




Hay una amplia diversidad de familias y grupos de antimicrobianos de interés clínico. Los mecanismos por los que los compuestos con actividad antibacteriana inhiben el crecimiento o causan la muerte de las bacterias son muy variados, y dependen de las dianas afectadas.
La pared celular (una estructura singular de la inmensa mayoría de las bacterias, ausente en células eucariotas) puede verse afectada en la síntesis (fosfomicina, cicloserina) o el transporte de sus precursores (bacitracina, mureidomicinas), o en su organización estructural (β-lactámicos, glucopéptidos). Los principales derivados que afectan a la membrana citoplásmica son las polimixinas y la daptomicina. La síntesis proteica puede bloquearse por una amplia variedad estructural de compuestos que afectan a algunas de las fases de este proceso: activación (mupirocina), iniciación (oxazolidinonas, aminoglucósidos), fijación del complejo aminoácido-ARNt al ribosoma (tetraciclinas, glicilciclinas) o elongación (anfenicoles, lincosamidas, macrólidos, cetólidos, estreptograminas o ácido fusídico). El metabolismo de los ácidos nucleicos puede verse afectado en la ARN polimerasa dependiente de ADN (rifamicinas) o en el proceso de enrollamiento/desenrollamiento del ADN (quinolonas); algunos compuestos afectan directamente al ADN (nitroimidazoles, nitrofuranos). El trimetoprim y las sulfamidas (con frecuencia usados en combinación) son los representantes de los antimicrobianos que bloquean las vías metabólicas de la bacteria. Algunos compuestos, aun siendo incapaces de inhibir o matar las bacterias, pueden bloquear sus mecanismos de resistencia, por lo que usados en combinación con otros antimicrobianos potencian la acción de estos últimos; de este grupo de sustancias sólo se emplean en clínica algunos inhibidores de β-lactamasas.
A large number of families and groups of antimicrobial agents are of clinical interest. The mechanisms by which compounds with antibacterial activity inhibit growth or cause bacterial death are varied and depend on the affected targets.
The bacterial cell wall—a unique structure in most bacteria that is absent in eukaryotic cells—can be affected in several ways: at different stages of synthesis (fosfomycin, cycloserine) or transport (bacitracin, mureidomycins) of its metabolic precursors, or by a direct action on its structural organization (β-lactams, glycopeptides). The main drugs affecting the cytoplasmic membrane are polymyxins and daptomycin. Protein synthesis can be blocked by a large variety of compounds that affect any of the phases of this process, including activation (mupirocin), initiation (oxazolidinones, aminoglycosides), binding of the tRNA amino acid complex to ribosomes (tetracyclines, glycylcyclines) and elongation (amphenicols, lincosamides, macrolides, ketolides, streptogramins, fusidic acid). The metabolism of nucleic acids can be altered at the DNA-dependent RNA polymerase or in the process of DNA coiling (quinolones); some compounds affect DNA directly (nitroimidazoles, nitrofurans). Trimethoprim and sulfamides (often used in combination) are examples of antimicrobial agents that block bacterial metabolic pathways. Some compounds are unable to inhibit or kill bacteria in themselves, but can block bacterial mechanisms of resistance, enhancing the activity of other antimicrobials administered in combination. Among this group of agents, only certain β-lactamase inhibitors are currently in clinical use.
El descubrimiento de la penicilina en 19291 y su posterior introducción en clínica supuso una verdadera revolución en el tratamiento de la patología infecciosa. Desde entonces, se han incorporado a la práctica clínica decenas de familias de antimicrobianos, con actividad frente a bacterias, hongos, parásitos y virus. En este artículo se abordarán los aspectos microbiológicos del mecanismo de acción de los compuestos con actividad antibacteriana.
Con excepción de la pared celular, la práctica totalidad del resto de las dianas de los antimicrobianos se encuentran también en células eucariotas. Las diferencias estructurales entre las bacterias y las células superiores hacen que la afinidad de los antimicrobianos de interés clínico por las dianas procarióticas sea mucho mayor que por las de sus homólogas eucariotas, disminuyendo así el riesgo de efectos adversos. La principales diferencias entre las células bacterianas y las eucariotas incluyen en las primeras2: a) existencia de un único cromosoma en la bacteria, que no está rodeado de membrana nuclear y se halla en contacto directo con el citoplasma (por tanto, muy accesible a los antibióticos que actúan sobre la síntesis de ADN); b) presencia de ribosomas del tipo 70S, y c) presencia de una pared celular con peptidoglicano (excepto en Mycoplasma spp.), estructura que confiere forma y rigidez a la bacteria.
Para que los antimicrobianos alcancen su diana deben atravesar la cubierta bacteriana, salvo cuando la diana es la propia envoltura externa de los gramnegativos. Las bacterias gramnegativas ofrecen mayor resistencia que las grampositivas a la entrada de antimicrobianos, pues poseen una membrana celular externa, que rodea la capa de peptidoglucano. Esa membrana es una bicapa de lipídica que, a diferencia de las membranas eucariotas, contiene lipolisacárido, y desempeña un importante papel de barrera frente a determinados antimicrobianos3. En la misma existen un gran número de proteínas, que representan en torno al 40% de su peso total, entre las cuales se encuentran las porinas, proteínas triméricas o monoméricas que forman conductos o poros hidrófilos que permiten el acceso al peptidoglucano. A través de estos poros difunden de forma pasiva pequeñas moléculas hidrofílicas (menores de 600Da), pero se impide el paso de otras mayores, por ejemplo los glucopéptidos (peso molecular >1.000Da). Por el contrario, los antibióticos más lipofilicos difunden a través de la bicapa lipídica, y algunos utilizan un mecanismo de transporte con gasto de energía. En las bacterias grampositivas, que carecen de membrana externa, se estima que el límite de exclusión es de 100kDa, mucho mayor que el tamaño de la mayoría de los antimicrobianos.
Ya en el interior del microorganismo los antimicrobianos deben evitar su hidrólisis o su transformación en un producto inactivo y reconocer de forma efectiva una diana antes de que algún sistema de expulsión lo lance de nuevo fuera de la bacteria.
