tratamiento:- Conseguir una cifra de colesterolLDL, según el riesgo. Si enfermedad coronaria o riesgo equivalente, LDL<70mg/dl; si RCV≥20% (Framingham), LDL<100mg/dl- Si triglicéridos >500mg/dl, se tratará la hipertrigliceridemia independientemente de la concentración de colesterolLDLTratamiento:- En primer lugar, medidas generales: dieta (consulta a experto en nutrición), ejercicio físico, abstinencia de tabaco, y sobre todo valoración individualizada de la retirada de IP y/o de los análogos de la timidina.Tratamiento farmacológico (si las medidas previas no son eficaces):- Estatinas. Utilizar preferentemente atorvastatina, pravastatina, rosuvastatina o pitavastatina; precaución por interacciones con IP. Valorar ezetimiba o la combinación ácido nicotínico/laropiprant solas (si no se pueden usar estatinas) o asociadas a estatinas (si no se consiguen los objetivos de tratamiento con estatinas solas)- Fibratos si hipertrigliceridemia aislada o junto a elevación moderada de colesterolLDL. En caso de hipertrigliceridemia intensa y refractaria pueden añadirse ácidos grasos omega-3.- Precaución con la coadministración de fibratos y estatinas (mayor riesgo de toxicidad muscular) |
Evaluación y tratamiento de la diabetes mellitus
| Evaluación | Tratamiento |
| Realizar analítica en ayunasDiagnóstico:Determinación de la hemoglobina glucosiladaTolerancia oral a la glucosa (si glucosa basal alterada)Tratamiento encaminado a evitar las complicaciones metabólicas a corto plazo (hipoglucemia, cetosis, cetoacidosis, y estado hiperosmolar) y las complicaciones micro y macrovasculares a largo plazo | Valorar individualizadamente la sustitución de análogos de la timidina y otros fármacos no antirretrovirales que pudieran estar implicadosObjetivo terapéutico guiado por hemoglobina glucosilada (HbA1c)<6,5%Consulta con endocrinólogoAntidiabéticos orales. Metformina, si sobrepeso u obesidad abdominal, o sulfonilureas. Pioglitazona si lipoatrofiaInsulina en los pacientes con diabetes de inicio e insulinopenia (cetosis o cetoacidosis), pérdida de peso y cuando no sea posible conseguir el objetivo de HbA1c<7% con antidiabéticos oralesDebería considerarse la administración de 75-150mg/día de AAS a todos los pacientes con diabetes |
Evaluación y tratamiento de la alteración de la distribución de la grasa corporala
| Evaluación | Tratamiento |
| Diagnóstico clínicoSería deseable la realización periódica de alguna medida objetiva de la composición corporal (según disponibilidad y posibilidades económicas de cada centro) | No hay ninguna medida que haya demostrado resolver satisfactoriamente los cambios corporales. Las que a continuación se describen han mostrado, en el mejor de los casos, una eficacia parcial y algunas de ellas no están exentas de riesgos:Medidas generales (dieta, ejercicio físico): evitar modificaciones de peso >5% del peso ideal; el ejercicio físico aeróbico mejora las alteraciones metabólicas y la lipoacumulación intraabdominalSustitución de FAR (IP, ITIAN): la retirada de los IP clásicos puede mejorar las alteraciones metabólicas y la lipoacumulación intraabdominal; la retirada de análogos de timidina mejora la lipoatrofia (existen más estudios con d4T que con AZT)Fármacos con efectos metabólicos (metformina, glitazonas, hormona del crecimiento). La hormona del crecimiento puede disminuir la lipoacumulación intraabdominal pero provoca hiperglucemia y otros efectos secundarios. Se han comunicado buenos resultados en la lipoacumulación visceral con tesamorelina (análogo del factor estimulante de la hormona de crecimiento). Los efectos de todos estos fármacos revierten con el cese del mismo y ninguno de ellos tiene indicación para el tratamiento de la lipodistrofiaCirugía plástica (relleno facial en lipoatrofia, cirugía reductora en lipoacúmulos accesibles): es el único tratamiento actual con resultados inmediatos |
En la tabla 11 se exponen los posibles efectos adversos debidos al daño mitocondrial producido por la inhibición de la enzima ADN-polimerasa mitocondrial. El más grave de ellos —una combinación de acidosis láctica y esteatosis hepática (ALEH)— es inducido por d4T y, en menor medida, por ZDV y ddI. Su incidencia es baja, pero si no se diagnostica a tiempo puede ser mortal486. Las manifestaciones clínicas, subagudas e inespecíficas (astenia, disnea e insuficiencia hepática), o la acidosis, aparecen cuando las concentraciones plasmáticas de lactato son >5-10mmol/l. Debe tenerse un alto grado de sospecha y realizar el diagnóstico antes de que aparezca acidosis, pues en esta fase (hiperlactatemia asintomática) la mortalidad es muy inferior a la de la ALEH. Su tratamiento consiste en retirar los ITIAN responsables y monitorizar los niveles plasmáticos de lactato.
La lipoatrofia, aunque también es secundaria a toxicidad mitocondrial, se comenta en el apartado de anomalías de la distribución de la grasa corporal487.
Reacciones de hipersensibilidadAunque todos los FAR pueden provocar RHS, son mucho más frecuentes con ABC y los ITINN. La reacción de hipersensibilidad a ABC se ha descrito más arriba113,254.
Las RHS frente a los ITINN suelen presentarse con un exantema cutáneo y rara vez con un cuadro sistémico grave o con síndrome de Stevens-Johnson o necrólisis epidérmica tóxica (tabla 12).
HepatotoxicidadEs, junto al exantema, el efecto adverso específico de grupo más importante de los ITINN, si bien su intensidad suele ser leve o moderada y son raros los casos de hepatitis sintomática. NVP provoca elevación de transaminasas con mayor frecuencia y se desaconseja su uso en mujeres con >250CD4/μl y varones con >400CD4/μl debido al mayor riesgo de hepatotoxicidad grave363,365. No obstante, en pacientes pre-tratados con CVP indetectable, generalmente en el contexto de una simplificación del TAR, ese mayor riesgo no existe362,488, lo que ha llevado a un cambio en la ficha técnica de NVP. Los IP potenciados que se usan actualmente y los FAR de otras familias tienen tasas de hepatotoxicidad más reducidas que los ITINN489–493(tabla 12).
Trastornos neuropsiquiátricosEFV produce diversos síntomas neuropsiquiátricos (mareo, somnolencia, insomnio, sueños vividos, confusión, ansiedad, despersonalización, etc.) en más del 50% de los casos, los cuales, aunque suelen remitir en las primeras 2-4semanas, obligan a interrumpir el tratamiento en un porcentaje de los casos494 (tabla 12). Se aconseja evitar este fármaco en pacientes con trastornos psiquiátricos mayores, aunque en un estudio aleatorizado el riesgo de depresión fue similar con EFV que con los IP495. Aunque otros FAR, como ZDV, pueden provocar síntomas neurológicos, son mucho menos frecuentes y predecibles que los de EFV.
NefrotoxicidadTDF puede inducir toxicidad renal en una pequeña proporción de pacientes. Su incidencia en estudios de cohortes496 es mayor que la observada en ensayos clínicos, en los que solamente se ha encontrado una reducción discreta (≈10%) y no progresiva del filtrado glomerular244,497. Esta complicación consiste en una disfunción tubular y se manifiesta como un síndrome de Fanconi acompañado de una disminución del filtrado glomerular498. Es más frecuente cuando coincide con otros factores de riesgo (insuficiencia renal previa o concomitante, diabetes, hipertensión arterial, fármacos nefrotóxicos, edad avanzada, bajo peso corporal y cifras bajas de linfocitos CD4+)244,247,498–500. Su incidencia es mayor en combinación con IP/r y ddI501. La nefrotoxicidad por TDF suele revertir al retirar el fármaco, aunque la reversión puede no ser completa502. IDV y ATV, este último con mucho menor frecuencia, producen nefrolitiasis por depósito tubular de cristales (tabla 12).
Trastornos metabólicos y riesgo cardiovascularEste grupo de trastornos incluye dislipidemia, resistencia a la insulina y diabetes mellitus, todos los cuales son más frecuentes con los IP clásicos que con los IP potenciados actualmente utilizados y con los análogos de timidina que con el resto de ITIAN. Asimismo, todas estas anomalías son más comunes en los pacientes con redistribución de la grasa corporal.
La dislipidemia —caracterizada por aumento de los niveles plasmáticos de colesterol total, colesterolLDL y, sobre todo, triglicéridos— es la anomalía metabólica que con mayor frecuencia se asocia al TAR. Aunque tradicionalmente se ha relacionado esta dislipidemia con los IP, no todos ellos tienen el mismo impacto sobre los lípidos, y otros FAR, especialmente los análogos de timidina, también inducen dislipidemia285,306,434,503,504. ABC o la formulación a dosis fija ABC/3TC aumentan los lípidos plasmáticos, mientras que TDF o la formulación a dosis fija TDF/FTC los disminuye, aunque el cociente colesterol total/colesterolHDL se mantiene por igual con ambos248,385. Respecto a los IP, diversos ensayos clínicos han revelado que ATV y FPV, sobre todo sin potenciar, tienen un mejor perfil lipídico; ATV/r, SQV/r (1.500/100 BID o 1.500/100 QD) y DRV/r (600/100 BID u 800/100 QD) presentan perfiles intermedios; mientras que FPV/r (1.400/100 QD o 700/100 BID), LPV/r (400/100 BID) y TPV/r (500/200 BID) tienen perfiles más desfavorables, sobre todo en lo que respecta a los triglicéridos266,303,305,307,374,375,386,435,436,443,505,506. Sin embargo, cuando se compara el cociente CT/HDL en pacientes tratados con DRV/r, ATV/r o LPV/r no existen diferencias entre los distintos regímenes; se desconoce la importancia real de este hecho. El manejo de la dislipidemia se comenta en la tabla 13383,507,508.
Los IP pueden disminuir la disponibilidad de la glucosa periférica y la secreción pancreática de insulina de forma aguda y pueden descompensar o desencadenar una diabetes en pacientes ya diabéticos o en personas predispuestas. En general, estas alteraciones no se mantienen a medio-largo plazo, por lo que deben existir mecanismos compensadores de las mismas. IDV y RTV a dosis plenas son los que se han relacionado con el desarrollo de resistencia a la insulina509,510, mientras que los demás IP parecen tener poco o ningún efecto clínicamente significativo sobre la homeostasis de la glucosa. La diabetes mellitus es menos común (tabla 14) y su asociación con los IP no es constante en todos los estudios508,511. El tratamiento con análogos de la timidina (en particular d4T) y ddI constituye un factor de riesgo para el desarrollo de resistencia a la insulina y diabetes mellitus509,512. Los inhibidores de la integrasa y los antagonistas del CCR5 tienen un perfil metabólico favorable171,312.
Al igual que en la población general, los factores de riesgo tradicionales son los que determinan principalmente el riesgo de desarrollar enfermedad cardiovascular en los pacientes infectados por el VIH-1. El efecto de la inflamación crónica de bajo grado y de la activación inmune puede poseer un papel relevante en la patogenia de la enfermedad cardiovascular de los pacientes con infección VIH-1513. La infección por VIH-1 no controlada y ciertos regímenes de TAR incrementan también dicho riesgo, aunque es muy probable que la contribución de estos últimos sea menor que la de los otros factores anteriormente mencionados. Es destacable la elevada frecuencia de factores de riesgo tradicionales modificables, lo que hace recomendable un manejo más agresivo de ellos514. Aunque algunos estudios de cohortes han puesto de manifiesto que la duración del tratamiento con IP es un factor de riesgo independiente para el desarrollo de cardiopatía isquémica515–517, otros, en su mayoría retrospectivos y con poco tiempo de seguimiento, han obtenido resultados contrapuestos517–520. El efecto sobre el riesgo cardiovascular de los IP como familia está mediado, al menos en parte, por la dislipidemia asociada con el uso de estos fármacos. Además, un análisis de la cohorte D:A:D ha revelado que determinados IP (IDV, LPV/r) están relacionados con un mayor riesgo de infarto de miocardio, que no puede ser exclusivamente justificado por dicha anomalía metabólica521. Este mismo estudio ha mostrado también que el uso reciente (últimos 6meses) de ABC o ddI se asocia con un mayor riesgo de infarto agudo de miocardio, especialmente en los pacientes que tienen un riesgo cardiovascular más elevado521. No obstante, la relación entre ABC e infarto de miocardio o enfermedad cerebrovascular es un motivo de controversia, pues aunque ha sido constatada también en el estudio SMART256, en sendos estudios de casos y controles258 y en otros estudios observacionales522, ni un análisis conjunto de 52ensayos clínicos aleatorizados que incluían un brazo de tratamiento con ABC260 ni otros ensayos clínicos y estudios de cohorte han observado tal asociación523,524, así como tampoco ninguna relación de ABC con potenciales mecanismos patogénicos que pudieran explicarla388. Dos metaanálisis han mostrado que no existe asociación epidemiológica entre el uso de ABC y el riesgo de infarto agudo de miocardio259,261. La controversia existente pone de manifiesto que no es posible controlar de forma adecuada posibles sesgos en los estudios que han implicado a ABC o ddI y ponen en duda la contribución causal de los mencionados ITIAN en el desarrollo de la enfermedad cardiovascular.
Envejecimiento prematuro en la infección por el virus de la inmunodeficiencia humanaLa eficacia del tratamiento ha permitido que muchos pacientes alcancen una supervivencia similar a la de la población general. De hecho, una elevada proporción de pacientes se encuentran en la actualidad alrededor de la edad de 50años. Existen similitudes biológicas en términos de senescencia del sistema inmune de pacientes con infección por el VIH jóvenes y las observadas en el envejecimiento fisiológico525; por otra parte, en pacientes VIH-positivos relativamente jóvenes se describen enfermedades observadas con frecuencia en edades más avanzadas, como osteoporosis, deterioro cognitivo o sarcopenia526. Para estas enfermedades crónicas se han reconocido factores de riesgo similares a los de la población general pero sobrerrepresentados en pacientes VIH-positivos (como el tabaco) y otros propios de la infección por VIH, como la inmunodeficiencia (nadir de CD4), la inmunoactivación y la inflamación527. Están justificadas la monitorización y las intervenciones de prevención, al menos similares a las recomendadas para la población general.
Anomalías de la distribución de la grasa corporalEl síndrome de lipodistrofia se caracteriza por la presencia, combinada o no, de pérdida de grasa periférica (lipoatrofia) y de acumulación de grasa perivisceral y/o en abdomen, mamas y cuello (lipoacumulación). La lipoatrofia, el efecto adverso más temido por los pacientes, se relaciona particularmente con los análogos de la timidina243,244,247,383,434,487,497,508. Aunque en su aparición pueden influir múltiples factores, las pautas con d4T y ZDV (particularmente junto con IP clásicos y EFV) han evidenciado un mayor riesgo de lipoatrofia, y su sustitución por ABC o TDF o por pautas sin ITIAN se asocia a una mejoría de la misma244,247,383,385,434,497,500,507,508 (tabla 12). Los resultados de 2estudios comparativos entre EFV y LPV/r sugieren que el uso de EFV puede asociarse a una mayor pérdida de grasa subcutánea en comparación con LPV/r cuando el régimen incluye análogos de timidina326,528. Esta diferencia no se observa en el estudio ACTG 5224s (subestudio metabólico del ACTG 5202), que compara EFV con ATV/r asociado a 2ITIAN (ABC/3TC o FTC/TDF) en el que hay una ganancia global de grasa, aunque hasta el 16,3% tienen una pérdida de grasa igual o superior al 10%, pero sin diferencias en función del tratamiento529. El que no se haya observado lipoatrofia en otros ensayos en los que EFV tampoco se asociaba a análogos de timidina244,247,383,386,497 indica que EFV no parece estar directamente implicado en un mayor riesgo de lipoatrofia. En el seguimiento a un año del estudio MONARK (comentado antes) la presencia de lipoatrofia en la rama de monoterapia con LPV/r era del 5% frente al 27% en los pacientes en triple terapia530. En la mayoría de ensayos clínicos en pacientes que nunca habían recibido tratamiento llevados a cabo en los últimos años con ITIAN no timidínicos y EFV, RAL, ATV/r o LPV/r, la incidencia de lipoatrofia, definida como la pérdida de >20% de la grasa en extremidades a las 48-96 semanas, por densitometría, es inferior al 10% de los casos308,531. En la tabla 15 se comentan la evaluación y las opciones terapéuticas para las anomalías de la distribución de la grasa corporal532–535.
Osteoporosis y riesgo de fracturas óseasLa osteoporosis es más frecuente en pacientes con infección por el VIH-1, especialmente si reciben TAR536,537. El número de fracturas también es superior en pacientes con infección VIH-1 que en sujetos de similares características sin esta infección. La base patogénica que justifica esta reducción en la densidad mineral ósea (DMO) es un incremento del remodelamiento óseo538 que condiciona una pérdida progresiva de la DMO. Los mecanismos que justifican esta reducción de la DMO son similares a los descritos en población general, entre los que se incluyen la edad, el tabaquismo, el bajo peso corporal, la insuficiencia renal, la diabetes, la hepatitis C, el uso de opiáceos, el consumo excesivo de alcohol o la menopausia. Algunos aspectos propios de la infección por VIH-1, como valores bajos de linfocitos CD4+539 o el efecto de algunos fármacos, pueden influir en la mayor incidencia de fracturas. Los regímenes basados en TDF reducen la DMO en mayor medida que los que contienen ABC540,541 o que los regímenes sin ITIAN. Esta reducción de la DMO se ha documentado en varones no infectados por el VIH que toman TDF/FTC como profilaxis pre-exposición en sus relaciones homosexuales542,543. Se desconoce la importancia clínica real de la osteoporosis en los pacientes con infección VIH-1, pero dado que los pacientes irán envejeciendo, es razonable esperar un incremento significativo del número de fracturas óseas en las próximas décadas. No se conoce cuál es el mejor abordaje de este problema, la importancia de las escalas de predicción del riesgo de fracturas y el papel de los fármacos antirreabsortivos (bisfosfonatos, en particular). Es aconsejable influir en los factores de riesgo tradicionales, mantener una adecuada ingesta de calcio y unos niveles de vitamina D dentro de límites normales. Existen documentos específicos publicados por GESIDA/PNS referidos al manejo de los trastornos óseos en pacientes con infección por el VIH-1544.
Recomendaciones- •
Se deben monitorizar la tolerancia y las reacciones adversas agudas del TAR durante las primeras 2-4 semanas, particularmente en los pacientes que tengan comorbilidades predisponentes o tomen otros fármacos cuyas interacciones puedan tener consecuencias clínicas, y llevar a cabo una eventual modificación del tratamiento según la gravedad de la reacción adversa y el fármaco implicado. Para ello debe facilitarse el contacto entre el paciente y los profesionales. (A-III)
- •
Se deben evitar fármacos que puedan reagudizar o empeorar enfermedades preexistentes. (A-III)
- •
Se recomienda monitorizar la glucemia y los lípidos plasmáticos (colesterol total, colesterolHDL, colesterolLDL y triglicéridos) en ayunas en cada visita de control. (A-II)
- •
Se recomienda calcular el riesgo cardiovascular al menos una vez al año. (B-III)
- •
Se recomienda efectuar un estudio elemental de orina con proteinuria y calcular la tasa de filtrado glomerular renal (fórmula MDRD o Cockroft-Gault) en la primera visita y luego una vez al año si no hay factores de riesgo para el desarrollo de nefrotoxicidad o cada 6meses si estos están presentes, así como también antes de iniciar el TAR (B-III). En los pacientes que ya reciben TAR se recomienda efectuar este estudio en todas las revisiones (A-III), en especial si toman TDF (A-II). Si el filtrado glomerular es <50ml/min o hay proteinuria manifiesta no se deben usar TDF ni IDV y se deben ajustar las dosis o intervalos de los ITIAN, excepto ABC (A-III). No se recomienda usar TDF en los pacientes en que el deterioro de la función renal sea agudo o esté directamente relacionado con este fármaco. (A-III)
- •
En pacientes con riesgo de osteoporosis (mujeres posmenopáusicas, fumadores, bajo peso corporal, mayores de 50años, déficit de vitaminaD, hepatitisC, insuficiencia renal, diabetes, CD4<250células/μl o toma crónica de esteroides) se aconseja analizar la densidad mineral ósea mediante densitometría al inicio del tratamiento y posteriormente de manera periódica. (B-III)
- •
Se aconseja realizar balance metabólico, incluida 25-OH vitaminaD, en pacientes con sospecha de pérdida de masa ósea, osteoporosis y fracturas óseas por fragilidad. (B-III)
Las interacciones de los FAR entre sí o con otros medicamentos constituyen un problema de primera magnitud en el tratamiento de los pacientes con infección por el VIH-1, ya que sus consecuencias pueden tener una importante repercusión clínica545–551. Las más relevantes suelen ser las interacciones farmacocinéticas, especialmente a nivel del metabolismo de los fármacos. Diferentes sistemas enzimáticos están implicados en dicho metabolismo, y en todos ellos pueden producirse interacciones. Los FAR son sustratos de uno o varios de estos sistemas enzimáticos, y a la vez pueden comportarse como inductores y/o inhibidores de cualquiera de ellos. La inducción del metabolismo producirá una disminución de las concentraciones del fármaco en el lugar de acción, pudiendo disminuir la eficacia del tratamiento, mientras que la inhibición ocasionará un aumento de las concentraciones con un mayor riesgo de toxicidad. En general la inducción se produce por un aumento de la síntesis proteica (enzimas) y es un proceso lento que requiere días o semanas, mientras que la inhibición suele ser competitiva y se produce de manera rápida, dependiendo de la concentración del inhibidor, pudiendo aparecer los efectos tóxicos de los sustratos en pocas horas. Dado que ambos mecanismos son diferentes e independientes, algunos fármacos pueden ser inhibidores e inductores al mismo tiempo, predominando uno u otro efecto.
El sistema metabólico más importante es el citocromo P450 (CYP) y su principal isoenzima, el CYP3A4. Muchos FAR, especialmente los IP e ITINN, y muchos otros fármacos que a menudo reciben los pacientes con infección por el VIH-1 son inhibidores o inductores de diferentes isoenzimas de CYP. La potente inhibición enzimática que produce RTV se utiliza para potenciar la farmacocinética de otros IP (sustratos de CYP3A4), logrando concentraciones plasmáticas más eficaces y menos susceptibles al efecto inductor de otros fármacos (por ejemplo, ITINN) y al mismo tiempo pautas más simples, con menos restricciones dietéticas. Se están investigando nuevos potenciadores farmacocinéticos que carecen de eficacia antirretroviral, de los cuales el que se encuentra en fases más avanzadas de investigación es cobicistat (COBI). La potenciación ejercida sobre ATV y DRV con 150mg/24h de COBI fue similar a la obtenida con 100mg/24h de RTV. COBI se está desarrollando fundamentalmente como potenciador del nuevo inhibidor de la integrasa elvitegravir (EVG). La formulación QUAD contiene en un solo comprimido EVG 150mg/COBI 150mg/FTC 200mg y TDF 300mg551–556. COBI es sustrato del CYP3A4 y minoritariamente del 2D6. Asimismo, es inhibidor de CYP3A y CYP2D6 y de los transportadores: glucoproteína-P (P-gp), BCRP, OATP1B1 y OATP1B3. La asociación de QUAD con fármacos que sean sustratos de las anteriores enzimas y transportadores puede dar lugar a un aumento de sus concentraciones plasmáticas557.
Otra vía metabólica es la conjugación de los FAR o de sus metabolitos procedentes de la oxidación del fármaco por el CYP. Diversos FAR son inductores o inhibidores del complejo enzimático de las uridindifosfato-glucuroniltransferasas (UDPGT) (glucuronización). A menudo los inductores del CYP son también inductores de la UDPGT y los inhibidores del CYP son inhibidores de la UDPGT, pero algunos inhibidores del CYP son inductores de la glucuronización, y viceversa. Así, por ejemplo, RTV y, en menor medida, NFV inhiben varias subfamilias del citocromo P450 y son inductores de las UDPGT. La combinación TPV/r muestra un efecto inductor de la glucuronización. ATV inhibe ambos sistemas enzimáticos.
Cada vez están adquiriendo mayor protagonismo una serie de proteínas transportadoras transmembrana, tales como la glucoproteína-P (P-gp), capaces de alterar la biodisponibilidad de diversos FAR y su distribución por el organismo. Estas proteínas pueden ser inducidas o inhibidas por diversos fármacos. Habitualmente la inducción o la inhibición del CYP y de la P-gp van en el mismo sentido, pero al igual que sucede con la glucuronización, ambos efectos pueden ser discordantes.
En las tablas 5–9 se detallan las características farmacocinéticas y asociaciones contraindicadas de los diferentes FAR. No se han incluido todas las posibles interacciones con los FAR, dado que existen diversas páginas web dedicadas a esta finalidad que pueden facilitar la búsqueda: www.interaccionesvih.com (en español)545 y www.hiv-druginteractions.org (en inglés)548. Debido a que la información científica relacionada con los FAR se renueva constantemente, se recomienda consultar también la ficha técnica de los fármacos y la información actualizada ofrecida por las distintas compañías farmacéuticas y las autoridades sanitarias. A continuación se detalla el metabolismo de los distintos FAR y su comportamiento inductor/inhibidor sobre las enzimas, que pueden ser útiles para predecir posibles interacciones cuando no se disponga de información.
- •
ITIAN. Tienen pocas interacciones metabólicas. ZDV y ABC se glucuronizan. 3TC, FTC, d4T y TDF se eliminan principalmente por vía renal y son poco susceptibles de padecer interacciones metabólicas relevantes. Se ha descrito aumento del riesgo de toxicidad renal al asociar TDF con algunos IP/r558–565.
- •
ITINN. NVP es metabolizada principalmente por el CYP3A4 y se comporta como inductor del CYP3A4 y del CYP2B6; EFV es sustrato de CYP2B6 y 3A4 y es fundamentalmente un inductor del CYP3A4, 2B6 y de la UGT1A1566. Existen discrepancias sobre su comportamiento frente al CYP2C19, dado que EFV ha demostrado in vitro un efecto inhibidor frente a CYP2C9 y 2C19, mientras que en voluntarios sanos se ha observado efecto inductor del CYP2C19567. ETR actúa como sustrato e inductor del CYP3A4 y como inhibidor débil del CYP2C9 y 2C19568–570. RPV es sustrato del CYP3A4 y, por tanto, los medicamentos inductores o inhibidores de CYP3A pueden afectar al aclaramiento de rilpivirina, no recomendándose su uso conjunto con estos571. A las dosis empleadas en terapéutica no se estima que RPV pueda tener efecto inductor o inhibidor clínicamente importante sobre otros fármacos eliminados mediante el citocromo P450571.
- •
IP. RTV es sustrato de CYP3A4>2D6 y es un inhibidor potente del CYP3A4 y moderado del CYP2D6, y también tiene un efecto inductor de varias isoenzimas del CYP (1A2, 2B6, 2C8, 2C9, 2C19)572, así como de la glucuronización; además es capaz de autoinducir su propio metabolismo. NFV es sustrato e inhibidor del CYP3A4 e inhibe de forma más débil CYP2C19, 2D6, 1A2 y 2B6; en cambio, es inductor de la glucuronización. IDV es sustrato e inhibidor del CYP3A4. SQV es sustrato e inhibidor débil del CYP3A4. FPV es sustrato e inhibidor del CYP3A4. LPV/r inhibe el CYP3A4; in vivo induce su propio metabolismo, los CYP2C9 y 2C19573 y la glucuronización. ATV es sustrato e inhibidor del CYP3A4 y de la UDPGT1A1 (enzima encargada de la glucuronización de la bilirrubina). TPV/r es sustrato del CYP3A4; in vivo y en estado de equilibrio es un inductor de CYP2C9, CYP1A2 y de la glucuronización e inhibidor del CYP3A4 y 2D6574. Respecto a su efecto sobre la P-pg, los datos sugieren que el efecto neto de la combinación TPV/r en estado de equilibrio es de inducción leve de la P-gp. Debido al efecto antagónico de inhibición del CYP3A4 e inducción de la P-gp, es difícil predecir el efecto neto de TPV/r sobre fármacos que sean sustratos de ambos.
- •
Inhibidores del correceptor CCR5. MVC es sustrato de CYP3A4, pero no es inhibidor ni inductor575. Los inhibidores e inductores de CYP3A4 alteran los parámetros farmacocinéticos de MVC, recomendándose cambios en su dosis (tabla 8). En general se ajustarán las dosis como sigue: 150mg BID cuando se administra con inhibidores del CYP3A4, como por ejemplo IP/r (con excepción de TPV/r y FPV/r); 600mg BID cuando se administra con fármacos inductores como EFV o rifampicina (con excepción de NVP), en ausencia de inhibidores potentes, en cuya presencia predomina el efecto inhibidor, y se administrarán 150mg BID; 300mg BID con otros fármacos (incluyendo TPV/r, FPV/r y NVP)575–578.
- •
Inhibidores de la integrasa. RAL no es sustrato ni influye en la actividad del CYP. Se metaboliza por glucuronización, sin inhibir ni inducir esta enzima. Los inhibidores e inductores de UGT1A1 modifican los parámetros farmacocinéticos de RAL, pero en la mayoría de los casos no se recomienda cambio en su dosificación por su amplio margen terapéutico. In vivo puede existir cierta inhibición de la UGT1A1 basándose en los efectos observados en la glucuronización de la bilirrubina. Sin embargo, parece improbable que la magnitud de los efectos tenga como resultado interacciones farmacológicas clínicamente importantes (tabla 8)579–583. EVG es sustrato mayoritario del CYP3A4 y minoritario de UGT1A1/3. Los inductores del CYP3A4 pueden reducir su eficacia antirretroviral (asociación no recomendada), mientras que los inhibidores del CYP3A4 pueden aumentar los niveles plasmáticos de EVG/COBI. EVG es inductor leve del CYP2C9557. EVG se emplea potenciado con COBI en la asociación QUAD, por lo que es importante considerar el efecto de COBI (véase el segundo párrafo de este capítulo).
- •
Inhibidores de la fusión. ENF se metaboliza a través de las vías catabólicas de las proteínas y aminoácidos. No es sustrato ni influye en la actividad de ninguno de los sistemas metabólicos de los otros FAR. No es susceptible de presentar interacciones metabólicas relevantes.
En la tabla 16 se especifican los ajustes de dosis de los FAR en caso de insuficiencia renal, hemodiálisis o diálisis peritoneal o insuficiencia hepática583-636.
Ajuste de dosis de los antirretrovirales en la insuficiencia renal y hepática
| Antirretrovirales | Insuficiencia renal | Hemodiálisis/diálisis peritoneal | Insuficiencia hepática |
| Inhibidores TI, análogos nucleósido | |||
| Abacavir (ABC) | No requiere ajuste de dosisNo administrar Combivir® y Trizivir® en pacientes con Cl<50ml/min (por separado, ajustar dosis adecuadamente) | Dosis habitualHD: administrar independientemente de la sesión de HD, ya que se elimina mínimamente | IH leve (Child-Pugh 5 a 6): 200mg c/12h. Utilizar la solución oral de Ziagen® (10ml c/12h)IH moderada-grave: la seguridad, eficacia y propiedades farmacocinéticas no han sido evaluadas. Evitar en lo posible su uso |
| Didanosina, cápsulas entéricas (ddI) | ≥ 60kgCl ≥ 60: 400mg c/24hCl 30-59: 200mg c/24hCl 10-29: 125mg c/24hCl<10: 125mg c/24h | HD/CAPD: 125mg c/24h; los días de HD administrar post-HD/CAPD (no requiere suplemento) | Riesgo elevado de toxicidad hepática y descompensaciónEn pacientes cirróticos no se recomienda el uso de didanosinaEn pacientes en tratamiento del VHC didanosina no debe administrarse conjuntamente con ribavirina |
| <60 kgCl ≥ 60: 250mg c/24hCl 30-59: 125mg c/24hCl 10-29: 125mg c/24hCl<10: emplear Videx® polvo para solución pediátrica 75mg/24h | HD/CAPD: emplear Videx® polvo para solución pediátrica 75mg/24h | ||
| Emtricitabina (FTC) | En cápsulasCl ≥ 50: 200mg c/24hCl 30-49: 200mg c/48hCl 15-29: 200mg c/72hCl <15: 200mg c/96hEn solución (10mg/ml)a:Cl ≥50: 240mg (24 ml) c/24hCl 30-49: 120mg (12ml) c/24hCl 15-29: 80mg (8ml) c/24hCl<15: 60mg (6ml) c/24hTruvada®: no administrar a pacientes con Cl<30ml/min | HD: en comprimidos 200mg c/96h, en solución (10mg/ml) 60mg (6ml) c/24hLos días de HD administrar post-HDNo se ha estudiado en diálisis peritonealTruvada®: no administrar a pacientes en HD (administrar los componentes por separado, ajustando la dosis adecuadamente) | Dosis habitual (no hay datos, pero en base a su mínimo metabolismo hepático, es poco probable que requiera ajuste de dosis) |
| Estavudina (d4T) | ≥ 60 kgCl ≥ 50: 40mg c/12hCl 26-49: 20mg c/12hCl ≤25: 20mg c/24h | HD: 20mg c/24h; los días de HD administrar post-HD | Dosis habitual. Utilizar con precaución por el riesgo de toxicidad mitocondrial y esteatosis hepática |
| <60 kgCl ≥ 50: 30mg c/12hCl 26-49: 15mg c/12hCl≤25: 15mg c/24h | HD: 15mg c/24h; los días de HD administrar post-HD | ||
| Lamivudina (3TC) | Cl ≥ 50: 150mg c/12h o 300mg c/24hCl 30-49: 150mg c/24h (primera dosis de 150mg)Cl 15-29: 100mg c/24h (primera dosis 150mg)Cl 5-14: 50mg c/24h (primera dosis 150mg)Cl<5: 25mg c/24h (primera dosis 50mg)No administrar Combivir® y Trizivir® si Cl<50 ml/min (administrar los componentes por separado, ajustando la dosis adecuadamente) | HD: 25mg c/24h (primera dosis 50mg) Los días de la HD, administrar post-HD | Dosis habitual |
| Tenofovir (TDF) | Cl ≥ 50: no requiere ajuste de dosisCl 30-49: 300mg c/48hCl 10-29: 300mg c/72 a 96hNo hay recomendaciones disponibles para pacientes con Cl<10 sin HD | HD: habitualmente 300mg una vez por semana, después de una de las sesiones (asumiendo 3 sesiones de diálisis semanales de 4h) | Dosis habitual |
| Zidovudina (AZT) | Puede acumularse el metabolito glucurónido (GAZT)Cl 10-50: 250-300mg c/12hCl<10: 250-300mg c/24hNo administrar Combivir® y Trizivir® en pacientes con Cl<50ml/min (administrar los componentes por separado, ajustando la dosis adecuadamente) | 300mg c/24hHD/CAPD: no afecta la eliminación de AZT y aumenta la eliminación de GAZT. Por precaución, se recomienda administrar la dosis diaria post-HD/CAPD | Se ha observado una reducción del aclaramiento oral de zidovudina el 32, al 63 y al 70%, respectivamente, en pacientes con IH leve, moderada-grave o cirrosis comprobada por biopsia, en comparación con sujetos sin alteración hepáticaAlgunos autores sugieren reducir la dosis a 200mg c/12h en pacientes con IH grave. Se recomienda monitorizar estrechamente la aparición de toxicidad hematológica |
| Inhibidores de la TI, no análogos | |||
| Efavirenz (EFV) | No requiere ajuste de dosisAtripla®: en pacientes con Cl<50 ml/min, utilizar los principios activos por separado | HD: no parece necesario ajustar la dosisCAPD: un estudio farmacocinético preliminar indica que no se requiere ajuste de dosis (datos de un solo paciente) | IH leve a moderada: dosis habitual. Dada la elevada variabilidad interindividual, se recomienda monitorizar niveles plasmáticos y aparición de efectos adversos, especialmente a nivel del SNCIH grave: datos escasos. Evitar en lo posible su uso/monitorizar niveles plasmáticos. En un paciente con Child Pugh gradoC, la semivida de EFV se duplicó. En 2 pacientes con IH (uno de ellos con cirrosis) el AUC de EFV aumentó 4 veces. En 3 pacientes con fibrosis >12kPa (Fibroscan®) la Cmin fue 2,5 veces superior a la obtenida en 15 pacientes coinfectados por VHC con fibrosis <12kPaEn otro estudio el 31% de los pacientes cirróticos presentaron concentraciones >4.000ng/ml, en comparación con un 3% en los coinfectados no cirróticos |
| Etravirina (ETR) | NRAD | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | IH leve o moderada: no requiere ajuste de dosisIH grave: no hay datos; evitar en lo posible su uso |
| Nevirapina (NVP) | No requiere ajuste de dosis | HD: los días de HD se recomienda administrar la dosis después de la HD o un suplemento de 200mg post-HD | IH leve a moderada (Child-Pugh≤7): dosis habitual. Sin embargo, en pacientes con IH moderada se recomienda monitorizar estrechamente niveles plasmáticos y aparición de efectos adversos. En un estudio en 4 pacientes con IH moderada (Child-PughB), el AUC de NVP aumentó 41%IH grave: evitar en lo posible su uso (hepatotoxicidad)En el estudio NEVADOSE el 66% de los pacientes con un mayor grado de fibrosis (F4; Fibroscan®) presentaron una Cmin de NVP por encima del límite superior de normalidad (>6.000ng/ml). En otro estudio el 50% de los pacientes cirróticos presentaron concentraciones >8.000ng/ml, en comparación con el 27% en los coinfectados no cirróticosSe ha observado un aumento de riesgo de hepatotoxicidad en pacientes sin tratamiento previo con CD4>250células/μl (mujeres) o >400células/μl (hombres) |
| Rilpivirina | IR leve-moderada: no requiere ajuste de dosisIR grave: no hay datos. Emplear con precaución | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | IH leve a moderada: no requiere ajuste de dosis. (En 8 voluntarios con insuficiencia hepática leve la Cmin, la Cmax y el AUC fueron el 31, el 27 y el 47% mayores en comparación con 8 voluntarios sanos. En 8 voluntarios con insuficiencia hepática moderada la Cmin, la Cmax y el AUC fueron comparables con los de 8 voluntarios sanos. Estos cambios no se consideran clínicamente relevantesIH grave: no hay datos |