Desde el punto de vista molecular, los antimicrobianos de uso clínico ejercen su acción en algunas de las siguientes estructuras o funciones bacterianas: inhibiendo la síntesis de la pared bacteriana, alterando la integridad de la membrana citoplásmica, impidiendo la síntesis proteica o bloqueando la síntesis o las funciones de ácidos nucleicos. Hay también otros antimicrobianos cuya función es proteger otros compuestos de las enzimas hidrolíticas bacterianas, como es el caso de los inhibidores de β-lactamasas4,5.
Atendiendo a su efecto antibacteriano, los antimicrobianos se han clasificado tradicionalmente en bactericidas (ejercen una acción letal para la bacteria) o bacteriostáticos (sólo inhiben transitoriamente el crecimiento bacteriano). Los límites de ambos conceptos se consideran en la actualidad un tanto difusos, como ya se recoge en otro número de EIMC6. Cada grupo de antibióticos actúa preferentemente de una forma u otra, aunque un mismo antibiótico puede comportarse como bactericida o bacteriostático, dependiendo de la concentración que alcance en la diana, o de su afinidad por la diana de un determinado microorganismo. En general, son bactericidas los antimicrobianos que actúan inhibiendo la síntesis de la pared, alterando la membrana citoplásmica o interfiriendo con algunos aspectos del metabolismo del ADN, y bacteriostáticos los que inhiben la síntesis proteica, excepto los aminoglucósidos.
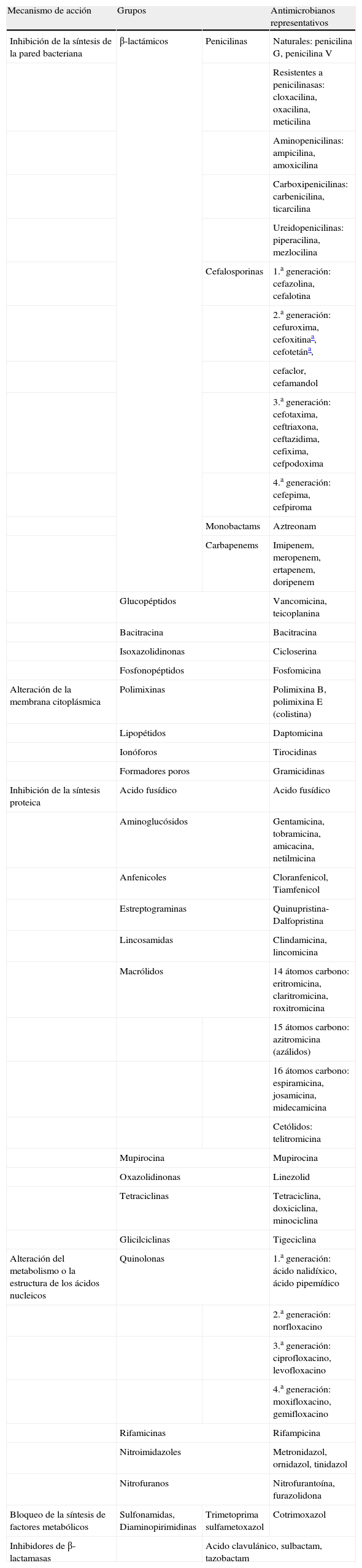
Atendiendo a su mecanismo de acción y estructura química, los principales grupos de antimicrobianos de interés clínico y sus principales representantes se recogen en la tabla 1.
Principales grupos de antimicrobianos y representantes de éstos
| Mecanismo de acción | Grupos | Antimicrobianos representativos | |
| Inhibición de la síntesis de la pared bacteriana | β-lactámicos | Penicilinas | Naturales: penicilina G, penicilina V |
| Resistentes a penicilinasas: cloxacilina, oxacilina, meticilina | |||
| Aminopenicilinas: ampicilina, amoxicilina | |||
| Carboxipenicilinas: carbenicilina, ticarcilina | |||
| Ureidopenicilinas: piperacilina, mezlocilina | |||
| Cefalosporinas | 1.a generación: cefazolina, cefalotina | ||
| 2.a generación: cefuroxima, cefoxitinaa, cefotetána, | |||
| cefaclor, cefamandol | |||
| 3.a generación: cefotaxima, ceftriaxona, ceftazidima, cefixima, cefpodoxima | |||
| 4.a generación: cefepima, cefpiroma | |||
| Monobactams | Aztreonam | ||
| Carbapenems | Imipenem, meropenem, ertapenem, doripenem | ||
| Glucopéptidos | Vancomicina, teicoplanina | ||
| Bacitracina | Bacitracina | ||
| Isoxazolidinonas | Cicloserina | ||
| Fosfonopéptidos | Fosfomicina | ||
| Alteración de la membrana citoplásmica | Polimixinas | Polimixina B, polimixina E (colistina) | |
| Lipopétidos | Daptomicina | ||
| Ionóforos | Tirocidinas | ||
| Formadores poros | Gramicidinas | ||
| Inhibición de la síntesis proteica | Acido fusídico | Acido fusídico | |
| Aminoglucósidos | Gentamicina, tobramicina, amicacina, netilmicina | ||
| Anfenicoles | Cloranfenicol, Tiamfenicol | ||
| Estreptograminas | Quinupristina-Dalfopristina | ||
| Lincosamidas | Clindamicina, lincomicina | ||
| Macrólidos | 14 átomos carbono: eritromicina, claritromicina, roxitromicina | ||
| 15 átomos carbono: azitromicina (azálidos) | |||
| 16 átomos carbono: espiramicina, josamicina, midecamicina | |||
| Cetólidos: telitromicina | |||
| Mupirocina | Mupirocina | ||
| Oxazolidinonas | Linezolid | ||
| Tetraciclinas | Tetraciclina, doxiciclina, minociclina | ||
| Glicilciclinas | Tigeciclina | ||
| Alteración del metabolismo o la estructura de los ácidos nucleicos | Quinolonas | 1.a generación: ácido nalidíxico, ácido pipemídico | |
| 2.a generación: norfloxacino | |||
| 3.a generación: ciprofloxacino, levofloxacino | |||
| 4.a generación: moxifloxacino, gemifloxacino | |||
| Rifamicinas | Rifampicina | ||
| Nitroimidazoles | Metronidazol, ornidazol, tinidazol | ||
| Nitrofuranos | Nitrofurantoína, furazolidona | ||
| Bloqueo de la síntesis de factores metabólicos | Sulfonamidas, Diaminopirimidinas | Trimetoprima sulfametoxazol | Cotrimoxazol |
| Inhibidores de β-lactamasas | Acido clavulánico, sulbactam, tazobactam | ||
La pared celular protege la integridad anatomofisiológica de la bacteria y soporta su gran presión osmótica interna (mayor en las bacterias grampositivas). La ausencia de esta estructura condicionaría la destrucción del microorganismo, inducida por el elevado gradiente de osmolaridad que suele existir entre el medio y el citoplasma bacteriano7. Los antibióticos que inhiben la síntesis de la pared necesitan para ejercer su acción que la bacteria se halle en crecimiento activo, y para su acción bactericida requieren que el medio en que se encuentre la bacteria sea isotónico o hipotónico, lo que favorece el estallido celular cuando la pared celular se pierde o se desestructura. Suelen ser más activos sobre las bacterias grampositivas por su mayor riqueza en peptidoglucano. En general, son poco tóxicos por actuar selectivamente en una estructura que no está presente en las células humanas.
La síntesis de la pared celular se desarrolla en 3 etapas, sobre cada una de las cuales pueden actuar diferentes compuestos: la etapa citoplásmica, donde se sintetizan los precursores del peptidoglucano; el transporte a través de la membrana citoplásmica, y la organización final de la estructura del peptidoglucano, que se desarrolla en la parte más externa de la pared.
Inhibidores de la fase citoplásmicaEn el citoplasma bacteriano se sintetizan los precursores del peptidoglucano a partir de diferentes elementos: uridindifosfato-N-acetil-glucosamina (UDP-NAG), ácido fosfoenolpirúvico, uridintrifosfato (UTP) y NADH, a partir de los cuales se forma el ácido uridindifosfato-N-acetilmurámico (UDP-NAM). Después se unen al azúcar una cadena de aminoácidos (frecuentemente 5) en la que se alternan las formas L y D y en la que los dos últimos conforman el dipéptido D-alanin-D-alanina.
En esta etapa de síntesis de precursores de peptidoglucano actúan la fosfomicina y la cicloserina.
1. Fosfomicina. Actúa inhibiendo la piruviltransferasa, enzima causante de la adición del fosfoenolpiruvato a la molécula de UDP-NAG para formar el precursor UDP-NAM. Esta reacción se inhibe porque la fosfomicina, que es un análogo estructural del fosfoenolpiruvato, se une covalentemente con la enzima. La fosfomicina atraviesa la membrana externa mediante las porinas; debido a su pequeño tamaño pasa la barrera de peptidoglicano sin dificultad y finalmente atraviesa la membrana citoplásmica a través de sistemas de transporte activo, uno de los cuales es de expresión inducible, que se favorece en presencia de glucosa-6-fosfato. Por eso, a los medios o discos para estudiar la sensibilidad de las bacterias a la fosfomicina debe añadirse glucosa-6-fosfato8.
Es un antibiótico de amplio espectro que incluye bacilos gramnegativos y grampositivos y Staphylococcus spp. (excepto S. saprophyticus y S. capitis).
2. Cicloserina. Actúa sobre la base de su analogía estructural con la D-alanina, inhibiendo competitivamente la actividad de la L-alanina-racemasa (transforma L-ala en D-ala) y la D-alanin-D-alanina-sintetasa (forma dímeros de D-ala). Por su elevada toxicidad sólo se usa como fármaco antimicobacteriano de segunda línea.
Inhibidores de la fase de transporte de precursoresEn esta fase, que se desarrolla en la membrana citoplásmica, un transportador lipídico tomará a su cargo el precursor formado en el citoplasma y lo hará atravesar la membrana citoplásmica. Se trata de un fosfolípido de 55 átomos de carbono, el undecaprenilfosfato. También en la membrana citoplásmica, termina de formarse el precursor mediante la adición de una molécula de N-acetilglucosamina, que se enlaza al átomo C1 del ácido murámico, formándose así un polímero lineal de peptidoglucano constituido por unidades de NAG y NAM-pentapétido. Una vez que este precursor disacárido-pentapéptido es transferido a un lugar aceptor en la pared preexistente, el transportador queda pirofosforilado y se separa, y debe presentar una defosforilación para convertirse en su forma monofosfato activa, que puede transportar ya nuevos precursores a la capa de peptidoglucano.
BacitracinaEste antimicrobiano se une al transportador y bloquea su defosforilación, e impide que pueda utilizarse de nuevo en el transporte de los polímeros lineales de disacárido-pentapéptido a través de la membrana citoplásmica, hasta la pared en formación. Este antibiótico es activo contra cocos grampositivos (excepto estreptococos del grupo B), Neisseria spp. y de forma variable C. difficile.
MureidomicinasSon un nuevo grupo de antimicrobianos producidos por Streptomyces flavidivirens, que por su analogía estructural con el precursor disacárido pentapéptido, se unen competitivamente con el transportador lipídico, bloqueando el transporte de los precursores a través de la membrana citoplásmica. No existen derivados de uso clínico.
Inhibidores de la organización estructural del peptidoglucanoEn esta etapa, los precursores de peptidoglucano se ensamblan con la ayuda de enzimas situados en su superficie conocidos como proteínas fijadoras de penicilina (penicillin binding proteins [PBP]). En esta etapa tienen su acción los glucopéptidos y los β-lactámicos.