| Inhibidores de la proteasa | |||
| Atazanavir (ATV) | No requiere ajuste de dosis | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPDHD: se recomienda su uso potenciado (ATV/r 300/100) para compensar el descenso de concentración de ATV (reducción del 28% en el AUC de ATV los días sin HD y del 42% los días de HD; la eliminación a través de la HD es de solo el 2%). Monitorizar niveles plasmáticos cuando sea posible | IH leve: según un estudio en pacientes coinfectados con VHC con IH leve a moderada, el ATV no potenciado (400mg/24h) puede no llegar a alcanzar la Cmin deseada (6/9 pacientes con niveles subterapéuticos). Se recomienda usar ATV potenciado con RTV (300/100mg/24h), IH moderada-grave: la Agencia Europea del Medicamento desaconseja el uso de atazanavir potenciado en este contexto por falta de datos. Sin embargo, diversos estudios han demostrado un buen perfil de seguridad de atazanavir (potenciado o no) en pacientes cirróticos, incluso con insuficiencia hepática avanzada (Child-Pugh B o C) o descompensación de su hepatopatíaEn 9 pacientes con fibrosis >12kPa (Fibroscan®) que recibieron ATV/r, la Cmin fue comparable a la obtenida en 26 pacientes coinfectados por VHC con fibrosis <12kPa. En un estudio en 12 pacientes coinfectados por VHC tratados con ATV/r, el AUC fue solo un 36% superior en los pacientes cirróticos (n=7)En un estudio en el que se compararon los niveles plasmáticos de ATV en pacientes VIH+/VHC–, VIH+/VHC+ sin cirrosis y VIH+/VHC+ con cirrosis, no se observaron diferencias en los 3 grupos en los que recibieron ATV/r 300/100mg/24h; sin embargo, en los que recibieron ATV 400mg/24h, los niveles fueron significativamente mayores en los coinfectados por el VHC con o sin cirrosis que en los no coinfectadosMonitorizar niveles plasmáticos cuando sea posible |
| Darunavir (DRV) | IR leve, moderada o grave: no requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | IH leve o moderada: no requiere ajuste de dosisIH grave: no hay datos; evitar en lo posible su uso |
| Fosamprenavir (FPV) | No requiere ajuste de dosis | HD/CAPD: debido a su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | IH leve (Child Pugh 5-6): FPV 700mg c/12h + RTV 100mg c/24hIH moderada (Child Pugh 7-9): FPV 450mg c/12h + RTV 100mg c/24hEn IH grave (Child Pugh 10-15): FPV 300mg c/12h + RTV 100mg c/24h (usar solución oral de FPV)En un estudio en 2 pacientes con fibrosis >12kPa (Fibroscan®) la Cmin fue comparable a la obtenida en 6 pacientes coinfectados por VHC con fibrosis <12kPa. En otro estudio los pacientes cirróticos (n=6) presentaron una Cmin 2veces mayor y AUC 43% mayorSe recomienda monitorizar los niveles plasmáticos cuando sea posible |
| Indinavir (IDV) | No requiere ajuste de dosis | HD: probablemente no requiera ajuste de dosis si la función hepática está conservada (datos de un solo paciente). Se elimina mínimamente a través de la HD | IDV (no potenciado):IH leve a moderada: 600mg c/8hIH grave: no hay datosSe recomienda monitorizar los niveles plasmáticos cuando sea posibleIDV/r:Algunos pacientes coinfectados con VHC pueden requerir reducción de dosis, habitualmente IDV/r 400/100mg c/12h o, incluso IDV/r 200/100mg c/12hSe recomienda monitorizar los niveles plasmáticos cuando sea posible |
| Lopinavir (LPV/r) | No requiere ajuste de dosis | HD: El AUC de LPV/r en 13 pacientes en HD fue equivalente a la de pacientes con función renal normal. No ajuste de dosisCAPD: no hay datos. Debido a la elevada unión a proteínas plasmáticas de lopinavir y ritonavir, no es de esperar que se elimine en las sesiones de CAPD | Según la ficha técnica de Kaletra®, en pacientes infectados por VIH con insuficiencia hepática leve a moderada se ha observado un aumento en la exposición a lopinavir del 30% aproximadamente, aunque no se espera que sea clínicamente relevante. No hay datos en pacientes con insuficiencia hepática grave, por lo que Kaletra no puede administrarse a estos pacientesUn estudio abierto en 65 pacientes VIH+ demostró que la farmacocinética de lopinavir/ritonavir no cambiaba significativamente en los pacientes coinfectados por el VHC sin cirrosisEn otro estudio, los pacientes coinfectados por el VHC con cirrosis hepática sin signos de IH (n=7) presentaron un aumento del 100% en el AUC y la Cmin de ritonavir, pero sin diferencias en la exposición a lopinavirUn estudio en pacientes con IH leve (n=6) o moderada (n=6) mostró un aumento significativo en la exposición tanto a lopinavir como a ritonavir. Sin embargo, la relevancia clínica de estos cambios no está clara, por lo que no se aconseja modificar las dosis de lopinavirSe recomienda monitorizar niveles plasmáticos cuando sea posible |
| Nelfinavir (NFV) | No requiere ajuste de dosis | HD: no es probable que NFV se elimine significativamente a través de la HD. Datos de un paciente con ERCA e IH mostraron la ausencia de eliminación de NFV durante una sesión de HD de 4hCAPD: no es probable que NFV se elimine significativamente a través de la CAPD. En un paciente tratado con 1.250mg c/12h de NFV las concentraciones en el líquido de diálisis fueron inferiores al límite de detección | Dosis habitual (aunque en presencia de IH el AUC de NFV aumenta entre un 49 y un 69%, los datos no parecen indicar que se produzca un aumento de toxicidad)Se recomienda monitorizar niveles plasmáticos cuando sea posible |
| Ritonavir (RTV) | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de ritonavir, no es de esperar que se elimine en las sesiones de HD/CAPD | No usar RTV a dosis plenas por el riesgo de hepatotoxicidadRitonavir como potenciador farmacocinético:IH leve a moderada: dosis habitualIH grave: según la ficha técnica de Norvir, no se debe administrar ritonavir como potenciador farmacocinético en aquellos pacientes que tengan descompensada la función hepática. En ausencia de estudios farmacocinéticos en pacientes con insuficiencia hepática grave estable (Child Pugh Grado C) no descompensada, se debe tener cuidado cuando el ritonavir se utilice como potenciador farmacocinético, ya que se puede producir un aumento de los niveles del inhibidor de la proteasaConsultar datos específicos sobre el inhibidor de la proteasa potenciado con el ritonavir |
| Saquinavir (SQV) | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de ritonavir, no es de esperar que se elimine en las sesiones de HD/CAPD. Datos de un paciente indican escasa eliminación a través de HD | IH leve-moderada: dosis habitualIH grave: no hay datos. Evitar en lo posible su uso. Contraindicado en pacientes con insuficiencia hepática descompensadaSe recomienda monitorizar niveles plasmáticos cuando sea posible |
| Tipranavir (TPV) | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de TPV/RTV, no es de esperar que se eliminen en las sesiones de HD/CAPD | Datos limitados. Elevado riesgo de toxicidad hepática: TPV/r se ha relacionado con casos de hepatitis clínica y descompensación hepática, incluyendo algunos casos mortales. Se recomienda un estrecho seguimiento de los pacientes coinfectados por VHB o VHC, por el aumento de riesgo de hepatotoxicidad que presentanIH leve (Child-Pugh A): dosis habitualIH moderada o grave (Child-Pugh B y C): contraindicado |
| Inhibidores de la fusión | |||
| Enfuvirtida (T-20) | No requiere ajuste de dosis | HD: No requiere ajuste de dosis | No hay datos. Algunos autores recomiendan utilizar la dosis habitual |
| Inhibidores de la integrasa | |||
| RAL (raltegravir) | No requiere ajuste de dosis | HD: no es probable que RAL se elimine significativamente a través de la HD. Datos de 2 pacientes con ERCA mostraron la ausencia de eliminación de RAL durante una sesión de HD de 4h | IH leve-moderada: no requiere ajuste de dosisIH grave: no requiere ajuste de dosis. A pesar de los aumentos de niveles plasmáticos observados (aumento del 72% en al AUC y de 6 veces la Cmin), raltegravir fue bien tolerado en pacientes con cirrosis hepática avanzada (Child-Pugh C) |
| Elvitegravir (EVG)/cobicistat/TDF/FTC (QUAD) | Cobicistat reduce leveme nte el filtrado glomerular renal estimado de creatinina (aunque no altera el filtrado glomerular real), debido a la inhibición de la secreción tubular de creatininaLa asociación QUAD no debe iniciarse en pacientes con Cl<70ml/min. Si el Cl durante el tratamiento se reduce a Cl<50ml/min debe suspenderse, dado que no se puede realizar el ajuste de dosis adecuado para FDF/FTC | No prodece. La combinación QUAD no debe emplearse si Cl<50ml/min | IH leve-moderada: no requiere ajuste de dosisIH grave: no hay datos, por lo que no se recomienda su empleo |
| Inhibidores correceptor CCR5 | |||
| MVR (maraviroc) | En ausencia de inhibidores potentes del CYP3A4 no requiere ajuste de dosisSolo se recomienda un ajuste de dosis en pacientes con Cl<80ml/min y que están recibiendo inhibidores potentes del CYP3A4, como los IP (excepto TPV/r), ketoconazol, itraconazol, claritromicina o telitromicina: en estos casos administrar 150mg c/24h. Si el Cl es <30ml/min se recomienda mucha precaución debido al aumento de riesgo de hipotensión posturalCon Cl<80ml/min y en combinación con FPV/r administrar 150mg c/12hCon Cl<80 ml/min y en combinación con TPV/r no se requiere ajuste de dosis (300mg c/12h)(Estos ajustes de dosis se recomiendan basándose en los datos de un estudio en insuficiencia renal y simulaciones farmacocinéticas, sin que su seguridad y eficacia hayan sido evaluadas clínicamente, por lo que se recomienda una estrecha monitorización) | HD: en ausencia de inhibidores potentes del CYP3A4 no se requiere ajuste de dosis. En presencia de los mismos, dosificar igual que para Cl<80ml/min (datos limitados)Escasa eliminación a través de la HD | Datos de un estudio con dosis únicas de 300mg MVR. En comparación con los voluntarios con función hepática normal:IH leve: +25% AUCIH moderada: +45% AUCIH grave: no hay datosSe desconoce la importancia clínica que estos aumentos pueden suponerSe ha descrito un caso de posible hepatotoxicidad precedido de una reacción alérgica sistémica |
AN: análogos de nucleósidos; ANt: análogos de nucleótidos; CAPD: diálisis peritoneal ambulatoria; CH: cirrosis hepática; Cl: aclaramiento de creatinina (en ml/min); ERCA: enfermedad renal crónica avanzada, HD: hemodiálisis; IH: insuficiencia hepática; IP: inhibidores de la proteasa; IR: insuficiencia renal; NN: inhibidores de la transcriptasa inversa no análogos de nucleósidos.
Algunas interacciones farmacodinámicas son de interés, como el incremento del riesgo de toxicidad mitocondrial que se produce al asociar ribavirina con ddI. La incidencia de esta se quintuplicó en comparación con el uso de ribavirina y otros ITIAN. Tres de los 23casos comunicados a la FDA fueron mortales, por lo que se recomienda evitar esta asociación. En lo posible se evitará también el uso simultáneo de ribavirina con ZDV o d4T, por toxicidad hematológica o mitocondrial. Este aspecto es especialmente importante con el uso de los nuevos inhibidores de la proteasa del VHC boceprevir y telaprevir, con los que aumenta la incidencia de anemia.
Se han descrito interacciones importantes entre los inhibidores de la proteasa del VHC y los FAR. Existe una web específica donde pueden consultarse las interacciones de los fármacos para el tratamiento de la hepatitis C: http://www.hep-druginteractions.org/637, así como un documento elaborado por la Agencia Española del Medicamento en el que participaron un grupo de expertos y establece una serie de recomendaciones sobre el empleo de antirretrovirales en pacientes tratados con boceprevir y telaprevir638.
Recomendaciones- •
Se deben reseñar en la historia clínica todos los medicamentos, productos naturales y medicinas alternativas, para evaluar posibles interacciones. (B-III)
- •
Se deben tener en cuenta las contraindicaciones y realizar los ajustes de dosis correspondientes cuando sea necesario. (B-III)
- •
Se debe considerar la monitorización de los niveles plasmáticos cuando se administren 2 o más fármacos con posibles interacciones farmacocinéticas relevantes para evitar toxicidad o ineficacia terapéutica. (B-II)
La hepatopatía crónica por virus de la hepatitis es la comorbilidad más relevante de las que presentan los sujetos infectados por el VIH en España, por su frecuencia, por la rápida progresión a enfermedad hepática terminal que experimenta y por aumentar la hepatotoxicidad del TAR.
Infección por virus de la inmunodeficiencia humana, tratamiento antirretroviral e historia natural de la hepatitis crónica por virus de la hepatitis C y BEn pacientes coinfectados por VIH-1 y virus hepatotropos la buena situación inmunológica, el control de la replicación viral del VIH-1 y el recibir TAR se asocian con un mejor pronóstico global, menor velocidad de progresión de la enfermedad hepática y menor riesgo de complicaciones y muerte por causa hepática639–645. Por ello, aun en ausencia de ensayos clínicos que valoren el impacto del TAR en la evolución de la fibrosis hepática en pacientes coinfectados, las evidencias anteriormente descritas respaldan el control precoz de la replicación del VIH-1 y el mantenimiento de una buena situación inmunológica en estos pacientes.
Recomendaciones- •
En pacientes coinfectados por el VHC se debe recomendar el inicio de TAR, independientemente de la cifra de linfocitos CD4+, individualizando la decisión en función de variables virológicas, histológicas y de motivación del paciente. (B-II)
- •
En pacientes coinfectados por el VHB con criterios de tratamiento de la hepatitis se debe iniciar un TAR que contenga tenofovir, independientemente de la cifra de linfocitos CD4+. (B-II)
- •
En pacientes sin necesidad de tratamiento frente al VHB, si se inicia el TAR se recomienda que este incluya tenofovir. (B-II)
La toxicidad hepática se ha descrito con todas las familias de FAR, aunque con incidencia y mecanismos patogénicos diferentes646–649. La incidencia real es difícil de estimar por problemas metodológicos650. En primer lugar el diagnóstico de toxicidad hepática por un fármaco implica una relación temporal y la exclusión de otras causas de elevación de enzimas hepáticas (EEH) y estos criterios no se cumplen en la mayoría de pacientes coinfectados por VIH-1 y VHC/VHB con EEH. En segundo lugar, la definición de los distintos grados de hepatotoxicidad no es uniforme. En los ensayos clínicos se suele definir como hepatotoxicidad grave a una elevación mayor de 5veces el límite superior de la normalidad de ALT y/o AST651. Sin embargo, esta definición está limitada al no considerar el fallo hepático, no tener en cuenta la hepatotoxicidad colestásica o mixta y ser más sensible para detectar hepatotoxicidad en pacientes con valores basales de transaminasas elevados. Para resolver este último problema se acepta considerar EEH grave asintomática a los incrementos de ALT y/o AST superiores a 3,5veces la cifra basal para los pacientes con niveles elevados transaminasa basales652.
No está completamente aclarado si la presencia de fibrosis significativa (≥F2), avanzada (≥F3) o cirrosis aumenta el riesgo de toxicidad hepática por TAR, y es posible que ello dependa de los FAR usados. Así, mientras en un estudio de cohortes la frecuencia de hipertransaminemia grave en pacientes tratados con NVP o EFV fue significativamente mayor en los que presentaban fibrosis avanzada653, en otras cohortes y en estudios observacionales específicos con FPV, ATV o RAL, e incluso con EFV654, este hecho no se ha confirmado613,655–657.
En pacientes coinfectados por VIH-1 y VHC la consecución de respuesta viral sostenida con el tratamiento de la hepatitisC reduce de modo notable el riesgo de toxicidad hepática658.
Recomendaciones- •
No se contraindica ningún FAR en caso de coinfección con VHC o VHB si la función hepática está preservada (B-II), pero se debe priorizar el uso de los que tienen el menor potencial de hepatotoxicidad. (C-III)
- •
Se debe retirar el TAR en caso de hepatitis sintomática, y en la asintomática si se sospecha que se debe a toxicidad mitocondrial o a una reacción de hipersensibilidad, o si existe hipertransaminemia de grado4. (B-III)
- •
En caso de hepatitis asintomática con hipertransaminemia de grado3 se debe considerar la suspensión del TAR en función de la situación clínica, inmunológica y virológica, de los fármacos utilizados y de la historia previa de exposición a FAR. (B-III)
La hepatopatía crónica puede alterar el metabolismo y la biodisponibilidad de los FAR, con incremento de toxicidad o alteración de la actividad antiviral. La hepatitis crónica sin insuficiencia hepatocelular es una situación muy frecuente, y la experiencia acumulada sugiere que se pueden usar los FAR a las dosis habituales y que su eficacia y seguridad no están comprometidas. Sin embargo, en la insuficiencia hepatocelular se reduce el metabolismo de fármacos por la vía del citocromo P450 y la glucuronoconjugación. Los niveles plasmáticos de EFV aumentan en pacientes cirróticos en mayor medida que los de los inhibidores de la proteasa586,659, por lo que, si se usa EFV en pacientes con insuficiencia hepatocelular, debería hacerse con monitorización de los niveles del fármaco para evitar la sobrexposición al mismo o, al menos, con vigilancia estrecha de efectos adversos.
En el caso de los IP/r solamente se dispone de datos farmacocinéticos del FPV/r cuya dosificación se ha estudiado en pacientes con Child-PughA, B y C. En el caso de la insuficiencia hepática moderada o avanzada la dosis de FPV o RTV debe disminuirse o aumentar el intervalo entre dosis (tabla 16), aunque las diferencias individuales aconsejan llevar un estrecho control para prevenir y evitar efectos adversos o fracaso virológico660. Se estima que esta misma conducta debe seguirse con otros IP/r.
Un estudio con un número reducido de pacientes demostró que, en sujetos con cirrosis hepática claseC de Child, la administración de RAL se asoció a niveles plasmáticos del fármaco más elevados que los que se observaron en los controles661. No obstante, la magnitud de esta elevación es similar a la que se observa en pacientes que reciben RAL junto a ATV661.
No hay datos sobre el uso del TAR en casos de hepatitis aguda.
Recomendaciones- •
Se debe evaluar el grado de fibrosis hepática y el grado de función hepática en todos los pacientes coinfectados por virus hepatotropos, ya que pueden condicionar la elección del TAR, las dosis prescritas de los FAR y la estrategia de monitorización de su eficacia y toxicidad. (C-III)
- •
Los FAR se pueden usar a las dosis habituales en caso de hepatitis crónica sin insuficiencia hepatocelular o con insuficiencia hepatocelular leve (Child A), aumentando la vigilancia por el mayor riesgo de toxicidad. (B-II)
- •
En caso de hepatopatía crónica con signos de insuficiencia hepatocelular se deberá ajustar la dosis de los fármacos, idealmente mediante la determinación de niveles plasmáticos, o en su ausencia mediante las recomendaciones de la tabla 16(B-III). El margen terapéutico de los IP es superior al de EFV en este escenario (B-II). FPV/r, en dosis ajustadas al estadioC de Child, y RAL, sin necesidad de ajuste de dosis, pueden considerarse las opciones preferentes en estos pacientes. (B-II)
- •
En caso de hepatitis aguda grave debe interrumpirse el TAR y reintroducirlo una vez superado el problema. (B-III)
El tratamiento recomendado de la hepatitis crónica por VHC de genotipos2, 3 y 4 en pacientes coinfectados por el VIH-1 sigue siendo aún la combinación de interferón pegilado y ribavirina662–666. En cambio, para la hepatitis crónica por VHC de genotipo1 el nuevo estándar de tratamiento es la triple terapia basada en boceprevir o telaprevir667. Actualmente existe en España la posibilidad de tratar con terapia triple, incluyendo los referidos inhibidores de la proteasa del VHC telaprevir y boceprevir, a enfermos infectados por el VHC con genotipo1 que tienen fibrosis avanzada638. Los ensayos en faseii con estos fármacos han deparado resultados muy superiores a los de la biterapia con interferón pegilado y ribavirina668,669.
Por otro lado, se han descrito brotes de hepatitis aguda C que afectan esencialmente a HSH infectados por el VIH-1670–672, para cuyo tratamiento se recomienda interferón pegilado junto con ribavirina ajustada a peso, en caso de que la viremia C persista detectable a las 12 semanas del diagnóstico672–676. Igualmente, una caída de la viremia menor de 2 unidades logarítmicas a las 4 semanas del diagnóstico tiene un alto valor predictivo positivo de evolución a la cronicidad, por lo que puede considerarse un indicador para iniciar el tratamiento de la hepatitis aguda C677.
Un aspecto importante, cuando se inicia tratamiento con interferón y ribavirina en pacientes coinfectados por el VIH-1, es la selección de los FAR. Estos pueden contribuir al incremento del riesgo de efectos adversos por la vía de toxicidades aditivas o sinérgicas, como por ejemplo anemia y/o neutropenia con ZDV678, aumento de la toxicidad mitocondrial con ddI y d4T679,680 o pancreatitis, acidosis láctica y descompensación de la cirrosis con ddI679–681. Un segundo mecanismo potencial por el que los ITIAN pueden influir en el tratamiento de la hepatitis C es por la vía de interferencia con la acción de la ribavirina. En este contexto, en algunos estudios se ha observado que el uso de ABC se asocia a una menor respuesta al tratamiento con interferón pegilado y ribavirina682–684, hallazgo que no se ha confirmado en otros trabajos685–687.
El uso de EFV concomitantemente con interferón pegilado aumenta la frecuencia de efectos adversos del SNC, probablemente por adición de toxicidades, aun cuando no compromete la eficacia del tratamiento de la hepatitis C688. Por ello, si es preciso usar simultáneamente estos 2fármacos, se debe vigilar especialmente la aparición de estos efectos adversos.
Por otra parte, se ha comunicado que una sustancial proporción de pacientes en tratamiento estable con ATV experimentan hiperbilirrubinemia e ictericia tras el inicio de tratamiento para la hepatitisC. Este hecho podría ser debido al incremento de bilirrubina asociado a la hemolisis por ribavirina y al compromiso del normal aclaramiento de bilirrubina debido a la inhibición competitiva de ATV sobre el sistema de la UDP glucuronil-transferasa689. Por último, en pacientes infectados por el VHC de genotipos1 y 4 el tratamiento con ATV se ha asociado con una mayor carga viral basal del VHC. Sin embargo no se conoce la trascendencia clínica de este hallazgo690.
Los inhibidores de la proteasa del VHC se eliminan también por la vía del citocromo P-450. Debido a ello, pueden interaccionar tanto con los ITINN como con los IP/r. Estudios farmacocinéticos han demostrado que EFV reduce las concentraciones plasmáticas de telaprevir, pero si este fármaco se administra a dosis de 1.125mg/8h se consiguen concentraciones plasmáticas adecuadas de ambos fármacos. La administración de telaprevir reduce de forma significativa los niveles plasmáticos de DRV, FPV y LPV potenciados y, a su vez, DRV y FPV potenciados causan una disminución de los niveles plasmáticos de telaprevir que puede comprometer su eficacia. Por el contrario, cuando se administran simultáneamente telaprevir y TDF o ATV/r, no se producen alteraciones significativas de la farmacocinética de ninguna de estos FAR691, lo que permite su coadministración sin ajuste de dosis, si bien, debido a que telaprevir produce un cierto incremento de las concentraciones de ATV, podría haber una mayor incidencia de hiperbilirrubinemia o que esta fuera más intensa cuando se administran conjuntamente los 2fármacos. No existen interacciones clínicamente significativas entre telaprevir y ETR. La administración conjunta de 750mg/8h de telaprevir y de 25mg/día de RPV a 16voluntarios sanos mostró un incremento de la exposición a RPV, con un incremento del 50% en la Cmax y del 80% en al ABC. La exposición a telaprevir no experimentó modificaciones significativas por la asociación con RPV692. Se ha observado prolongación del intervalo QT con cualquier dosis de RPV, aunque una prolongación significativa, con riesgo de torsade de pointes, solo se produce con dosis de 75mg/día o más. Kakuda et al. consideran que el incremento de exposición a RPV al asociarse a telaprevir no es clínicamente significativo y que carece de relevancia desde el punto de vista del riesgo de prolongación del intervalo QT. En consecuencia no recomiendan ajustar las dosis cuando se administra RPV y telaprevir, lo cual tampoco se incluye en la ficha técnica de RPV. Sin embargo, hay que tener en cuenta que muchos de los estudios farmacocinéticos están realizados con un número pequeño de casos, voluntarios sanos o pacientes muy seleccionados, donde el significado clínico de la prolongación del intervalo QT es difícil de establecer. Por tanto, el uso de RPV en situaciones que aumentan su exposición, como en el caso de la administración con telaprevir, debe realizarse con precaución, tanto más cuando se administren conjuntamente otros fármacos que también incrementan el intervalo QT. En este sentido debe tenerse una especial precaución en los pacientes coinfectados por VIH y VHC que reciben tratamiento sustitutivo con metadona.
La administración de RTV no aumenta de forma significativa los niveles de boceprevir, mientras que cuando se dan EFV y boceprevir juntos, se produce una reducción de los niveles valle de este último que podría comprometer su eficacia. Los datos farmacocinéticos disponibles tampoco sugieren ninguna interacción significativa de boceprevir con ETR693. Cuando se administran simultáneamente ETR y boceprevir, las concentraciones de ETR descienden alrededor del 25%. Aunque el significado clínico de esta interacción no está totalmente aclarado, en principio boceprevir y ETR podrían administrarse conjuntamente. LPV y DRV potenciados presentan una interacción bidireccional con boceprevir, de tal modo que se reducen de forma significativa las concentraciones plasmáticas, tanto de boceprevir, como de estos IP del VIH. Cuando boceprevir se coadministra con ATV, los niveles plasmáticos de boceprevir no se modifican de modo relevante, pero la concentración valle de ATV cae un 49%694. Sin embargo, en el estudio P05411 los pacientes tratados con terapia triple del VHC que incluía boceprevir, y, a la vez, un IP del VIH, respondieron igual que los que recibieron RAL y en ellos no hubo más repuntes de viremia VIH695. Por ello, se precisa más información acerca del significado clínico de las interacciones antes mencionadas.
Debido a las peculiares vías de metabolización de RAL, cuando este FAR y telaprevir o boceprevir son administrados a un mismo paciente, no se producen modificaciones de las concentraciones plasmáticas de ninguno de dichos fármacos que comprometan su eficacia o su seguridad696,697.
Recomendaciones- •
El TAR no debe iniciarse simultáneamente con el tratamiento del VHC. (B-III)
- •
Cuando se traten simultáneamente el VIH-1 y el VHC, debe realizarse un seguimiento estrecho del paciente para detectar reacciones adversas. (B-III)
- •
No se debe asociar ribavirina con ddI. (A-I)
- •
Se evitará el uso simultáneo de ribavirina y ZDV. (A-I)
- •
Si se usan simultáneamente interferón pegilado y EFV, debe vigilarse estrechamente la aparición de efectos adversos del SNC. (B-III)
- •
Si telaprevir se administra a un paciente que requiere TAR, se podrá administrar, sin riesgo de interacciones significativas con TDF, ABC, 3TC, FTC, ATV/r, RAL, ETR, RPV (con monitorización del intervalo QT y aumentando la vigilancia de los niveles plasmáticos de bilirrubina) o EFV, aunque en este último caso se debe aumentar la dosis de telaprevir a 1.125mg/8h. (B-I)
- •
En caso de iniciarse tratamiento con boceprevir en un paciente que requiere a la vez TAR, pueden administrarse TDF, ABC, 3TC, FTC, RAL y ETR (A-I). En pacientes con CVP del VIH indetectable, sin antecedentes de fallos virológicos a IP del VIH, y en los que no existe otra opción terapéutica, puede administrarse también ATV/r. (C-I)
Los FAR activos frente al VIH-1 y al VHB son 3TC, FTC y TDF698–701. Si en pacientes coinfectados por el VHB se retirara alguno de tales fármacos de un TAR que hubiese fracasado, puede aparecer un rebrote del VHB, con el consiguiente daño hepatocelular702. El tratamiento de la hepatitis crónica B con 3TC o FTC en monoterapia facilita la aparición de resistencias al VIH-1701,703,704, por lo que deben usarse siempre en combinación. El uso de 3TC o FTC como único fármaco activo frente al VHB dentro de la combinación de FAR proporciona escaso beneficio clínico sobre la hepatopatía, por lo que se aconseja el uso de TDF siempre que sea posible705. Entecavir, fármaco activo frente al VHB, ha demostrado actividad frente a VIH-1 y capacidad para inducir igualmente mutaciones, como la M184V706.
Recomendaciones- •
Se recomienda iniciar el TAR usando la asociación de TDF+FTC (o 3TC) como ITIAN en los pacientes coinfectados que requieran tratamiento del VIH-1 o del VHB. (A-III)
- •
Si se requiere tratamiento del VHB y se decide no tratar el VIH-1, se recomienda usar fármacos que no induzcan resistencias al VIH-1. (A-III)
- •
No debe usarse entecavir en pacientes infectados por el VIH-1, salvo que su replicación esté controlada con otros fármacos. (B-II)
- •
En pacientes coinfectados en los que por cualquier motivo se suspenda 3TC, FTC o TDF, se debe incluir en el TAR otro fármaco con actividad anti-VHB. (A-II)
Los pacientes infectados por el VIH-1 presentan un mayor riesgo de enfermedad renal crónica (ERC) que la población general707–713. Entre las causas que pueden motivar este hecho se encuentran la propia infección por el VIH-1, las enfermedades asociadas, el tratamiento de estas y el propio TAR714–716. Además, debido al progresivo envejecimiento de la población infectada por el VIH-1 la frecuencia de enfermedades comunes relacionadas con el desarrollo de enfermedad renal crónica ha aumentado en la misma. Por todo ello, se prevé que a medio y largo plazo la frecuencia de nefropatía se incremente entre pacientes infectados por el VIH-1715,716. En los pacientes con ERC es esencial prevenir que esta progrese, evitando la utilización de fármacos potencialmente nefrotóxicos y efectuando, siempre que ello sea necesario, un ajuste de dosis de los mismos. El ajuste de las dosis de los FAR tiene una gran importancia clínica, pues su omisión puede originar complicaciones e interacciones farmacológicas potencialmente graves717. Diversos estudios han demostrado que los errores en la dosificación de FAR en caso de ERC son más frecuentes de lo esperado718,719.
Los ITIAN, exceptuando ABC, cuyo metabolismo es casi exclusivamente hepático, requieren ajustes de dosis en la insuficiencia renal, ya que su excreción se realiza por esta vía (tabla 16). Sin embargo tanto los ITINN como los IP se metabolizan por vía hepática, por lo cual no precisan ajuste de dosis en caso de ERC (tabla 16). Los parámetros farmacocinéticos de RAL son muy similares en individuos sanos y en pacientes con ERC, por lo cual tampoco es necesario ajustar su dosis. Los datos disponibles sobre MVC en la ERC son muy escasos; sin embargo, dado que fundamentalmente se metaboliza por vía hepática, es muy probable que no requiera modificación de dosis en la ERC en ausencia de coadministración con inhibidores potentes del CYP3A4, como los IP (tabla 16). No es necesario ajustar la dosis de ENF en caso de insuficiencia renal.
En la tabla 16 se detallan los ajustes de dosis de cada uno de los FAR en caso de hemodiálisis o diálisis peritoneal.
La creatinina plasmática es un marcador poco sensible para detectar descensos ligero-moderados del filtrado glomerular (FG) y consecuentemente estadios precoces de enfermedad renal720. Por ello, reducciones leves o moderadas del FG pueden pasar desapercibidas si utilizamos como única herramienta para su cribado la determinación de la creatinina plasmática. En esta situación son de gran utilidad la estimación del FG mediante diversas fórmulas como la Modified Diet for Renal Disease (MDRD)720, de Cockcroft-Gault721 o la Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI)722.
Ecuaciones utilizadas para la estimación del filtrado glomerular718,720,721:FG (ml/min)=[140–edad (años)×peso (kg)×0,85 (si mujer)/CrP (mg/dl)×72
Calculadora automática en: http://nephron.org/cgi-bin/CGSI.cgi
MDRD (Modification of Diet for Renal Diseases)FG (ml/min/1,73 m2=186×[CrP (mg/dl)]–1.154×[edad (años)]–0,203×0,742 (si mujer)×1.212 (si raza negra)
Calculadora automática en: http://nephron.org/cgi-bin/MDRD_GFR/cgi
CKD-EP I (Chronic Kidney Disease Epidemiology Collaboration)FG (ml/min)=141×min (CrP/κ, 1)α×max (CrP/κ, 1)–1.209×0.993edad×1.018 (si mujer)×1.159 (si raza negra)
Calculadora automática en: http://qxmd.com/calculate-online/nephrology/ckd-epi-egfr
Notas: α: −0,329 para mujeres y −0,411 para varones; CrP: creatinina plasmática; FG: filtrado glomerular; κ: 0,7 para mujeres y 0,9 para varones; Max: valor máximo de CrP o 1; Min: valor mínimo de CrP/k o 1.
- •
En pacientes infectados por el VIH-1 con ERC es esencial realizar un ajuste de dosis de los FAR, pues su omisión puede originar complicaciones e interacciones farmacológicas potencialmente graves. (B-I)
- •
Los IP, los ITINN y RAL no precisan ajuste de dosis. (A-I)
Recientemente GeSIDA, junto con la SPNS, ha elaborado un documento de consenso sobre TAR en pacientes con TB en el que se analiza con detalle cuándo debe comenzarse el TAR en los pacientes infectados por el VIH con TB activa, qué regímenes de TAR deben utilizarse en tales casos y el síndrome inflamatorio de reconstitución inmune que pueden presentar tras el inicio del TAR los mencionados pacientes que tengan una inmunodeficiencia celular profunda723. Por ello, solo se exponen a continuación las recomendaciones correspondientes a los temas anteriormente señalados y se remite al mencionado documento específico a quienes necesiten o deseen ampliar la información al respecto.
Cuándo comenzar el tratamiento antirretroviral en pacientes infectados por el virus de la inmunodeficiencia humana con tuberculosis activaRecomendaciones- •
En pacientes infectados por el VIH con TB, cualquiera que sea su recuento de linfocitos CD4+, se debe iniciar el TAR durante el tratamiento de la TB, ya que reduce el riesgo de muerte. (A-I)
- •
En pacientes infectados por el VIH con TB y cifras de linfocitos CD4+<50células/μl se debe iniciar el TAR a las 2semanas del tratamiento de la TB y una vez comprobada la buena tolerancia al tratamiento antituberculoso, ya que ello reduce el riesgo de muerte y el desarrollo de sida. (A-I)
- •
En pacientes infectados por el VIH con TB y cifras de linfocitos CD4+>50células/μl se debe iniciar el TAR una vez finalizada la fase intensiva del tratamiento de la tuberculosis, ya que ello reduce el riesgo de efectos adversos y de SIRI sin comprometer la supervivencia. (A-I)
- •
Aunque se desconoce el momento óptimo de inicio del TAR en pacientes con meningitis tuberculosa, se recomienda utilizar en este contexto los criterios anteriores. (A-III)
- •
En pacientes infectados por el VIH con TB en tratamiento estándar se recomienda como régimen de TAR de uso preferente la combinación de TDF/FTC (a dosis habituales) o de ABC/3TC (a dosis habituales)+EFV a dosis de 600mg/día. (A-I)
- •
Se recomiendan como regímenes alternativos por orden de preferencia: 1)TDF/FTC o ABC/3TC+NVP (a dosis habituales); 2) TDF/FTC o ABC/3TC+RAL a dosis de 800mg/12h. (A-II)
- •
Se recomienda que otros regímenes de TAR, como TDF/FTC o ABC/3TC+MVC (a dosis de 600mg/12h) solo sean usados en situaciones especiales en las que no sea posible utilizar ninguno de los regímenes anteriores. (C-III)
- •
En presencia de SIRI no debe discontinuarse ni el tratamiento antituberculoso ni el TAR. (A-III)
- •
Las formas clínicas leves o moderadas de SIRI deben tratarse con antiinflamatorios no esteroideos. (A-III)
- •
El tratamiento con corticosteroides del SIRI con manifestaciones clínicas moderadas-graves mejora los síntomas sin provocar efectos adversos añadidos. (A-II)
El VIH-2 posee una organización genómica similar al VIH-1, aunque presenta respecto a este diferencias estructurales que influyen de forma significativa en su patogenicidad y sensibilidad a los FAR724.
No existen ensayos clínicos controlados que evalúen el momento óptimo para el inicio del TAR, ni la elección del régimen de TAR más apropiado para la terapia inicial o siguientes líneas de tratamiento frente al VIH-2. Pese a ello, parece razonable asumir que los objetivos del TAR en pacientes infectados por el VIH-2 son similares a los descritos en la infección por el VIH-1.