GlucopéptidosLos glucopéptidos (vancomicina y teicoplanina) actúan en un paso previo al de los β-lactámicos. Impiden la transferencia del disacárido pentapéptido, unido al transportador lipídico de la membrana citoplásmica, al aceptor de la pared celular. Esto se debe a que estos compuestos recubren el extremo D-alanin-D-alanina del disacárido-pentapéptido, evitando así la acción de las glucosiltransferasas y transpeptidasas, y en consecuencia evitando la elongación del peptidoglucano9.
El gran tamaño de estas moléculas impide su paso a través de la pared de las bacterias gramnegativas, de forma que sólo resultan activas frente a grampositivos (incluyendo con gran frecuencia cepas multirresistentes). Aunque son bactericidas frente a Staphylococcus spp., sólo tienen actividad bacteriostática frente a Enterococcus spp.
β-lactámicosRepresentan el grupo más numeroso y de mayor uso en clínica. Su nombre deriva de la presencia de un anillo lactámico en su estructura, con un oxígeno en posición β con respecto a un nitrógeno. En función de los radicales que se unen a este anillo se distinguen varios subgrupos, de los que los más importantes son: penicilinas, cefalosporinas, monobactamas y carbapenemas10.
Los β-lactámicos son compuestos bactericidas que inhiben las fases finales de la síntesis del peptidoglucano, en la que intervienen activamente las ya citadas encimas PBP. Las PBP tienen actividad transpeptidasa, transglucosilasa y carboxipetidasa, por lo que pueden entrelazar los componentes del peptidoglucano11. Los β-lactámicos bloquean estas enzimas porque el anillo β-lactámico tiene una estructura espacial similar a la del residuo acil-D-alanin-D-alanina de las cadenas del peptidoglicano, que es el sustrato natural de las PBP.
Las bacterias poseen varias PBP, cuyas funciones difieren unas de otras. Por esta razón, las consecuencias de su bloqueo también son distintas. Las PBP-1a y PBP-1b actúan como transpeptidasas causantes de la elongación, y su bloqueo provoca la formación de esferoplastos que rápidamente se lisan. La PBP-2 determina la forma bacteriana, y su inhibición da lugar a formas ovoideas que se lisan fácilmente. La PBP-3 interviene en la división bacteriana, y su bloqueo provoca la aparición de formas filamentosas sin septos. Las PBP-4, PBP-5 y PBP-6 tienen actividad carboxipeptidasa, e intervienen en la liberación del quinto aminoácido del pentapéptido, necesaria para la polimerización del peptidoglucano.
Pese a tener el mismo mecanismo de acción, hay diferencias en la actividad de los diferentes β-lactámicos, y ello se debe principalmente a 3 factores: rapidez en la difusión de los antibióticos al espacio periplásmico, resistencia a las β-lactamasas, capacidad para escapar a los sistemas de expulsión activa y afinidad variable por las distintas PBP. Así, un β-lactámico que difunda rápidamente, tenga una gran estabilidad frente a las β-lactamasas, no sea sustrato de las bombas de expulsión y tenga una alta afinidad por las PBP más críticas, será un antibiótico de gran actividad como es el caso de las cefalosporinas de cuarta generación y los carbapénemes.
La acción bactericida de los β-lactámicos no está relacionada directamente con la inhibición de la síntesis de peptidoglucano impidiendo su crecimiento (efecto bacteriostático), sino con la activación de un sistema de enzimas líticos (autolisinas) que, a la inversa de las PBP, están implicadas en la degradación del peptidoglucano12. El peptidoglucano está en continua renovación, resultante del equilibrio entre los procesos de síntesis (PBP) e hidrólisis (autolisinas a bajo nivel de actividad); la rotura de este equilibrio por la acción de los β-lactámicos (inhibición de las PBP y activación de las autolisinas) provoca la muerte bacteriana.
Las bacterias que carecen de autolisinas son inhibidas pero no destruidas, por lo que se dice que son tolerantes. En clínica, se define el fenómeno de tolerancia como la necesidad de una concentración al menos 32 veces mayor a la CMI para que un antimicrobiano destruya una cepa bacteriana13.
El espectro de los β-lactámicos abarca las bacterias grampositivos, gramnegativas y espiroquetas. No son activos frente a micoplasmas por carecer de pared celular, ni frente a bacterias intracelulares como Chlamydia spp. y Rickettsia spp.
Antimicrobianos que bloquean mecanismos de resistenciaLos más importantes son los inhibidores de β-lactamasas de serina, que incluyen ácido clavulánico, sulbactam y tazobactam14. Carecen (habitualmente) de acción antibacteriana intrínseca de verdadera importancia clínica, pero se unen irreversiblemente a algunas β-lactamasas, protegiendo de su acción a los antibióticos β-lactámicos. El sulbactam, además, es activo frente a A. baumannii.
Aunque se conocen sustancias que bloquean in vitro las bombas de expulsión activa o las enzimas modificadoras de aminoglucósidos, ninguna de ellas ha podido introducirse en terapéutica.
Antibióticos activos en la membrana citoplásmicaLa membrana citoplásmica es vital para todas las células, ya que interviene activamente en los procesos de difusión y transporte activo, y de esta forma controla la composición del medio interno celular. Las sustancias que alteran esta estructura modifican la permeabilidad, y provocan la salida de iones potasio, elementos esenciales para la vida bacteriana, o la entrada de otros que a altas concentraciones alteran el metabolismo bacteriano normal.
Los antimicrobianos que actúan en esta estructura se comportan como bactericidas, incluso en bacterias en reposo, y pueden tener alta toxicidad sobre las células humanas, al compartir algunos componentes de la membrana citoplásmica.
A este grupo pertenecen las polimixinas, los lipopétidos, los antibióticos poliénicos (activos frente a hongos) y 2 grupos de escaso interés clínico (ionóforos y formadores de poros).
PolimixinasSon antibióticos polipeptídicos, cíclicos y policatiónicos, con una cadena de ácido graso unido al péptido y se comportan como detergentes catiónicos. Tiene una parte hidrofílica (el péptido) con alta carga positiva que por atracción electrostática se une a la superficie de la membrana, cuya carga neta es negativa. Por otro lado, el extremo lipofílico (la cadena lateral de ácido graso) por interacciones hidrofóbicas se une a los fosfolípidos de la membrana. Como resultado se desorganiza la estructura de la membrana y aumenta su permeabilidad, con la pérdida de metabolitos esenciales15. La mayor presencia de fosfolípidos en la membrana de las bacterias gramnegativas hace que éstas sean más sensibles que las grampositivas a la acción de estos agentes.
Son activos exclusivamente frente a bacilos gramnegativos aerobios, incluidos P. aeruginosa y A. baumannii multirresistentes. No son activos frente a microorganismos anaerobios, Proteus spp., Providencia spp., Serratia spp., Neisseria spp. y B. cepacia.
DaptomicinaLa daptomicina es un lipopéptido cíclico de reciente introducción en clínica que posee una gran actividad frente a bacterias grampositivas. Actúa en la membrana citoplásmica de las bacterias grampositivas, sin entrar en la célula, y se produce una rápida despolarización de la membrana con alteración del potencial eléctrico y salida de iones potasio exterior. Como consecuencia de ello, se produce un bloqueo de la síntesis proteica y de ácidos nucleicos, que provoca la muerte bacteriana16. La daptomicina es una gran molécula cíclica peptídica (parte hidrofílica), al que se une una cadena lateral de ácido graso (parte lipofílica). Se sabe que su actividad antibacteriana depende de la presencia de iones de calcio que es óptima a las concentraciones normales presentes en suero (50mg/l). Probablemente, el calcio favorece la unión de la parte lipofílica de la molécula de daptomicina a la membrana citoplásmica, donde la estructura de la daptomicina presentará cambios conformacionales que provocará su inserción en la membrana citoplásmica. La unión de varias moléculas en la membrana forma canales por los que saldrán los iones potasio. El resultado es una potente actividad bactericida, con efecto postantibiótico.
Su espectro antimicrobiano se reduce a las bacterias grampositivas, ya que no puede atravesar la pared de los gramnegativos.
Ionóforos y formadores de porosLos ionóforos son antibióticos polipeptídicos cíclicos como la valinomicina o las tirocidinas A y B. Estos compuestos tiene una estructura circular peculiar, es hidrofóbica en el exterior e hidrofílica o polar en el interior. Los ionóforos incorporan cationes monovalentes en su interior, y les permite cruzar la bicapa lipídica. La penetración elevada de potasio altera el potencial eléctrico y el gradiente químico existente en la membrana, alterando su función.
Los antibióticos formadores de poros incluyen las gramicidinas, que a diferencia de los ionóforos, son cadenas lineales de aminoácidos (polipéptidos acíclicos) con un mecanismo de acción distinto. Varias moléculas de gramicidina se acomodan una sobre otra, enroscándose y formando un túnel que atraviesa la membrana, constituyendo un poro que permite el paso selectivo de moléculas según su tamaño y características. La gramicidina se encuentra en formulaciones tópicas para tratamiento de conjuntivitis bacterianas, y resulta efectiva frente a bacterias grampositivas. Es un agente hemolítico potente, y su alta toxicidad lo descarta para uso sistémico. Además se inactiva en suero y líquidos orgánicos.
Antibióticos inhibidores de la síntesis proteicaLa síntesis proteica es uno de los procesos más frecuentemente afectados por la acción de los antimicrobianos, y su inhibición selectiva es posible gracias a las diferencias estructurales entre los ribosomas bacterianos y eucariotas. Los ribosomas bacterianos están formados por dos subunidades (30S y 50S), que contienen ARN ribosómico (ARNr 16S en la subunidad 30S, y ARNr 5S y ARNr 23S en la subunidad 50S) y diversas proteínas llamadas S (small o pequeña, en la subunidad 30S) o L (large o grande, en la subunidad 50S). En esta estructura diferentes componentes pueden ser lugares de unión para los antimicrobianos (p. ej., determinados nucleótidos para las oxazolidinonas, algunas proteínas S para las tetraciclinas o proteínas L para el cloranfenicol).
La mayoría de los antibióticos de este grupo tienen actividad bacteriostática, aunque los aminoglucósidos se comportan como bactericidas. La acción bactericida o bacteriostática también va a depender de las concentraciones del antimicrobiano, y del microorganismo afectado.
La síntesis proteica se desarrolla en diferentes fases17, en las cuales actúan diferentes antimicrobianos, como se explica a continuación.
Inhibidores de la fase de activaciónLos aminoácidos son transportados a la cadena peptídica en formación en el ribosoma, por moléculas de ARN de transferencia (ARNt) que se unirán al ARNm codificante de la proteína en formación. Para ello, cada aminoácido se une con su ARNt específico mediante una enzima también específica de aminoácido (aminoacil ARNt sintetasa). En las bacterias, el primer aminoácido de la cadena peptídica es la metionina, es decir, la síntesis proteica se inicia con la formación del complejo formilmetionil-ARNt que reconocerá el codón de iniciación AUG del ARNm (adenosina-uracilo-guanosina).
MupirocinaAntibiótico bacteriostático obtenido de especies de Pseudomonas spp., que inhibe competitivamente la enzima isoleucil-ARNt sintetasa, con lo cual no puede incorporarse el aminoácido isoleucina al péptido en formación y la síntesis de proteínas se interrumpe18.
Su acción es especialmente potente frente a grampositivos, aunque debido a las altas concentraciones que se alcanzan en piel y mucosa nasal tras su aplicación tópica, puede tener también alguna actividad frente a microorganismos gramnegativos. Se usa fundamentalmente en el tratamiento tópico de infecciones cutáneas o para erradicación del estado de portador de S. aureus.
Inhibidores del inicio de la síntesis proteicaEl ARNm dispone de un codón específico para la fijación del ARNt que porta el aminoácido formilmetionina. Ambos se unen en la subunidad 30S, y posteriormente a la subunidad 50S, y constituye el complejo de iniciación de la síntesis de proteínas. En este complejo hay 2 sitios activos, el locus A, en el que se fijan los aminoacil-ARNt, y el locus P, donde se engarza el péptido en formación y donde se ubicará el formilmetionil-ARNt que inicia la cadena peptídica. En esta fase de inicio de la síntesis actúan las oxazolidinonas y los aminoglucósidos.