El VIH-2 presenta diferencias importantes respecto al VIH-1 en su perfil de sensibilidades a los FAR. El VIH-2 presenta resistencia intrínseca a los ITINN725, por lo que estos FAR están contraindicados. El VIH-2 es sensible a los ITIAN, aunque su barrera genética frente a ellos es más baja que para el VIH-1726,727. Por otro lado, el VIH-2 presenta una sensibilidad variable frente a los IP/r, siendo LPV/r, SQV/r y DRV/r los más activos728–731. RAL es activo frente al VIH-2732. Sin embargo, el uso de MVC en el tratamiento de la infección por el VIH-2 está limitado por la no disponibilidad de una adecuada prueba de tropismo y por la capacidad del VIH-2 para utilizar, además del CCR5 y el CXCR4, otros correceptores distintos733. Por último, el VIH 2 presenta resistencia intrínseca a ENF734.
Diversas circunstancias provocan que la toma de decisiones respecto al TAR en pacientes infectados por el VIH-2 tenga una mayor dificultad. En primer lugar, la historia natural del VIH-2 difiere de la del VIH-1 y, en ausencia de ensayos clínicos controlados, carecemos de las evidencias necesarias que permitan identificar el momento óptimo de inicio del TAR en estos pacientes. En segundo lugar los algoritmos genotípicos utilizados para predecir resistencias a FAR en la infección por el VIH-1 pueden no ser aplicables al VIH-2. Por último, una dificultad añadida es que para el seguimiento de los pacientes no disponemos en el momento actual de una prueba comercial que permita medir la carga viral del VIH-2.
Pese a que no disponemos de ensayos clínicos aleatorizados que permitan identificar regímenes de uso preferencial o alternativos en TAR de inicio en el escenario de la infección por VIH-2, por los motivos anteriormente expresados, y hasta no disponer de evidencias, parece razonable recomendar como TAR de inicio preferente en pacientes infectados por el VIH-2 la combinación de 2ITIAN y un IP/r.
Recomendaciones- •
El TAR de inicio de uso preferente en la infección por el VIH-2 es la combinación de 2ITIAN más un IP/r. (C-III)
- •
El uso de regímenes de TAR basados en ITINN, MVC o ENF están contraindicados en el tratamiento de la infección por VIH-2. (B-III)
Más del 50% de la población mundial infectada por el VIH-1 son mujeres. En Europa las nuevas infecciones están aumentando en la población femenina, sobre todo por vía sexual y en los estratos sociales más desfavorecidos, como en los inmigrantes735. En España, datos de la cohorte CoRis muestran una discreta disminución (24,3%) en los nuevos casos, aunque confirman cambios en los patrones de infección que afectan especialmente a la población inmigrante. Según datos recientes, el porcentaje de nuevos casos entre mujeres inmigrantes en España alcanza el 43%736, y en los casos de transmisión heterosexual las mujeres siguen incrementando su proporción, siempre superior a la masculina737.
Las incógnitas por resolver son muchas, dado que las mujeres no han sido incorporadas a los ensayos clínicos hasta 1993, y aun en los más recientes su número no sobrepasa el 30%. Las diferencias biológicas y de composición corporal, por citar algunas, podrían conllevar diferencias farmacocinéticas738 que implicasen cambios, tanto en la evolución de la enfermedad como en la respuesta a la terapia739 o en las toxicidades farmacológicas, que no han sido suficientemente explorados740,741.
El VIH-1 puede afectar a la mujer en todas y cada una de las etapas de su vida: infancia, adolescencia, edad fértil, menopausia y envejecimiento. Las mujeres con infección por el VIH-1 afrontan situaciones peculiares que suponen con frecuencia desafíos clínicos específicos y diferentes a los de los hombres infectados. Aunque van sumándose nuevos datos, somos conscientes de la necesidad de responder a muchas preguntas sobre esas diferencias de sexo y su repercusión en el manejo clínico de la mujer infectada por el VIH-1742–746.
GeSIDA y la SPNS han publicado, de forma casi simultánea con estas directrices, un documento de consenso sobre la asistencia sanitaria integral de las mujeres infectadas por el VIH-1. Remitimos a los clínicos a este prolijo y actualizado documento que contempla de forma extensa y detallada todos los aspectos esenciales en el abordaje interseccional y multidisciplinar de las mujeres infectadas por el VIH-1747.
Consideraciones especiales del tratamiento antirretroviral en la mujerVarios estudios de cohortes han descrito en las mujeres datos discordantes, tanto en los parámetros inmuno-virológicos como en la progresión de la enfermedad748–750. No obstante, no se han observado diferencias por sexo en la eficacia de los FAR, como se describe en diferentes metaanálisis750 y en los análisis específicos de eficacia según el sexo de grandes ensayos clínicos282,303,751–756, aunque no de forma unánime754–758. El inicio y la elección del TAR en la mujer tienen las mismas indicaciones y objetivos que en el varón, con la única salvedad del número de linfocitos CD4+ (<250células/μl), si la combinación antirretroviral incluye NVP.
Existen, sin embargo, aspectos específicos del sexo que obligan a modificaciones en el TAR y deben ser tenidos en cuenta. Así, el potencial reproductivo de la mujer759, en sus distintas etapas, condiciona una cuidadosa aproximación multidisciplinar, con el fin no solo de evitar la transmisión vertical, sino además de prevenir otras ITS, embarazos no deseados y comorbilidades específicas (infección por HPV y carcinoma de cérvix)760,761.
Hasta hace muy poco se creía que la infección por el VIH-1 y su terapia incidían en una menopausia más precoz y con síntomas más intensos760,761. No obstante, estudios recientes han señalado que, exceptuando la asociación con la inmunodepresión profunda, la menopausia precoz en mujeres con infección por el VIH se relaciona con los factores clásicos ya descritos en las mujeres VIH-negativas, tales como el uso de drogas y alcohol o la pertenencia a determinadas etnias762.
Diferencias en efectos adversosSe han descrito diferencias entre ambos sexos en cuanto a los efectos adversos del TAR, que sin duda es un tema preocupante. A las evidencias aportadas por el grupo ICONA y otras cohortes741 se suman los resultados de ensayos clínicos como el FIRST751,753,754. Sin embargo, dada la baja representatividad de las mujeres en los ensayos, son necesarios datos que confirmen estas diferencias y su repercusión a largo plazo282,303,753–755,763.
De sobra conocido y con reflejo específico en las directrices es el incremento de la susceptibilidad femenina al exantema y la hepatotoxicidad grave con NVP, así como la mayor incidencia de acidosis láctica con ITIAN de primera generación, sobre todo durante el embarazo764.
En cuanto a las alteraciones metabólicas, incluida la resistencia insulínica, también se han descrito diferencias entre ambos sexos, aunque los datos son menos concluyentes. La redistribución grasa765,766 tiene características especiales en las mujeres con una mayor repercusión psicológica767–770.
También los efectos tóxicos del EFV sobre el SNC son más frecuentes en las mujeres y condicionan, en ocasiones, un abandono precoz de la terapia771,772. Por último, aunque la osteopenia/osteoporosis es más prevalente en las mujeres con infección por el VIH-1773,774, su menor densidad ósea es clínicamente irrelevante antes de la menopausia775–777, cobrando mayor importancia en la posmenopausia777,778. Los cambios en la densidad mineral ósea son multifactoriales y están en relación directa con la composición corporal779, con el consumo de tóxicos780, con la dieta, con el ejercicio, etc., además de la acción del propio VIH y del TAR. Este aspecto no es baladí en una población que envejece con mayor velocidad y con mayor concomitancia de factores de riesgo que la de referencia, y puede determinar cambios en el TAR.
Otras diferencias de sexo que pueden afectar la eficacia del tratamiento antirretroviralAunque no se han identificado peores resultados en la eficacia virológica del TAR 282,304,736,752–756, se ha observado una menor durabilidad del TAR tanto en ensayos clínicos753–755 como en estudios observacionales772,781–783.
Podemos identificar factores de confusión potenciales que explicarían parte de estas diferencias: aspectos socioculturales (la desigualdad de la mujer en algunos sectores como inmigrantes, minorías étnicas, etc.), superior incidencia de problemas psicológicos/psiquiátricos784, mayor temor a la estigmatización, excesiva preocupación por la confidencialidad, etc., todo lo cual contribuye a dificultades en el acceso al sistema sanitario, al retraso en el inicio del TAR y a una peor adherencia785,786. En resumen, el éxito en el cuidado a la mujer con infección por el VIH depende en gran medida de la capacidad de entender y subsanar todas las barreras específicas de género para mitigar los peores resultados observados en la clínica diaria787,788.
Mujer en periodo fértil: interacciones con anticonceptivos hormonalesAproximadamente el 70% de las mujeres que participan en estudios sobre el VIH-1 son sexualmente activas, muchas de ellas en edad fértil. El uso del preservativo es el método anticonceptivo de elección tanto por su eficacia (con una tasa de fallos del 3%)789 como por prevención simultánea de otras ITS. En la práctica real, el uso de métodos anticonceptivos es variable y la anticoncepción hormonal puede ser más frecuente en muchas pacientes debido a influencias culturales u otros factores. Además, en ocasiones son prescritos por médicos de familia o ginecólogos desconocedores de los riesgos potenciales, sin consultar con un especialista en VIH-1. Los métodos anticonceptivos hormonales e intrauterinos en general se han mostrado seguros en mujeres con infección VIH-1790–793, incluso a largo plazo, como es el caso de los dispositivos intrauterinos que liberan levonorgestrel794. No obstante, se han descrito embarazos no deseados con el uso de implantes de etonorgestrel en pacientes con EFV795. Los anticonceptivos hormonales inyectables son en algunas cohortes más eficaces que los orales y mucho más que los preservativos796. Los autores preconizan la importancia de los planes de prevención familiar y de los métodos anticonceptivos duales.
La anticoncepción hormonal no interfiere con la eficacia del TAR, pero los datos son discordantes en cuanto a su relación con la progresión de la enfermedad792. Muchos de los FAR de uso común interaccionan con los anticonceptivos hormonales797–804, aumentando o disminuyendo los niveles en sangre de etinilestradiol y/o de noretindrona, conduciendo a la intensificación de su toxicidad (como, por ejemplo, el tromboembolismo) o a la disminución de su eficacia respectivamente (para más detalles, véase el capítulo de interacciones y las tablas 5-9). Incluso la escasa experiencia que existe con levonorgestrel en la contracepción de urgencia indica que la inducción por EFV podría hacer necesario un aumento de dosis805. Cuando se usan estos fármacos se recomienda la utilización de medios alternativos o adicionales de contracepción.
En cuanto al acetato de medroxiprogesterona depot (DMPA o Depo-Progevera), aunque hay pocos estudios, no parece ver afectados sus niveles por NFV, NVP o EFV806,807. Hay que tener en cuenta su efecto negativo sobre la densidad mineral ósea808 en pacientes con osteoporosis establecida o con factores de riesgo. Un aspecto especialmente controvertido es el que se refiere a la asociación entre el uso de contraceptivos hormonales, como la DMPA, y el incremento del riesgo de adquisición y transmisión del VIH. Así se observó en un estudio que incluyó a casi 3.800 parejas serodiscordantes en las que el riesgo de transmisión se duplicó809. A pesar de los posibles sesgos del estudio810, su plausibilidad biológica y su concordancia con otros estudios obligan a una investigación más amplia811–813.
Hay muy pocos datos respecto a otros tipos de anticoncepción hormonal combinada (parches de larga duración, anillos vaginales con componente hormonal, etc.)796, por lo que se recomienda también el uso de medios combinados de contracepción.
Recomendaciones sobre tratamiento antirretroviral en mujeres- •
El inicio y los objetivos del TAR son los mismos en las mujeres que en los hombres. (A-III)
- •
La mayor incidencia de exantema y hepatotoxicidad por NVP condiciona modificaciones en su uso en las mujeres (cifras de linfocitos CD4+<250células/μl). (A-II)
- •
La elección de la combinación del TAR de inicio en la mujer debe tener en cuenta su deseo o riesgo de embarazo y el uso de anticonceptivos orales. Estos fármacos interaccionan con muchos FAR, por lo que deben tenerse en cuenta sus posibles interacciones y complementar su uso con un método de barrera (protección dual). (A-III)
La transmisión maternofetal es la causa de prácticamente la totalidad de los casos de infección por VIH-1 en niños. Estamos, sin duda, ante una nueva etapa de la epidemia, en la cual el deseo de la maternidad forma parte de los derechos de la paciente y su adecuada planificación entra dentro de su atención integral814. El periodo fetal más vulnerable es durante la gestación temprana, por lo que todas las mujeres deben recibir información previa sobre la teratogenicidad de los fármacos como parte de su control clínico y adecuar el TAR ante el deseo de embarazo815–818.
A continuación se resumen las evidencias sobre las que se basan las recomendaciones del TAR en el embarazo y la prevención de la transmisión vertical. Si se requiere más información, se aconseja la lectura de documentos más amplios815,818.
Se recomienda realizar la serología frente al VIH-1 a todas las embarazadas independientemente de sus antecedentes epidemiológicos. El riesgo de transmisión vertical depende de varios factores (maternos, virales, placentarios, obstétricos, lactancia, fetales, neonatales, etc.), pero la CVP de la madre durante el embarazo y en el parto es el factor determinante, sin que exista un dintel mínimo que evite la transmisión815.
Ante una mujer infectada por el VIH-1, la prevención de la transmisión vertical se basa en los siguientes principios: realización de TAR, CVP indetectable o lo más baja posible, prueba de resistencias, cesárea programada (si la CVP es >1.000copias/ml), tratamiento con ZDV del niño durante 6semanas y evitar la lactancia materna815,819. El tratamiento con ZDV intravenosa durante el parto está ahora cuestionado, recomendándose solo si la CVP es >400copias/ml820,821.
El objetivo del TAR en el embarazo es conseguir y mantener la CVP indetectable. La prueba de resistencia genotípica está recomendada para todas las embarazadas antes de comenzar el TAR, así como para aquellas que, estando en TAR, tengan CVP detectable. En ocasiones es necesario iniciar el TAR aun sin disponer del resultado de la prueba, sujeto a modificación posterior en el caso de que este detecte mutaciones de resistencias a los FAR elegidos819.
Consideraciones a tener en cuenta respecto al tratamiento antirretroviral en el embarazoEl embarazo en la mujer con infección por el VIH-1 implica unas elecciones específicas que afectan tanto a los FAR y sus dosis (los niveles de algunos de ellos disminuyen en el segundo y tercer trimestre) como al momento del inicio del TAR, a los efectos adversos por toxicidad durante el embarazo y a los potenciales riesgos para el neonato, muchos de los cuales son desconocidos con la mayoría de los FAR. Son controvertidos tanto la necesidad como el acceso a la monitorización de los niveles de fármacos durante la gestación, así como el aumento de su dosis, no siendo este recomendado de forma universal si la CVP está por debajo de los niveles de detección.
Los datos de seguridad de los FAR para el feto son limitados (tabla 17). De los datos disponibles, se puede destacar:
- 1.
Está en plena discusión el momento del inicio del TAR en la paciente embarazada sin TAR previo. Las respuestas de las directrices no son homogéneas al respecto, pero se acumulan evidencias acerca de que el momento de inicio y la duración del TAR, sobre todo en mujeres con CVP>100.000copias/ml, son factores determinantes de la consecución de una CVP indetectable en el embarazo y de un recién nacido libre de infección822.
- 2.
ZDV es segura al menos a corto y medio plazo, aunque un estudio sugirió la existencia de riesgo de toxicidad mitocondrial en los niños.
- 3.
EFV es un fármaco potencialmente teratógeno. Puesto que su período de neurotoxicidad se restringe a las primeras 5-6semanas de gestación823, la mayoría de mujeres conocen su embarazo pasado ese tiempo. Por otro lado, en un reciente metaanálisis de 21estudios, el riesgo relativo de defectos en los recién nacidos de mujeres con EFV en relación con los de mujeres tratadas con otros FAR fue 0,85824. Teniendo en cuenta todo ello, no sería necesario retirar EFV a las embarazadas que ya lo estuvieran tomando, y así lo recogen las últimas directrices perinatales del DHHS y la BHIVA815,821.
- 4.
Los FAR más seguros en el embarazo son LPV816, ATV825,826, NVP, ZDV y 3TC. ATV ha pasado a ser un IP preferido en lugar de alternativo al disponerse de más información sobre su seguridad en el embarazo.
- 5.
En algunos estudios se ha asociado el uso de IP con bajo peso al nacer827 o prematuridad. En trabajos más recientes, la prematuridad y el bajo peso se relacionan tanto con el propio VIH828 como con las características sociodemográficas de la mujer y el TAR, y más específicamente con el inicio de la administración de RTV829.
- 6.
La seguridad de otros fármacos es aún peor conocida, y aunque se catalogan como categoría «B» o «C» de la FDA (tabla 17), los datos disponibles de seguridad, especialmente a largo plazo, son limitados. Con los datos que se van acumulando, DRV ha pasado a ser un FAR alternativo en el embarazo y RAL pasa de tener insuficientes datos a ser de uso recomendado en determinadas circunstancias815,821. Se tiene escasa información de TDF, FPV e IDV y muy escasa de TPV, DRV, RAL, ETR y MVC830–832. Por ello, se deben evitar estos FAR, salvo en pacientes embarazadas en fracaso virológico, en cuyo caso deberemos orientarnos en función de la escasa información disponible.
Seguridad de los antirretrovirales en el embarazo
| Fármaco | FDA | Paso a través de placenta (cociente recién nacido/madre) | Carcinogenicidad (animales) | Teratogenicidad (animales) |
| Zidovudina | C | 0,85 | Sí | Sí |
| Didanosina | B | 0,5 | No | No |
| Estavudina | C | 0,76 | Sí | No |
| Lamivudina | C | 1 | No | No |
| Abacavir | C | Sí (ratas) | Sí | Sí |
| Tenofovir | B | 0,95-0-99 | Sí | No |
| Emtricitabina | B | 0,4-0,5 | No | No |
| Saquinavir | B | Mínimo | No | No |
| Indinavir | C | Mínimo | Sí | No |
| Ritonavir | B | Mínimo | Sí | Sí |
| Nelfinavir | B | Mínimo/Variable | Sí | No |
| Fosamprenavir | C | ¿? | Sí | No |
| Lopinavir | C | 0,2 ± 0,13 | Sí | Sí |
| Atazanavir | B | Mínimo/Variable | Sí | No |
| Tipranavir | C | ¿? | No completado | No |
| Darunavir | C | ¿? | Sí | No |
| Nevirapina | B | 1 | Sí | No |
| Efavirenz | D | 1 | Sí | Sí |
| Etravirina | B | ¿? | No completado | No |
| Rilpivirina | B | ¿? | Sí | No |
| Enfuvirtida | B | No (basado en muy pocos datos) | No realizado | No |
| Maraviroc | B | ¿? | No | No |
| Raltegravir | C | Ratas (1,5-2,5) Conejos (0,02) | No completado | No |
También existe controversia respecto a la seguridad de TDF en el embarazo, cuestión de gran importancia habida cuenta de que cada vez son más las embarazadas cuyo régimen de TAR incluye este FAR833. Aunque la mayoría de los datos más recientes no reflejan alteraciones en el desarrollo óseo834, un importante estudio de cohortes (PHACS)835 observó una muy ligera menor longitud y circunferencia de la cabeza de los nacidos al año de seguimiento.