OxazolidinonasRepresentan una de las últimas familias de antimicrobianos incorporadas a la práctica clínica. Son compuestos obtenidos por síntesis, y su representante en uso es el linezolid19. Las oxazolidinonas inhiben la síntesis proteica e impiden la formación del complejo de iniciación 70S, formado por formilmetionil-ARNt, ARNm, diversas proteínas y las subunidades ribosómicos 30S y 50S. El linezolid se fija a la subunidad ribosómica 50S, en el centro peptidiltransferasa dentro del ARN ribosómico 23S (dominio V), distorsiona así el punto de unión del formilmetionil-ARNt y evita, por tanto, la formación del complejo de iniciación. Esta familia de antibióticos tiene un mecanismo de acción singular, y al actuar en una diana distinta no hay resistencia cruzada con otros antibióticos que también inhiben la síntesis proteica.
El linezolid es bacteriostático frente a bacterias grampositivas (incluidas cepas multirresistentes de S. aureus y Enterococcus spp.) y carece de actividad frente a la práctica totalidad de las bacterias gramnegativas.
AminoglucósidosSon compuestos naturales obtenidos de actinomicetos del suelo o productos semisintéticos derivados de ellos. Poseen un anillo aminociclitol al que se unen diferentes azúcares.
Aunque la diana primaria de actuación de los aminoglucósidos está en los ribosomas y sus procesos de síntesis proteica, su actividad sobre las bacterias no se entiende sin conocer los fenómenos que se producen en la membrana. Los aminoglucósidos son moléculas muy cargadas positivamente, lo que les permite concentrarse en torno a las bacterias por atracción de las cargas negativas de la superficie bacteriana, aportadas por los grupos fosfatos de los fosfolípidos de la membrana externa de las bacterias gramnegativas y de los ácidos teióicos unidos al peptidoglucano de las grampositivas. En consecuencia, desplazan los iones de magnesio y calcio que se enlazan a las moléculas de lipopolisacáridos adyacentes; este proceso desestructura la membrana externa y permite al paso de los aminoglucósidos. Una vez pasada fácilmente la barrera de peptidoglucano (grampositivos y gramnegativos), vuelve a concentrarse en torno a la membrana citoplásmica. La difusión a través de esta membrana ocurre en 2 fases: una inicial lenta y otra posterior rápida; ambas dependientes de la energía generada por el transporte de electrones que implica la participación de sistemas enzimáticos del metabolismo aerobio, que crea un gradiente eléctrico a ambos lados de la membrana20. Este hecho explica la ineficacia de estos compuestos frente a microorganismos anaerobios. La presencia de iones de magnesio y calcio en el medio y las situaciones que disminuyen el potencial transmembrana (pH ácido, ambiente anaerobio o hiperosmolaridad), reducen la difusión del aminoglucósidos al interior de la bacteria y aumentan las CIM de forma importante. Una vez que los aminoglucósidos han empezado a actuar en los ribosomas, comienzan a producirse muchos errores en la lectura del ARNm, que darán como resultado proteínas anómalas que se unirán a la membrana, deteriorando su integridad y acelerando la difusión de más moléculas de aminoglucósido (fase rápida). En consecuencia, una gran cantidad de aminoglucósidos alcanza los ribosomas, que llegan a bloquearse, y se detienen irreversiblemente la síntesis de proteínas.
En el ribosoma, los aminoglucósidos tienen su acción principalmente en la subunidad 30S, donde se unen a diferentes proteínas S y al ARN 16S. Bloquean la actividad normal del complejo de iniciación, impiden el inicio de la síntesis y provocan también una lectura errónea del ARNm.
Los aminoglucósidos tienen un efecto bactericida dependiente de su concentración y poseen un importante efecto postantibiótico21, es decir que una breve exposición de la bacteria a estos compuestos induce una supresión de su crecimiento, aun cuando el antimicrobiano no alcance ya concentraciones que inhiban o maten al microorganismo. Son activos frente a un amplio número de especies bacterianas, especialmente frente a microorganismos gramnegativos aerobios.
Inhibidores de la fijación del aminoacil-ARNt al ribososmaUna vez iniciada la síntesis proteica, el proceso continúa con la incorporación de nuevos aminoácidos al locus A, donde reconocerán los codones internos del ARNm a través de los nucleótidos complementarios del ARNt que porta el aminoácido. Esta fase se ve bloqueada por antibióticos bacteriostáticos como las tetraciclinas y sus derivadas, las glicilciclinas.
TetraciclinasSon moléculas naturales o semisintéticas con un núcleo hidronaftaceno, que contiene cuatro anillos fundidos al que se pueden unir distintos radicales que darán lugar a las diferentes tetraciclinas. Penetran en el citoplasma bacteriano por un proceso dependiente de energía y se unen de forma reversible a la subunidad 30S del ribosoma (proteínas S7, S14, S19), bloqueando el acceso de los complejos aminoacil-ARN-t, e impidiendo la continuación de la síntesis proteica22. Estos compuestos pueden también unirse (aunque menos selectivamente) a los ribosomas 80S de las células humanas y efectuar la misma acción; sin embargo, carecen de los medios de transporte específicos de la membrana bacteriana.
El compuesto más usado es la doxiciclina, y en España también están disponibles minociclina, oxitetraciclina y tetraciclina.
Son el mejor ejemplo de lo que se denomina antibióticos de amplio espectro, con actividad tanto para grampositivos como frente a gramnegativos, pero esta actividad depende de los grados de resistencia observados en cada especie, que son muy variables. Son también activas frente a micobacterias atípicas, rickettsias, micoplasmas, clamidias, espiroquetas, Coxiella burnetii y algunos protozoos.
GlicilciclinasSon compuestos sintéticos derivados de las tetraciclinas, de las cuales está comercializada la tigeciclina, un derivado de la minociclina. Poseen el mismo mecanismo de acción aunque se unen al ribosoma con una afinidad 5 veces superior que la minociclina23. Además, las glicilciclinas se fijan a la membrana citoplásmica y alteran su permeabilidad.
La tigeciclina posee el amplio espectro propio de las tetraciclinas, pero es más potente e incluso activa contra bacterias con modificaciones ribosómicas resistentes a las mismas, incluyendo enterococos resistentes a glucopéptidos, S. aureus resistente a meticilina, S. pneumoniae multirresistentes y diversas bacterias gramnegativas resistentes a otros compuestos.
Inhibidores de la elongaciónUna vez que el ARNt que porta un aminoácido se ha fijado al locus A, el centro peptidiltransferasa, situado en la subunidad 50S, cataliza la unión entre el aminoácido incorporado y el último aminoácido del péptido en formación (locus P), proceso denominado transpeptidación, que puede estar bloqueado por el cloranfenicol y las lincosamidas.
Una vez formado el enlace peptídico, el ARNt fijado al locus P se libera y se separa de su aminoácido correspondiente, quedando libre el locus P. Seguidamente se produce la translocación del peptidil-ARNt del locus A al locus P, desplazándose la subunidad 30S un codón a lo largo del ARNm. De esta manera queda libre el locus A, y preparado para recibir un nuevo ARNt con su correspondiente aminoácido. Este proceso, que conlleva gasto de energía, requiere la participación clave del factor de elongación G, que puede estar bloqueado por el ácido fusídico. El péptido en formación va pasando a través de un canal peptídico en la subunidad 50S y emerge por la parte posterior del ribosoma, y este proceso puede estar bloqueado por los antibióticos del grupo MLSB (macrólidos, lincosamidas y estreptograminas del grupo B) y por los cetólidos. La mayoría de los aminoglucósidos también ejercen su acción interfiriendo con la fase de elongación peptídica.
Todos los antimicrobianos que inhiben la transpeptidación y la translocación actúan sobre la subunidad ribosómica 50S.
AnfenicolesEl cloranfenicol y su derivado, el tiamfenicol, son antibióticos bacteriostáticos que bloquean la síntesis proteica bacteriana uniéndose reversiblemente a la proteína L16 localizada en la subunidad 50S. Esta proteína es la que media la fijación del ARNt a la enzima peptidiltransferasa, y por tanto, su bloqueo por el cloranfenicol evita la formación de los enlaces peptídicos.
Tiene un amplio espectro de actividad contra microorganismos grampositivos, gramnegativos y anaerobios. Su espectro incluye a neisserias, Haemophilus spp, clamidias, rickettsias, micoplasmas y espiroquetas.
LincosamidasLa principal lincosamida es clindamicina, un derivado semisintético de la lincomicina, que es un aminoácido unido a un aminoazúcar. Generalmente bacteriostáticos, pueden ser bactericidas dependiendo de su concentración y del microorganismo considerado.
Actúa inhibiendo la síntesis proteica tras unirse reversiblemente a la subunidad 50S del ribosoma, en un lugar próximo al del cloranfenicol o los macrólidos, impidiendo la acción de la peptidiltransferasa24.
Es activa frente a bacterias grampositivas, excepto enterococos y microorganismos anaerobios, incluido el grupo de B. fragilis. También es activa frente a algunos protozoos como Plasmodium spp. o Toxoplasma gondii. Al igual que los macrólidos y las estreptograminas, no son activas frente a enterobacterias, Pseudomonas spp. u otros gramnegativos aerobios, probablemente porque no pueden atravesar la pared bacteriana.
Macrólidos y cetólidosForman un grupo de antimicrobianos que se caracteriza por la presencia de un anillo lactónico macrocíclico al que unen uno o varios azúcares. La eritromicina fue el primer macrólido utilizado en clínica, a partir del cual se introdujeron modificaciones en su estructura química que dieron lugar a derivados semisintéticos con mejores propiedades farmacocinéticas aunque, salvo excepciones, no presentaban mejorías en su actividad antimicrobiana. Dependiendo del número de elementos contenidos en el anillo, se clasifican en: macrólidos de 14 átomos de carbono como eritromicina, claritromicina, roxitromicina. etc.; macrólidos de 15 átomos de carbono como la azitromicina, en la que se incorpora un átomo de nitrógenos entre los carbonos 9 y 10 que da lugar a una estructura nueva conocida como azálido; macrólidos de 16 átomos de carbono como espiramicina, josamicina, midecamicina, etc. Los cetólidos, como la telitromicina, son un grupo nuevo de antibióticos derivados de la eritomicina, en los que el azúcar unido al carbono 3 se sustituye con un grupo cetónico.
Se unen de forma reversible al dominio V del centro peptidiltransferasa, en el ARNr 23S de la subunidad 50S del ribosoma, interfiriendo así el proceso de elongación de la síntesis proteica. Además los cetólidos interacciona también con el dominio II del ARNr 23S por lo que la afinidad de los cetólidos por el ribosoma es mucho mayor que el resto de los macrólidos. Estos lugares de unión se sitúan en el orificio de entrada al túnel ribosómico por donde sale la proteína en formación, de manera que al unirse los macrólidos o los cetólidos, se bloquea este canal, impidiendo estéricamente el crecimiento del péptido25. También se ha descrito en los cetólidos, una inhibición de la formación de los ribosomas 50S al evitar el ensamblaje de los ARNr 5S y 23S con las riboproteínas, con lo que se impide el inicio de la síntesis26.
Son generalmente considerados como bacteriostáticos, aunque a altas concentraciones y con un bajo inóculo pueden mostrar acción bactericida especialmente en Streptococcus spp.
Son activos frente a bacterias grampositivas (incluyendo actinomicetos e incluso micobacterias), Bordetella pertussis, Haemophilus ducreyi, Moraxella spp, Neisseria spp., Campylobacter spp., Helicobacter pylori (claritromicina), treponemas, borrelias, Legionella spp., micoplasmas, clamidias y ricketsias. La azitromicina es algo menos activa que la eritromicina frente a microorganismos grampositivos, pero es más activa frente a gramnegativos. Apenas son activos frente a enterobacterias y P. aeruginosa, aunque parecen ser útiles (sobre todo, azitromicina) para el tratamiento de infecciones respiratorias crónicas por P. aeruginosa, tal vez por propiedades antiinflamatorias o inmunomoduladores o por inhibición de la síntesis de alginato, componente de los biofilms.