Existe un registro establecido en 1989 que recoge de forma prospectiva la prevalencia de malformaciones entre los expuestos a cualquier FAR durante el primer trimestre, que es del 2,9%, similar al de la población no expuesta (2,7%)836.
En cuanto a la madre, cabe reseñar que se han descrito casos de toxicidad mitocondrial y acidosis láctica en gestantes, las cuales pueden estar más predispuestas a esta complicación. Estos efectos secundarios se asociaron inicialmente a ZDV, pero posteriormente se han descrito casos relacionados con d4T. La FDA ha comunicado 3muertes maternas por acidosis láctica y 3muertes fetales en mujeres tratadas con d4T y ddI como ITIAN, por lo que desaconseja esta combinación765. Además se ha descrito que la hepatotoxicidad por NVP sería 12veces más frecuente en gestantes con una cifra de linfocitos CD4+>250células/μl837.
La elección del TAR en la embarazada y sus particularidades se explican detalladamente en las directrices específicas nacionales o internacionales815,818,821.
Los datos disponibles acerca de la eficacia de la cesárea programada (semana38) como instrumento potencial para reducir la transmisión vertical sugieren que si en el momento del parto la CVP es <1.000copias/ml, la cesárea programada no disminuye el riesgo de transmisión del parto vaginal (0,8% frente a 0,7%)815,838,839. La morbilidad relacionada con la cesárea en los países desarrollados no parece ser importante839.
Recomendaciones sobre tratamiento antirretroviral y gestación- •
La prueba de VIH-1 debe efectuarse a toda mujer embarazada (A-III). Si hay prácticas de riesgo, debe repetirse en el tercer trimestre. (A-III)
- •
En las mujeres que llegan al parto sin conocer su estado respecto al VIH-1 se debe hacer una prueba rápida, ya que la cesárea programada reduce la transmisión en un 50%. (A-II)
- •
El objetivo del TAR es conseguir CVP indetectable (A-II). La decisión de iniciar el TAR en el primer trimestre o demorarlo hasta la semana 12 dependerá del recuento de linfocitos CD4+, de la CVP y de las condiciones de cada mujer, tales como náuseas y vómitos (A-III). El inicio más precoz del TAR puede ser más efectivo en reducir la transmisión maternofetal. (B-III)
- •
Debe realizarse una prueba de resistencias en todas las mujeres con infección por el VIH-1 embarazadas sin TAR o en las que la CVP sea detectable. (A-III)
- •
Uno de los fármacos a incluir en el TAR de inicio de la mujer embarazada debe ser ZDV, que se administrará durante el embarazo y al recién nacido (A-I). En cualquier caso el régimen de TAR debe incluir 2ITIAN que atraviesen la barrera placentaria (ZDV, 3TC, FTC, TDF o ABC).
- •
En general, la mujer infectada por el VIH que ya realiza TAR y se queda embarazada debe mantener su régimen de TAR previo, siempre que sea bien tolerado y eficaz. Muchos expertos recomiendan no retirar EFV una vez pasadas 5 o 6 semanas de gestación (C-III). Si la mujer tomaba NVP y tiene CVP indetectable, debe mantenerse este FAR, independientemente del recuento de linfocitos CD4+. (A-III).
- •
Se desaconseja la combinación de d4T+ddI por riesgo de acidosis láctica. (B-III)
- •
Es fundamental planificar el control de la CVP antes del parto, hacia las semanas 32-36. Si no se consigue una CVP suficientemente baja (<1.000copias/ml), se debe indicar una cesárea programada en la semana 37-38. (A-II)
En el año 2010 la incidencia de nuevas infecciones se estabilizó o disminuyó en algunos países, pero se calcula que alcanzó los 2,7 millones840. En las últimas décadas se han realizado múltiples intervenciones biomédicas para disminuir la transmisión del VIH-1. Se han conseguido algunos éxitos con los programas de intercambio de jeringuillas y tratamiento sustitutivo con metadona en los UDVP, así como la disminución de nuevas infecciones en un 50-60% en varones heterosexuales mediante la circuncisión masculina, la difusión del uso del preservativo, el control de las ITS, sobre todo del herpes genital, la espectacular disminución de la transmisión vertical, y la disminución de la transmisión en parejas serodiscordantes16,152,841,842.
Las actuaciones destinadas a lograr modificaciones de conducta no han logrado sus frutos, fundamentalmente por la diferente percepción del riesgo en la transmisión sexual16. Una de las principales dificultades en la disminución de la transmisión sexual del VIH-1 estriba en la detección de la infección por el VIH-1 aguda por su elevada capacidad de transmisión843. Por otra parte, el diagnóstico tardío de la infección por el VIH-1 contribuye a la transmisión sexual.
En el año 2008 se estimó que el número de pacientes infectados nuevos era 2,5 veces superior al de pacientes que iniciaron TAR840. Una intervención biomédica combinada en la prevención del VIH-1 es una necesidad urgente16,844–846. En ausencia de una vacuna preventiva, se ha planteado que el TAR puede prevenir la transmisión del VIH-1: antes de la exposición mediante la profilaxis pre-exposición (PrPE), después de la exposición por medio de la profilaxis post-exposición (PPE), y la prevención secundaria con el tratamiento de la población infectada844–846. La lucha frente al VIH/sida ha alcanzado un gran desarrollo, y ahora es posible plantearse una próxima generación libre de la infección por el VIH847.
Papel del tratamiento antirretroviral en la prevención de la transmisión del virus de la inmunodeficiencia humana tipo 1El TAR disminuye la CVP en las secreciones genitales, lo que puede contribuir a la reducción de la transmisión sexual841. La mayor evidencia de la efectividad del TAR en la reducción de la transmisión sexual del VIH-1 proviene del estudio aleatorizado HPTN 052152 y de estudios observacionales realizados en parejas heterosexuales serodiscordantes. En un metaanálisis se observó un riesgo cero con CVP<400copias/ml bajo TAR. El riesgo de transmisión de una persona en TAR es de 0,5 por 100personas/año y de 5,6 por 100personas/año de los pacientes no tratados848.
Varios estudios observacionales realizados en África han demostrado una disminución de la transmisión en un 80-92% en 6.424 parejas serodiscordantes tras el inicio de TAR en las personas infectadas849–851, la cual fue cero en una cohorte de Madrid, a pesar de contabilizar más de 7.000 relaciones coitales no protegidas y 47 gestaciones naturales, mostrando que el TAR confirió una mayor capacidad preventiva que el uso adecuado del preservativo852. Estos estudios y el HPTN 052 fueron valorados en The Cochrane Collaboration, un estudio de la cual concluye que el TAR es una intervención potente en la prevención de la infección por el VIH-1 en las parejas heterosexuales serodiscordantes en las que el caso índice tenga una cifra de linfocitos CD4+<550células/μl (límite superior de linfocitos CD4+ en la población estudiada)853.
El estudio aleatorizado en faseiii (HPTN 052)152 incluyó 1.763parejas serodiscordantes cuyos casos índice presentaban cifras de linfocitos CD4+ comprendidas entre 350-550células/μl (el estudio tenía 2objetivos: evaluar la evolución de la infección según se iniciara el tratamiento de modo precoz o tardío y estudiar el riesgo de transmisión de la infección). Todas las parejas fueron instruidas en la prevención de la transmisión sexual y en la adherencia. Se objetivaron 39 casos de transmisión, 28 de los cuales estaban relacionados genéticamente con el VIH-1 del paciente infectado de la parejas. De los 28 casos de transmisión, 27 se produjeron en el grupo de inicio tardío del TAR (<250linfocitos CD4+/μl) y un caso de transmisión en el grupo de inicio precoz del TAR (>350linfocitos CD4+/μl). Se demostró una reducción de la transmisión sexual del 96% (HR: 0,04; IC95%: 0,01-0,27; p<0,001) en el grupo de TAR precoz y un retraso de la progresión clínica de la infección (HR: 0,59; IC95%: 0,40-0,88; p<0,01). La durabilidad del efecto del TAR en la prevención es un objetivo de la siguiente fase del estudio HPTN 052.
La eficacia demostrada del TAR en la prevención secundaria en las parejas heterosexuales serodiscordantes no puede ser extrapolada a otras situaciones. El comité científico del President's Emergency Plan for AIDS Relief (PEPFAR) ha concluido que no existen razones para pensar que el TAR no pueda suprimir la transmisión heterosexual en grupos de riesgo elevado854. En cambio, el Comité de Expertos de la OMS concluye que no hay razón para presuponer que con TAR la contagiosidad de los HSH sea menor855. Hay estudios específicos en parejas seodiscordantes de HSH. La biología de la transmisión en UDVP precisa nuevas investigaciones.
Estos datos sugieren que una difusión amplia de TAR podría reducir la transmisión por vía sexual. Un modelo matemático basado en la detección precoz de la infección, mediante una prueba universal voluntaria, y el inicio de TAR precoz en todas las personas recién diagnosticadas, mostró una disminución de la incidencia y de la mortalidad a menos de un caso por 1.000personas/año en 10años856. Otro modelo similar realizado en HSH de San Francisco mostró una disminución del 91% en la incidencia en 10años857. Sin embargo, en países con una elevada cobertura de TAR, como Estados Unidos, Holanda, Francia y Australia, la incidencia de transmisión entre HSH no ha disminuido858.
Recientemente se han comunicado efectos beneficiosos indirectos de TAR en la comunidad. Tanto en San Francisco como en Vancouver se observó una relación entre la generalización del TAR y la disminución de nuevos casos de infección VIH-1859,860. Por su contribución a la disminución de la infección en la comunidad, estos estudios han sido denominados «estudios ecológicos». Dichos estudios son interesantes en la medida que generan nuevas expectativas, pero también son problemáticos porque tienen sesgos en la metodología y resultados contradictorios, fundamentalmente porque se debería evaluar en ellos la accesibilidad y la adherencia al TAR861. En un estudio realizado en Sudáfrica con una cohorte longitudinal de 16.667individuos se observó que por cada aumento del 1% en la cobertura del TAR en la población infectada había una disminución del 1,7% en el riesgo de transmisión en la población no infectada862. Este resultado se presenta como uno de los más convincentes dentro de la estrategia de la prevención secundaria del VIH-1.
Los problemas de los modelos matemáticos basados en el TAR para disminuir la transmisión sexual del VIH-1 y de los estudios «ecológicos» comienzan cuando se confrontan con la realidad. Se calcula que en los países occidentales el 21-30% de la población infectada por el VIH-1 desconoce su situación y el porcentaje de diagnósticos tardíos alcanza el 15-45%, a pesar de las campañas realizadas en Estados Unidos para el uso de las pruebas rápidas para la detección precoz del VIH-1861,863. La detección de la infección aguda, y su importancia en la transmisión, es un problema difícil de abordar y habitualmente no está considerado en los modelos matemáticos. En Estados Unidos aproximadamente el 75% de los pacientes diagnosticados en los 12meses previos son controlados en el sistema sanitario y el 80-90% en un período de 3-5años. En los países occidentales, a pesar de las recomendaciones de las diferentes guías para el inicio de TAR, el 20-27% de pacientes con indicación de TAR no son tratados. En la práctica diaria, si obtuviéramos una detección del 90%, con el uso de TAR precoz, con una adherencia a la terapéutica del 90% y una fidelidad del 90% en el seguimiento se lograría la indetectabilidad de la CVP en el 90% de los pacientes tratados. En este escenario ideal, se calcula que el 34% de los pacientes infectados se mantendrían con CVP detectables, con el potencial riesgo de transmisión863.
En este campo quedan cuestiones importantes por determinar: la durabilidad de la protección, el balance de los beneficios y los efectos adversos de la terapia precoz, la adherencia a largo plazo, la posibilidad de transmisión de cepas resistentes y las dificultades de su implementación en los países en vías de desarrollo844,846.
Recomendaciones- •
Se recomienda el uso de TAR para prevenir la transmisión del VIH de una persona infectada a su pareja heterosexual. (A-I)
- •
Se recomienda el uso del TAR para prevenir la transmisión del VIH entre personas con otras prácticas de riesgo. (A-III)
La profilaxis pre-exposición (PrPE) está basada en estudios realizados con animales, en los que se demostró que los FAR pueden prevenir la transmisión del VIH-1. Está dirigida a grupos de personas no infectadas por el VIH-1 pero con un elevado riesgo de adquirir la infección por vía sexual, como trabajadores del sexo, mujeres y varones en parejas discordantes, HSH y UDVP.
El estudio CAPRISA-004, dirigido a mujeres sexualmente activas de parejas serodiscordantes, comparó TDF al 1% en gel vaginal, antes y después del coito, frente a placebo. Se demostró en él una disminución global en la incidencia de transmisión del 39%, que alcanzó hasta el 54% en las mujeres con una adherencia superior al 80%864. El estudio IPrEX, que comparó TDF/FTC por vía oral frente a placebo en HSH, mostró una disminución de la transmisión del VIH del 44%. La adherencia fue un factor fundamental en la eficacia, como indica el hecho de que se hallasen niveles plasmáticos detectables de TDF y FTC en el 51% de los HSH que permanecieron seronegativos frente al 9% de los que se infectaron865.
En 2012 se han publicado 3estudios que previamente habían sido comunicados en congresos. El estudio Partners-PrEP se realizó en África con 4.758parejas serodiscordantes (distribución por sexo de los miembros seronegativos: 38% mujeres y 62% varones). Se observó una reducción del riesgo de transmisión con TDF del 67% y con TDF/FTC del 75% respecto al grupo placebo. La eficacia fue similar para hombres y mujeres y no se objetivaron diferencias estadísticamente significativas entre TDF y TDF/FTC. Tampoco hubo diferencias en el porcentaje de efectos adversos ni en el de embarazos866. El estudio TDF-2 fue realizado en Botswana, donde la prevalencia de infección por el VIH es del 17,6% en la población general y del 40% en el estrato de personas de edad comprendida entre 30 y 44años, en 540mujeres y 660varones heterosexuales. Se consiguió una reducción del 62% con TDF/FTC frente a placebo, pero sin diferencias significativas entre las mujeres. Este estudio tuvo la limitación de que el 30% de los individuos aleatorizados no finalizaron el seguimiento867. En estos 2estudios, a pesar de una elevada adherencia, evaluada por la referencia de los pacientes y el recuento de comprimidos, se halló de forma retrospectiva una diferencia estadísticamente significativa en los niveles plasmáticos de TDF entre los pacientes que se infectaron y los que permanecieron sin infección. El estudio FEM-PrEP, que también fue realizado en África en 2.120mujeres de edades comprendidas entre 18 y 45años y con un alto riesgo de infección por vía sexual, fue suspendido por falta de eficacia, pues en el grupo de TDF/FTC se observó una tasa de incidencia de 4,7 infecciones por 100pacientes/año y en el grupo placebo de 5 infecciones por 100pacientes/año (HR: 0,94; IC95%: 0,59-1,52; p=0,81). Hubo un mayor porcentaje de embarazos durante el seguimiento en el grupo de TDF/FTC. Este estudio tuvo un problema muy importante en la adherencia, pues un porcentaje inferior al 40%, tanto de las mujeres que presentaron seroconversión como de las que permanecieron sin infectarse por el VIH, presentaban niveles plasmáticos detectables de TDF868. El estudio VOICE, que aún no ha sido publicado, fue igualmente realizado en África e incluyó 5.000mujeres de edades comprendidas entre 18 y 45años. Una parte del estudio pretendía comparar TDF, TDF/FTC y placebo por vía oral, y otra un gel de TDF al 1% con un gel de placebo, ambos administrados diariamente por vía vaginal. La rama de TDF por vía oral y la de TDF en gel vaginal fueron suspendidas por falta de eficacia869. En todos los estudios anteriormente comentados la tolerabilidad y la seguridad fueron excelentes. En el estudio TDF-2 se observó una disminución en la densidad mineral ósea en los pacientes tratados con TDF, aunque se desconoce su significación clínica. Los 3estudios reflejaron en sus seguimientos una disminución de las prácticas sexuales de riesgo.
Las diferencias observadas en estos estudios probablemente estén relacionadas con la falta de adherencia y con niveles insuficientes de FAR en el tracto genital femenino. Se ha observado que las concentraciones alcanzadas en las secreciones con TDF por vía oral son muy inferiores a las obtenidas con TDF en forma de gel tópico. Las concentraciones intracelulares de TDF-DP fueron 100veces superiores en la mucosa rectal que a nivel cervicovaginal y, en cambio, las concentraciones de FTC-TP fueron 10-15veces superiores en el tejido vaginal y en el cérvix que a nivel rectal. No se conocen las concentraciones de TDF-DP y FTC-TP que son necesarias para inhibir la transmisión del VIH-1845,870.
Recientemente, la OMS y los CDC han elaborado unas recomendaciones para el uso de PrPE con TDF o TDF/FTC por vía oral en personas sin infección por el VIH-1 que tienen un elevado riesgo de adquirir la infección por vía sexual, fundamentalmente individuos heterosexuales con múltiples parejas, HSH y miembros de parejas heterosexuales serodiscordantes871–873. La FDA ha autorizado el uso de TDF/FTC en la PrPE del VIH-1. Durante la PrPE se recomienda la realización de la prueba del VIH al menos cada 3meses.
No hay datos específicos en mujeres embarazadas ni en UDVP. Se recomienda realizar estudios de adherencia de forma prospectiva y una búsqueda activa de infección aguda por el VIH-1, así como de toxicidad. Actualmente hay en marcha estudios de PrPE con otros FAR (MVC, RAL, RPV), otros fármacos inyectables, microbicidas vaginales, pautas intermitentes, etc.845,870,874.
La PrPE es un reto para los organismos internacionales. Teniendo en cuenta los escasos recursos existentes, se plantea la duda entre ofertar PrPE a personas no infectadas o TAR a las personas infectadas. La mejor estrategia de salud pública en la actualidad es la de ofertar TAR a todos los pacientes infectados y reservar la PrPE para casos muy específicos845,870,874. En el actual estado de conocimientos no se ha emitido ninguna recomendación a este respecto en Europa.
Profilaxis post-exposición ocupacionalEl uso de TAR tras una exposición profesional al VIH-1 reduce el riesgo de transmisión, aunque se han documentado transmisiones a pesar de una PPE correcta con 3FAR. El riesgo global de transmisión del VIH-1 tras una exposición percutánea con sangre infectada oscila entre el 0,24 y el 0,65%, y tras un contacto con mucosas o piel no intacta es del 0,09%875. Los factores asociados a un mayor riesgo de transmisión del VIH-1 son: pinchazo profundo (OR: 15; IC95%: 6,0-41), presencia de sangre visible en el dispositivo (OR: 6,2; IC95%: 2,2-21), con un sistema recién extraído de vena o arteria (OR: 4,3; IC95%: 1,7-12) o de una fuente de exposición con una infección por el VIH-1 avanzada y una CVP>1.500copias/ml (OR: 5,6; IC95%: 2-16). La PPE con monoterapia (ZDV) reduce la transmisión un 81% (OR: 0,19; IC95%: 0,06-0,52)876. No existen estudios comparativos que permitan establecer recomendaciones firmes sobre el momento de inicio, la duración o los FAR o combinaciones de FAR a emplear tras una exposición accidental.
Por modelos animales, y por estudios de casos y controles, sabemos que la PPE es tanto más eficaz cuanto antes se inicie. Su duración no está establecida, pero en modelos animales los periodos de 3-10días de tratamiento son menos eficaces que los de 28días, por lo que se ha consensuado recomendar 4semanas. No hay estudios que demuestren que la PPE con 3FAR sea mejor que con 2FAR, aunque prevalece la opción de indicar triple terapia, de modo similar al TAR de inicio. La elección de los FAR dependerá de los potenciales efectos adversos y de las preferencias de los sanitarios, teniendo en cuenta que la intolerancia a los FAR entre el personal sanitario es muy elevada (50%)877,878.
Para establecer la indicación de profilaxis debemos conocer el tipo de exposición y solicitar, tras consentimiento informado, una prueba serológica rápida frente al VIH-1, el VHB y el VHC del paciente-fuente y del trabajador. En la elección de PPE se debe tener en cuenta un posible embarazo y potenciales interacciones medicamentosas con fármacos que pudiera estar tomando la persona afectada. En la actualidad se están revisando las guías sobre PPE. Parece que se va a recomendar una pauta de 3FAR en todos los casos, dada la excelente tolerabilidad y la eficacia de los nuevos regímenes. En la revisión de octubre de 2012 de la guía del Estado de Nueva York se recomienda una combinación de TDF/FTC y RAL como régimen preferente, y de TDF/FTC y DRV/r (800/100mg QD) o ATV/r (300/100mg QD) o FPV/r (1.400/100 mg/QD) como regímenes alternativos. Estas mismas guías ya no recomiendan pautas de 2ITIAN ni el uso de ZDV. Sin embargo, otros organismos norteamericanos y europeos continúan recomendando el uso de TDF/FTC/LPV/r. NVP no es aconsejable por su potencial toxicidad cutánea o hepática875,877–881. Tampoco se recomienda ABC por la posibilidad de que ocurra una RHS.
La transmisión ocupacional del VIH-1 es muy rara, y la evidencia muestra que la PPE modifica los riesgos de transmisión. Conviene recordar que ninguna pauta de PPE será eficaz al 100%881. En el Reino Unido, un estudio de la Health Protection Agency mostró que en el 20% de los casos con exposición manifiesta a pacientes con infección por el VIH-1 no se realizó la PPE. El conocimiento de las recomendaciones por parte de los sanitarios era bueno, pero existían diferencias en la percepción del riesgo de la transmisión en los diversos estamentos sanitarios a la hora de iniciar una PPE tras una exposición882.
En caso de conocer o sospechar que el paciente fuente tiene un virus resistente, se deben seleccionar FAR sin resistencia cruzada. Se ha demostrado que una proporción no desdeñable de virus de los casos fuente pueden presentar mutaciones de resistencia, especialmente a ITIAN e ITINN. No se recomiendan MVC, RAL o ENF para PPE, salvo en casos excepcionales de multirresistencia. Una revisión demostró que en el 14% de los casos de PPE se utilizan FAR no recomendados en las guías: DRV/r (42%), RAL (36%), ATV/r (22%) y MVC (7%). La razón fundamental para ello era la presencia de resistencias a FAR en el paciente fuente883.
La PPE debe iniciarse lo antes posible, preferentemente en las primeras 2h y nunca más tarde de 24-36h tras la exposición875,877–881.
A toda persona que haya sido evaluada para PPE debe ofrecérsele un plan de seguimiento, que incluirá información, apoyo psicológico y control de los posibles síntomas de primoinfección. Si se prescribió PPE, se debe efectuar un control dentro de las primeras 72h para reevaluar la necesidad de profilaxis y conocer si el trabajador ha presentado reacciones adversas, interacciones o problemas de adherencia. Se programarán controles analíticos y serológicos a las 4semanas y a los 3 y 6meses. Algunos organismos recomiendan no prolongar el seguimiento más allá de las 12semanas, pues con un resultado negativo a las 12semanas con las pruebas serológicas de última generación se puede descartar razonablemente una seroconversión.
Recomendaciones- •
Los servicios sanitarios deben disponer de un protocolo escrito sobre las actuaciones y derivaciones a seguir en el caso de exposición al VIH-1, profesional o no, con disponibilidad de diagnóstico serológico rápido y con accesibilidad de 24h a los fármacos utilizados en la PPE. (A-III)
- •
Se debe valorar el caso fuente (VIH-1 confirmado o sospechoso), el estado serológico de la persona expuesta y las características de la exposición para indicar la PPE. (A-III)
- •
La administración de PPE debe iniciarse lo antes posible, mejor en las primeras 2h, y hasta las 24-36h. Su duración será de 4semanas. (A-II)
- •
No se recomienda iniciar PPE pasadas 72h de la exposición. (A-III)
- •
Cuando esté indicada la PPE, se recomienda una pauta convencional con 3FAR. Se recomienda una combinación fija de TDF/FTC asociada a un IP/r (A-III) o, como alternativa, raltegravir. (C-III)
- •
Si se sospecha que el virus del caso índice puede tener resistencia a uno o varios FAR, la profilaxis debe incluir medicamentos sin resistencia cruzada. (A-III)
- •
En caso de duda sobre la indicación de la PPE, se recomienda administrar la primera dosis de forma inmediata y valorar su continuidad en las 24h posteriores por un experto en infección VIH-1. (A-III)
- •
El seguimiento debe incluir la revaloración de la indicación a las 24-72h del inicio del TAR y control del cumplimiento y tolerabilidad del TAR, así como serología al VIH-1, VHB y VHC (estos en caso de fuente infectada o con sospecha) en los meses 1, 3 y 6 tras la exposición. (B-III)
La indicación de la PPE no ocupacional se basa en la patogenia de la infección por el VIH-1, en estudios observacionales, principalmente realizados en mujeres víctimas de violaciones y en HSH, y en la información procedente de otras profilaxis frente al VIH-1 (exposición ocupacional, transmisión vertical) y de datos de experimentación animal. No hay datos sobre la eficacia de esta estrategia, pero la información disponible sobre su accesibilidad y seguridad ha conducido a una amplia aceptación de la misma884.
La práctica sexual con mayor riesgo de transmisión del VIH es la relación anal receptiva no protegida con un varón infectado por VIH-1 (0,5-3%), seguida del intercambio de jeringuillas (0,67%), de la punción percutánea con una aguja usada por una persona infectada por el VIH-1 (0,3%), de la relación vaginal receptiva (0,05-0,8%) o de la relación vaginal o anal insertiva (0,05-0,065%). La relación orogenital receptiva e insertiva tiene un riesgo menor (0,005- 0,01%)884–886. Un estudio ha evaluado el riesgo de transmisión por acto sexual en 1.381HSH entre los años 2001-2004. La transmisión en la relación anal receptiva fue del 1,43% (IC95%: 0,48-2,85) si existía eyaculación y del 0,65% (IC95%: 0,15-1,53) en caso de retirada previa a la eyaculación. La probabilidad de transmisión por relación anal insertiva en varones circuncidados fue del 0,11% (IC95%: 0,02-0,24) y del 0,62% (IC95%: 0,07-1,68) en varones no circuncidados887. Es muy importante recordar que, en la era del TAR, la probabilidad de transmisión del VIH-1 por una relación anal receptiva con eyaculación es el doble que sin eyaculación y 10veces superior a la observada en la relación anal insertiva con un varón circuncidado, en las relaciones heterosexuales o en la exposición ocupacional. El riesgo de transmisión está igualmente aumentado en caso de ITS y en las agresiones sexuales.
Inicialmente existió cierta preocupación porque la extensión de la PPE no ocupacional pudiera reducir las medidas de prevención primaria al existir una supuesta «profilaxis para el día después» que relajara la seguridad en las prácticas sexuales o en la inyección de drogas. Sin embargo, se ha demostrado que si la PPE no ocupacional se acompaña de la oportuna intervención educativa se pueden reducir las prácticas de riesgo888. En este sentido es importante realizar una historia sexual en la primera visita y de forma periódica, junto con el cribado de ITS y discutir con el paciente los métodos para la prevención de la transmisión del VIH-1 en las relaciones sexuales. Los problemas de la PPE no ocupacional son que a menudo no se conoce la situación serológica de la fuente, el inicio de la profilaxis suele ser más tardío y el nivel de abandonos suele ser más alto, ya sea por efectos adversos de los FAR o por simples pérdidas del seguimiento878,884,885. En España, al igual que en otros países, la demanda de la PPE no ocupacional ha aumentado y en ocasiones se utiliza en escenarios no previstos en las recomendaciones existentes889.
Recientemente se ha publicado la revisión más extensa de PPE no ocupacional con datos de la cohorte suiza890. Desde el año 2006 no se recomienda ninguna profilaxis en la exposición sexual a un paciente con infección por el VIH-1 en TAR estable y con CVP<50copias/ml durante más de 6meses. En el referido estudio se observó un aumento progresivo de las consultas para PPE (1.223consultas y 910indicaciones de PPE), objetivándose un buen grado de conocimiento y facilidad de acceso a la PPE. El 72% de las solicitudes habían tenido una exposición heterosexual. En el 23% de los episodios el paciente fuente tenía una infección por el VIH-1 y se detectaron 11nuevos casos de infección desconocida. La mediana de tiempo desde la exposición hasta la consulta fue de 17h. Completó la PPE el 60%, presentó efectos adversos el 64% y se perdió el seguimiento del 16%. Se detectaron 2seroconversiones, pero no fueron fracasos de la profilaxis. Un estudio aleatorizado comparó la incidencia de seroconversión en los pacientes que habían recibido PPE tras un asesoramiento estándar (2sesiones) frente a un asesoramiento intensivo de 5sesiones. La incidencia de seroconversión al VIH-1 disminuyó en el grupo de mayor riesgo asignado al grupo de asesoramiento intensivo. La adherencia a la PPE fue similar en ambos grupos888. La incidencia no ha disminuido entre los HSH en Estados Unidos, Francia, Australia y Holanda. Además, la incidencia de transmisión en el año 2000 fue 4veces superior en un grupo de HSH que habían recibido PPE que la observada en la cohorte de Amsterdam891. Estos hallazgos reflejan más una elevada actividad de riesgo que un verdadero fracaso de PPE.
En las recomendaciones de los CDC877 y del PNS/GeSIDA879 se insiste en 2aspectos claves de la PPE no ocupacional: a)la actuación médica no debe ceñirse exclusivamente a valorar la indicación de PPE con FAR, sino que debe contemplar la oferta de la prueba para el VIH-1, educación sanitaria para la reducción del riesgo de adquisición del VIH-1, valoración del riesgo de transmisión de otras infecciones y seguimiento clínico, y b)la decisión de llevar a cabo PPE con FAR ha de ser tomada por el médico y el paciente de forma individualizada, valorando sus beneficios y riesgos y desaconsejándola en personas con exposiciones repetidas. La PPE debe considerase teniendo en cuenta el nivel de riesgo, la vía de exposición, el estado serológico y/o prácticas de riesgo de la persona fuente, así como el TAR recibido por el paciente fuente en caso de estar infectado por el VIH-1 y la existencia de factores de riesgo añadidos (tabla 18).
Recomendaciones de profilaxis post-exposición
| Exposición a: | PPE recomendada si: | |
| Tipo exposición | Fuente | |
| Sangre u otros fluidos potencialmente infectivos | Penetración s.c. o i.m. con aguja i.m./i.v. o sistema i.v. | VIH-1+, o desconocido pero con factores de riesgo |
| Accidente percutáneo con instrumento cortante o aguja i.m./s.c. o suturaContacto >15min con mucosas o piel no intacta | VIH-1+ | |
| Secreciones genitales | Sexo anal o vaginal | VIH-1+, o desconocido pero con factores de riesgo |
| Sexo oral receptivo con eyaculación | VIH-1+ | |
| UDVP | Intercambio de jeringuilla o agujas | VIH-1+ |
| Régimen terapéutico:TDF/FTC + IP/r (ver tabla 4)AlternativasaAZT+3TC + IP/rTDF/FTC + RALTDF/FTC/EFVAZT/3TC + EFVAZT/3TC + TDF | ||
La elección de los FAR, su inicio y su duración son similares a los de la PPE ocupacional. En la PPE no ocupacional se deberían valorar las concentraciones que alcanzan los FAR en las secreciones genitales respecto a las plasmáticas. La relación entre la penetración de los FAR en el tejido genital, la supresión de la carga viral en el tejido genital y la relevancia de la presencia de una replicación de baja intensidad a nivel genital no ha sido definitivamente resuelta845,870.
Históricamente la tolerabilidad y la adherencia de la PPE no ocupacional eran muy bajas, con abandonos del 22-76%, siendo la toxicidad la principal causa de abandono884. Por ello las guías norteamericanas y las de la OMS optan por 2FAR (2ITIAN) para mejorar la adherencia y recomiendan añadir un tercer FAR en los casos de mayor riesgo. En cambio, las guías europeas y las británicas recomiendan TDF/FTC+LPV/r892. La tolerabilidad ha mejorado con las nuevas pautas884. En el año 2010 se comunicó la experiencia francesa893, con 249individuos tratados con TDF/FTC+LPV/r y un 22% de abandonos, y la experiencia holandesa894, con 139 individuos que recibieron ZDV/3TC+ATV/r, de los que el 91% completó el tratamiento. Sin embargo, la toxicidad no es el único factor relacionado con la adherencia, dado que el estudio holandés mostró que los HSH que habían tenido una exposición con un individuo con infección por el VIH-1 conocida finalizaron la pauta, a pesar de que algunos presentaban efectos adversos más importantes que otros que la suspendieron. Se ha comunicado una experiencia española con 255 pacientes aleatorizados a recibir ZDV/3TC+LPV/r o ZDV/3TC+ATV (400mg QD). La tolerancia fue buena, pero las pérdidas de seguimiento fueron elevadas895. En 2012 se ha publicado una serie de 100individuos que recibieron TDF/FTC+RAL. Finalizaron la pauta de 28días el 57% de los mismos, el 28% suspendió o modificó la pauta y el porcentaje de pérdidas en el seguimiento fue del 15%896.
Recomendaciones- •
La PPE no ocupacional debe individualizarse y llevarse a cabo en el marco de una intervención médica integral. (A-III)
- •
La PPE no ocupacional debe recomendarse en las situaciones denominadas de «riesgo apreciable» si se dan las siguientes condiciones: a)instauración precoz (similar a la PPE ocupacional); b)ausencia de contraindicaciones para tomar FAR; c)exposición excepcional, y d)garantía de seguimiento clínico y analítico del paciente. (B-III)
- •
Los fármacos a emplear, su duración y el seguimiento de los pacientes será igual que en la PPE ocupacional. (A-III)
- •
En caso de exposición sexual, debe valorarse el riesgo de ITS y embarazo. (A-III)
En los últimos años, gracias al TAR, se ha reducido la mortalidad relacionada con el sida y la calidad de vida de los pacientes ha mejorado. Sin embargo, la TAR tiene un elevado coste, y en un entorno donde los recursos son limitados es necesario gestionar correctamente el gasto. Por ello se considera conveniente introducir en estas guías unas tablas comparativas de los costes de las diferentes combinaciones de TAR utilizadas como terapia de inicio, ya que existen diferencias sustanciales entre ellas (fig. 1): el gasto mensual con pautas de eficacia similar puede diferir en cantidades de hasta casi 500euros. En la tabla 19 se indica el precio de venta laboratorio (PVL) de los FAR disponibles en España en enero de 2013, así como el PVL con la deducción obligatoria del 7,5% sobre el precio de compra de medicamentos no genéricos y no afectados por el sistema de precios de referencia adquiridos con cargo a fondos públicos del Sistema Nacional de Salud, a través de los servicios de farmacia de los hospitales, centros de salud y estructuras de atención primaria (Real Decreto-Ley 8/2010, de 20 de mayo: Medidas extraordinarias para la reducción del déficit público). Cabe considerar que pueden existir variaciones entre los precios finales, que pueden variar entre comunidades autónomas e incluso entre distintos hospitales en una misma Comunidad. Sin embargo, se ha empleado el (PVL–7,5%)+4% IVA) como aproximación, por ser unitario en todo el Estado.
. * Solo estas pautas han sido consideradas como preferentes por la totalidad del panel de expertos. Calculado según (PVL – 7,5%) + 4% IVA (basándose en los combos Truvada®, Kivexa®, Atripla® y Eviplera®). Ordenado por tercer fármaco (ver tabla 4)")
Coste mensual de los regímenes de tratamiento antirretroviral de inicio (según la tabla 4).
* Solo estas pautas han sido consideradas como preferentes por la totalidad del panel de expertos.
Calculado según (PVL – 7,5%) + 4% IVA (basándose en los combos Truvada®, Kivexa®, Atripla® y Eviplera®).
Ordenado por tercer fármaco (ver tabla 4)
Coste mensual (PVL – 7,5% + 4% IVA en euros) de los antirretrovirales de uso más frecuentea
| Nombre genérico | Nombre comercial | Presentación | Coste (PVL) | Coste (PVL − 7,5% + 4% IVA)b | Coste/unidadb | Unidades/mes | Coste mensual de la pauta (PVL – 7,5% + 4% IVA)b | Pauta |
| Análogos de nucleósido y nucleótido | ||||||||
| Abacavir | Ziagen | 300mg 60 comp | 225,69 | 217,12 | 3,62 | 60 | 217,12 | ABC 300mg/12h |
| Didanosina | Videx | 400mg 30 caps | 154,38 | 148,52 | 4,95 | 30 | 148,52 | ddI 400mg/24h |
| Didanosina | Videx | 250mg 30 caps | 96,48 | 92,81 | 3,09 | 30 | 92,81 | ddI 250mg/24h |
| Emtricitabina | Emtriva | 200mg 30 caps | 147,35 | 141,75 | 4,72 | 30 | 141,74 | FTC 200mg/24h |
| Estavudina | Zerit | 40mg 448 caps | 1.167,15 | 1.122,80 | 2,51 | 60 | 150,38 | d4T 40mg/12h |
| Estavudina | Zerit | 30mg 448 caps | 1.129,47 | 1.086,55 | 2,43 | 60 | 145,52 | d4T 30mg/12h |
| Lamivudina | Epivir | 300mg 30 comp | 141,16 | 135,80 | 4,53 | 30 | 135,80 | 3TC 300mg/24h |
| Lamivudina | Lamivudina Normon | 300mg 30 comp | 62,76 | 65,27c | 2,18 | 30 | 65,27c | 3TC 300mg/24h |
| Tenofovir | Viread | 245mg 30 comp | 288,7 | 277,73 | 9,26 | 30 | 277,73 | TDF 300mg/24h |
| Zidovudina | Retrovir | 300mg 300 comp | 788,65 | 758,69 | 2,53 | 60 | 151,74 | AZT 300mg/12h |
| Zidovudina | Zidovudina Combinopharm | 300mg 300 caps | 414,06 | 430,62c | 1,44 | 60 | 86,12c | AZT 300mg/12h |
| Análogos de nucleósido y nucleótido en combinación | ||||||||
| Abacavir+Lamivudina | Kivexa | 600/300mg30 comp | 355,54 | 342,03 | 11,40 | 30 | 342,04 | Kivexa 1c/24h |
| Emtricitabina+Tenofovir | Truvada | 200/245mg30 comp | 432,73 | 416,29 | 13,88 | 30 | 416,29 | Truvada 1c/24h |
| Zidovudina+Lamivudina | Combivir | 150/300mg60 comp | 290,41 | 279,38 | 4,66 | 60 | 279,38 | Combivir 1c/12h |
| Zidovudina+Lamivudina+Abacavir | Trizivir | 300/150/300mg60 comp | 490,32 | 471,69 | 7,86 | 60 | 471,68 | TZV 1c/12h |
| Inhibidores de la transcriptasa inversa no nucleósidos | ||||||||
| Efavirenz | Sustiva | 600mg 30 comp | 265,03 | 254,96 | 8,50 | 30 | 254,96 | EFV 600mg/24h |
| Nevirapina | Viramune | 200mg 60 comp | 199,69 | 192,10 | 3,20 | 60 | 192,10 | NVP 200mg/12h |
| Nevirapina | Viramune lib. prolongada | 400mg 30 comp | 184,71 | 177,69 | 5.923 | 30 | 177,69 | NVP 400mg/24h (LP) |
| Etravirina | Intelence | 100mg 120 comp200mg 60 comp | 420 | 404,04 | 3,376,73 | 12060 | 404,04 | ETR 200mg/12h |
| Rilpivirina | Edurant | 25mg 30 comp | 238,53 | 229,47 | 7,65 | 30 | 229,47 | RPV 25mg/24h |
| Inhibidores de la transcriptasa inversa no nucleósidos en combinación con análogos de nucleósidos y nucleótidos | ||||||||
| Efavirenz+Emtricitabina+Tenofovir | Atripla | 600mg200mg245mg comp | 701,08 | 674,44 | 22,48 | 30 | 674,44 | Atripla 1c/24h |
| Rilpivirina+Emtricitabina+Tenofovir | Eviplera | 25mg200mg245mg comp | 627,87 | 604,01 | 20,13 | 30 | 604,01 | Eviplera 1c/24h |
| Nombre genérico | Nombre comercial | Presentación | Coste PVL) | Coste PVL −7,5% +4% IVA)b | Coste/unidadb | Unidades/mes | Coste/mesb | Coste/mes RTVb | Coste mensual de la pauta con descuento del 7,5%b | Pauta |
| Inhibidores de la proteasa | ||||||||||
| Atazanavir | Reyataz | 200mg 60 caps | 436,59 | 420,00 | 7,00 | 60 | 420,00 | 420,00 | ATV 400mg/24h | |
| Atazanavir | Reyataz | 300mg 30 caps | 436,59 | 420,00 | 14,00 | 30 | 420,00 | 21,61 | 441,61 | ATV/r 300/100mg/24h |
| Darunavir | Prezista | 600mg 60 comp | 640,81 | 616,46 | 10,27 | 60 | 616,46 | 43,22 | 659,68 | DRV/r 600/100mg/12h |
| Darunavir | Prezista | 400mg 60 comp | 427,21 | 410,98 | 6,85 | 60 | 410,98 | 21,61 | 432,59 | DRV/r 800/100mg/24h |
| Fosamprenavir | Telzir | 700mg 60 comp | 316,89 | 304,85 | 5,08 | 60 | 304,85 | 43,22 | 348,07 | FPV/r 700/100mg/12h |
| Indinavir | Crixivan | 400mg 180 caps | 261,75 | 251,80 | 1,40 | 120 | 167,87 | 43,22 | 211,09 | IDV/r 800/100mg/12h |
| Lopinavir+Ritonavir comp | Kaletra | 120 comp | 400,02 | 384,82 | 3,21 | 120 | 384,82 | 384,82 | LPV/r 2comp/12h | |
| Nelfinavir | Viracept | 250mg 270 comp | 295,11 | 283,89 | 1,05 | 300 | 315,44 | 315,44 | NFV 1.250mg/12h | |
| Saquinavir | Invirase | 500mg 120 comp | 303,96 | 292,40 | 2,44 | 120 | 292,40 | 43,22 | 335,62 | SQV/r 1.000/100mg/12h |
| Ritonavir | Norvir | 100mg 30 comp | 22,46 | 21,61 | 0,72 | 60 | – | 43,22 | 43,22 | RTV 100 mg/12h |
| Tipranavir | Aptivus | 250mg 120 caps | 705 | 678,21 | 5,65 | 120 | 678,21 | 86,44 | 764,65 | TPV/r 500/200mg/12h |
| Nombre genérico | Nombre comercial | Presentación | Coste (PVL) | Coste (PVL −7,5% +4% IVA)# | Coste/Unidad | Unidades/mes | Coste mensual de la pauta con descuento del 7,5%b | Pauta |
| Inhibidores de la fusión | ||||||||
| Enfuvirtida | Fuzeon | 90mg/ml 60viales | 1.525,36 | 1.467,40 | 24,46 | 60 | 1.467,39 | ENF 90mg/12h |
| Inhibidores de los correceptores CCR5 | ||||||||
| Maraviroc | Celsentri | 150 y 300mg 60comp | 706,8 | 679,94 | 11,33 | 60 | 679,94 | MVC 300 (o 150) mg/12h |
| Maraviroc | Celsentri | 150 y 300mg 60comp | 706,8 | 679,94 | 11,33 | 30 | 339,97 | MVC 300 (o 150) mg/24h |
| Inhibidores de la integrasa | ||||||||
| Raltegravir | Isentress | 400mg 60comp | 690 | 663,78 | 11,06 | 60 | 663,78 | RAL 400 mg/12h |
Incluye la deducción obligatoria del 7,5% sobre el precio de compra de medicamentos no genéricos y no afectados por el sistema de precios de referencia adquiridos con cargo a fondos públicos del Sistema Nacional de Salud, a través de los servicios de farmacia de los hospitales, centros de salud y estructuras de atención primaria (Real Decreto-ley 8/2010, de 20 de mayo: Medidas extraordinarias para la reducción del déficit público). Cabe considerar que normalmente los precios que se pagan son algo inferiores al PVL + 4% IVA − 7,5% y pueden variar entre comunidades autónomas e incluso entre distintos hospitales en una misma Comunidad, dependiendo de las negociaciones llevadas a cabo con la industria farmacéutica. Sin embargo, se ha empleado el PVL − 7,5% + 4% IVA como aproximación, por ser unitario en todo el Estado.
Una evaluación farmacoeconómica con objeto de determinar la eficiencia (relación entre coste y resultados) de nuevas estrategias o de nuevos medicamentos debe contemplar no solamente el coste, sino también la eficacia (ensayos clínicos) o efectividad (práctica clínica habitual) de forma conjunta. Así lo demuestra un reciente estudio farmacoeconómico que ha evaluado los costes y la eficiencia de iniciar TAR con las pautas preferentes en pacientes sin tratamiento previo, según el Documento de consenso de GeSIDA y el PNS de enero de 2012897. En este estudio no fueron más eficientes ni las pautas con mayor eficacia clínica ni las más baratas. Si analizamos los datos de forma parcial cometemos el riesgo de llegar a conclusiones erróneas. Un nuevo estudio farmacoeconómico, que evaluará las pautas de inicio recomendadas en el presente Documento de consenso, se publicará de forma simultánea.
En el momento actual disponemos de múltiples alternativas de elevada eficacia para el control crónico de la infección por el VIH-1. Aunque las decisiones de tratamiento en el caso de la terapia de rescate están muy condicionadas por factores tales como resistencias, adherencia o toxicidad, es probable que en el caso del paciente sin fracaso previo o en situación de primer fracaso pueda incluirse el concepto de eficiencia en la toma de decisiones para seleccionar la alternativa.
Conflicto de interesesKoldo Aguirrebengoa ha efectuado labores de consultoría en los laboratorios Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Abbott Laboratories, GlaxoSmithKline y Merck; ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, Merck, Roche y ViiV Healthcare, y ha recibido pago por desarrollo de presentaciones educacionales para Abbott Laboratories, Gilead Sciences y GlaxoSmithKline.
Antonio Antela ha efectuado labores de consultoría para Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences y Janssen-Cilag; ha recibido compensaciones económicas por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare, así como pagos por desarrollo de material educacional para Boehringer Ingelheim, Gilead Sciences y ViiV Healthcare.
José R. Arribas ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Janssen; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, y ha recibido pago por desarrollo de presentaciones educacionales para Janssen.
Víctor Asensi ha efectuado labores de consultoría para Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, Boehringer-Ingelheim, GlaxoSmithKline y ViiV Healthcare; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, Boehringer-Ingelheim, GlaxoSmithKline y ViiV Healthcare, y ha recibido pagos por desarrollo de presentaciones educacionales para Bristol-Myers Squibb.
Juan Berenguer ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Janssen, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Bristol-Myers Squibb, GlaxoSmithKline y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, Roche y ViiV Healthcare.
José R. Blanco ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck y ViiV Healthcare; ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Gilead Sciences y Bristol-Myers Squibb.
Vicente Boix ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, GlaxoSmithKline, Janssen, Merck, Pfizer y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Gilead Sciences, GlaxoSmithKline, Janssen y Merck; ha recibido compensación económica por charlas de Janssen, Bristol-Myers Squibb, Merck, Pfizer y ViiV Healthcare, y ha recibido pago por desarrollo de presentaciones educacionales para Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Janssen y Merck.
Pere Domingo ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare.
Vicente Estrada ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Gilead Sciences y Janssen; ha disfrutado de becas para investigación clínica de Abbott Laboratories y Janssen; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Abbott Laboratories.
Federico García ha efectuado labores de consultoría en los laboratorios GlaxoSmithKline, Merck y ViiV Healthcare, ha disfrutado de becas para investigación clínica de Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Merck y ViiV Healthcare.
José M. Gatell ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare.
Félix Gutiérrez ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Merck; ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Gilead Sciences.
Hernando Knobel ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Josep M. Llibre ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, Pfizer, Roche y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott, Boehringer-Ingelheim, Gilead Sciences, GlaxoSmithKline, Janssen-Cilag, Merck Sharp & Dohme, Pfizer, Roche y ViiV, así como pagos por desarrollo de presentaciones educacionales para Boehringer-Ingelheim, Merck y ViiV Healthcare.
José López Aldeguer ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare; ha disfrutado de becas para investigación clínica de ViiV Healthcare y Merck, y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Fernando Lozano ha efectuado labores de consultoría para Abbott Laboratories, Bristol-Myers Squibb, Boehringer Ingelheim, Gilead Sciences, GlaxoSmithKline, Janssen, Merck-Sharp & Dome, Pfizer, Roche Pharmaceuticals y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Boehringer Ingelheim, Gilead Sciences, GlaxoSmithKline, Janssen, Merck-Sharp & Dome, Pfizer, Roche Pharmaceuticals y ViiV Healthcare.
Josep Mallolas ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Merck, Janssen, Roche y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Roche, Merck y ViiV Healthcare.
Esteban Martínez ha efectuado labores de consultoría en los laboratorios Abbott, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck Sharp & Dohme, Theratechnologies, Tibotec y ViiV Healthcare; ha recibido compensaciones económicas por charlas de los laboratorios Abbott, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck Sharp & Dohme, Theratechnologies, Tibotec y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Abbott, Boehringer-Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline y ViiV Healthcare.
Celia Miralles ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha recibido compensaciones económicas por escritura de manuscritos de los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
José M. Miró ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Cubist, Gilead Sciences, Merck, Novartis, Pfizer y Theravance; ha disfrutado de becas para investigación clínica de Cubist, Novartis, Fondo de Investigaciones Sanitarias (FIS) del Instituto de Salud Carlos III (Madrid), Fundación para la Investigación y Prevención del Sida en España (FIPSE, Madrid), Ministerio de Sanidad, Política Social e Igualdad (MSPSI, Madrid), National Institutes of Health (NIH, Bethesda, MA, EE.UU.), y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Cubist, GlaxoSmithKline, Gilead Sciences, Janssen, Merck, Novartis, Pfizer, Roche, Schering-Plough, Theravance y ViiV Healthcare. El Dr. J.M. Miró tuvo durante el año 2011 una beca (INT 10/219) de Intensificación de la Actividad Investigadora del Sistema Nacional de Salud y del Departamento de Salud de la Generalitat de Cataluña (Programas I3 SNS y PRICS).
Santiago Moreno ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche; ha disfrutado de becas para investigación clínica de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche, y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche.
Rosario Palacios ha efectuado labores de consultoría en los laboratorios Boehringer Ingelheim y ha recibido compensación económica por charlas de Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, ViiV Healthcare y Roche.
María J. Pérez Elías ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de los laboratorios Abbott Laboratories, Gilead Sciences, ViiV Healthcare y Janssen; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Abbott Laboratories, Bristol-Myers Squibb, Janssen, Merck y ViiV Healthcare.
Juan A. Pineda ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Boehringer Ingelheim, GlaxoSmithKline, Gilead Sciences, Janssen, Merck, Pfizer, Schering-Plough y ViiV Healthcare, ha disfrutado de becas para investigación clínica de Abbott Laboratories, Bristol-Myers Squibb, Boehringer Ingelheim, GlaxoSmithKline, Gilead Sciences, Janssen, Merck, Pfizer, Roche, Schering-Plough y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Gilead Sciences, Janssen, Merck, Roche, Schering-Plough y ViiV Healthcare
Rosa Polo declara no tener conflictos de intereses.
Antonio Rivero ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de laboratorios Abbott Laboratories, Gilead Sciences, Merck y ViiV Healthcare; ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Jesús Santos ha efectuado labores de consultoría en los laboratorios Boehringer Ingelheim y Janssen; ha recibido compensación económica por charlas de Bristol-Myers Squibb y Gilead Sciences, así como pagos por desarrollo de presentaciones educacionales para Bristol-Myers Squibb.
Montse Tuset ha disfrutado de becas para investigación clínica de laboratorios Bristol-Myers Squibb, Gilead Sciences y Merck, y ha recibido compensación económica por charlas de Janssen, Merck y ViiV Healthcare.
Francesc Vidal declara no tener conflictos de intereses.
La Junta Directiva de GeSIDA y la Secretaría del Plan Nacional sobre el Sida agradecen las aportaciones y opiniones de Marisa Álvarez, Harkaitz Azkune, Pablo Bachiller, Manuel Cotarelo, Susana Fernández, Pedro Ferrer, Mar Franco, Henar Hevia, Eloy Gómez, Montse Jansá, Juan Emilio Losa, Jose Emilio Martín, Marta Palazuelos, Federico Pulido, Enrique Redondo, Felipe Rodríguez, Jorge del Romero y Nuria Sánchez, que han contribuido a mejorar la redacción y a enriquecer el contenido del documento.
Comité de redacción: Pere Domingoa, Rosa Polob, Fernando Lozanoc, José López Aldeguerd, Koldo Aguirrebengoae, Vicente Estradaf, Félix Gutiérrezg, Hernando Knobelh, Josep M. Llibrei, Celia Mirallesj, José M. Mirók, Antonio Riverol, Jesús Santosm, Montserrat Tussetk, Antonio Antelan, Víctor Asensio, José R. Arribasp, José R. Blancoq, Vicente Boixr, Federico Garcías, José M. Gatellk, Josep Mallolask, Esteban Martínezk, Santiago Morenot, Rosario Palaciosm, María J. Pérez-Elíast, Juan A. Pinedac, Francesc Vidalu y Juan Berenguerv
Autor para correspondencia: jlopezal@medynet.com (José López Aldeguer).
aHospital de la Santa Creu i Sant Pau, Barcelona.
bSecretaría del Plan Nacional del Sida, Ministerio de Sanidad, Servicios Sociales e Igualdad.
cHospital Universitario Virgen de Valme, Sevilla.
dHospital Universitario La Fe, Valencia.
eHospital de Cruces, Bilbao.
fHospital Clínico, Madrid.
gHospital General Universitario, Elche, Alicante.
hHospital del Mar, Barcelona.
iHospital Universitari Germans Trías i Pujol, Badalona, Barcelona.
jHospital Xeral, Vigo, Pontevedra.
kHospital Clínic, Barcelona.
lHospital Reina Sofía, Córdoba.
mHospital Universitario Virgen de la Victoria, Málaga.
nHospital Clínico Universitario, Santiago de Compostela, La Coruña.
oHospital Universitario Central de Asturias, Oviedo.
pHospital La Paz, IdiPAZ, Madrid.
qHospital San Pedro, Logroño.
rHospital General Universitario, Alicante.
s Hospital Universitario San Cecilio, Granada.
tHospital Ramón y Cajal, Madrid.
u Hospital Universitari Joan XXIII, Tarragona.
vHospital General Universitario Gregorio Marañón, Madrid.
| 3TC | Lamivudina |
| ABC | Abacavir |
| APV | Amprenavir |
| ATV | Atazanavir |
| AZT, ZDV | Zidovudina |
| BID | Pauta de tratamiento administrada dos veces al día |
| COBI | Cobicistat |
| CVP | Carga viral plasmática |
| d4T | Estavudina |
| ddI | Didanosina |
| DRV | Darunavir |
| DTG | Dolutegravir |
| EFV | Efavirenz |
| EMA | European Medicines Agency |
| ENF | Enfuvirtida |
| ERC | Enfermedad renal crónica |
| ETR | Etravirina |
| EVG | Elvitegravir |
| FAR | Fármacos antirretrovirales |
| FPV | Fosamprenavir |
| FTC | Emtricitabina |
| IDV | Indinavir |
| IF | Inhibidores de la fusión |
| InInt | Inhibidores de la integrasa |
| IP | Inhibidores de la proteasa |
| IP/r | Inhibidor de la proteasa (IP) potenciado con ritonavir |
| ITIAN | Inhibidor/es transcriptasa inversa análogos nucleósido o nucleótido |
| ITINN | Inhibidor/es transcriptasa inversa no nucleósidos |
| ITS | Infecciones de transmisión sexual |
| ITT | Análisis por intención de tratamiento |
| LPV | Lopinavir |
| MVC | Maraviroc |
| NFV | Nelfinavir |
| NVP | Nevirapina |
| OT | Análisis «en tratamiento» |
| PPE | Profilaxis post-exposición |
| PrPE | Profilaxis pre-exposición |
| QD | Fármaco o pauta de tratamiento administrada una vez al día |
| RHS | Reacción de hipersensibilidad |
| RAL | Raltegravir |
| RPV | Rilpivirina |
| RTV | Ritonavir |
| SIRI | Síndrome inflamatorio de reconstitución inmune |
| SQV | Saquinavir |
| TAM | Mutaciones asociadas con resistencia a los análogos de la timidina |
| TAR | Tratamiento antirretroviral; ídem. de alta eficacia |
| TB | Tuberculosis |
| TDF | Tenofovir (disoproxil fumarato) |
| TLOVR | Tiempo hasta la pérdida de la eficacia virológica |
| TPV | Tipranavir |
| VIH-1 | Virus de la inmunodeficiencia humana tipo 1 |
| VIH-2 | Virus de la inmunodeficiencia humana tipo 2 |
| ZDV, AZT | Zidovudina |