EstreptograminasTambién llamadas sinerginas, forman un grupo de antimicrobianos con un estructura compleja constituida por una macrolactona (estreptogramina grupo A) y un polipéptido cíclico (estreptogramina grupo B)27. Ambos compuestos actúan sinérgicamente de forma bactericida, bloqueando la acción de la peptidiltransferasa en diferentes puntos. Su principal representante es la asociación quinupristina-dalfopristina en proporción 3:7, con actividad fundamentalmente frente a bacterias grampositivas (excepto E. faecalis) y también frente a algunas bacterias fastidiosas (Moraxella spp., Neisseria spp., Mycoplasma spp., L. pneumophila) y algunos anaerobios (Prevotella y Porphyromonas spp.). Las estreptograminas A (dalfopristina) se unen al ARNr 23S (subunidad 50S), y modifican la estructura terciaria de ciertas proteínas ribosómicas, de manera que aumenta la afinidad por las estreptograminas del grupo B (quinupristina). Este hecho explica su acción bactericida cuando actúan juntos, ya que por separado son sólo bacteriostáticos.
Ácido fusídicoEs un antibiótico de estructura esteroide que se une al complejo causante de la translocación formado por el factor de elongación G, GDP y el ribosoma. Al unirse al complejo, lo estabiliza e impide la liberación del factor de elongación G para una nueva translocación.
Puede comportarse como bacteriostático o bactericida según la concentración y el microorganismo. Es de espectro reducido a los microorganismos grampositivos como S. aureus, S. epidermidis, Clostridium spp. y Corynebacterium spp., aunque también puede ser activo frente a meningococos, gonococos y algunos protozoos como Giardia lamblia y Plasmodium falciparum.
Antibióticos que actúan en el metabolismo o la estructura de los ácidos nucleicosEl genoma bacteriano contiene información para la síntesis de proteínas que se transmite a través del ARN mensajero producido a partir del molde de ADN (transcripción), y para la síntesis de ARN ribosómico que formará parte de los ribosomas bacterianos. La información del ADN debe duplicarse (replicación) cuando la bacteria se divide, para transmitir esta información a la descendencia. La replicación y la transcripción del ADN se realizan en varias fases con la participación de diferentes enzimas y sustratos, además del ADN molde, que constituyen dianas para la acción de diversos antibióticos.
Dentro de este grupo incluimos las rifamicinas y las quinolonas que actúan en enzimas que participan en los procesos de transcripción y replicación, y los nitroimidazoles y nitrofuranos que actúan directamente sobre el ADN, dañándolo. Por lo general, los antibióticos de este grupo no son particularmente selectivos en su acción y comportan cierta toxicidad para las células eucarióticas. La mayoría de los antibióticos que actúan sobre el ADN son bactericidas rápidos y normalmente independientes del inóculo y de la fase de crecimiento bacteriano.
RifamicinasInhiben la síntesis de ARN ribosómico y mensajero al bloquear la subunidad beta de la ARN polimerasa ADN-dependiente bacteriana codificada por el gen rpoB28. Impide el inicio del proceso de transcripción, pero carece de efecto antimicrobiano si la transcripción ya se ha iniciado.
La rifampicina, derivado semisintético de la rifamicina B, es el antibiótico representativo de este grupo y tiene actividad bactericida frente a microorganismos grampositivos, Neisseria spp., Chlamydia spp. y Mycobacterium spp.
QuinolonasEl cromosoma bacteriano está constituido por una doble cadena de ADN que es 1.000 veces más largo que la propia bacteria, por lo que se encuentra muy plegado sobre sí mismo en una forma superenrollada. Esta configuración no es accesible para que pueda realizarse la replicación y transcripción del ADN bacteriano, por lo que debe desenrollarse. Las topoisomerasas son las enzimas encargadas del superenrollamiento y desenrollamiento del ADN, así como del corte, unión y separación de las hebras de ADN, necesarias para los procesos de síntesis de ADN y de partición del cromosoma a las células hijas cuando la bacteria se divide. Las quinolonas ejercen su acción bloqueando las topoisomerasas II (ADN-girasa) y IV29. Ambas son enzimas tetraméricas compuestas por dos subunidades A y 2 subunidades B, codificadas respectivamente por los genes gyrA y gyrB en el caso de la ADN-girasa, y parC y parE en el caso de la topoisomerasa IV. Las topoisomerasas se acoplan al ADN, provocan un pequeño corte en las hebras de ADN que posteriormente es reparado, y quedan de nuevo libres. Las fluoroquinolonas, se unen al ADN roto y a la topoisomerasa formando un complejo ternario quinolona-ADN-topoisomerasa de forma irreversible, impidiendo que el proceso de transcripción o replicación continúen.
Su acción en las topoisomerasas no explica por sí sola su potente acción bactericida, sino que se debe a fenómenos secundarios mal conocidos30, entre los que la activación del sistema de reparación de mutaciones SOS parece desempeñar un papel importante.
Las quinolonas constituyen en la actualidad, junto a los β-lactámicos, los antibióticos de mayor uso. De forma semejante a lo que ocurre con las cefalosporinas, las quinolonas se han clasificado en generaciones, atendiendo a su espectro de actividad y propiedades farmacocinéticas. Las quinolonas de primera generación (ácido nalidíxico) tienen un espectro limitado a bacilos gramnegativos y sólo se utilizan para infecciones de tracto urinario. La introducción de un átomo de flúor ha permitido la síntesis de nuevas generaciones (fluoroquinolonas) con mejor actividad farmacocinética. Las de segunda generación (norfloxacino) son mucho más activas frente a gramnegativos, tienen alguna actividad frente a grampositivos, pero no frente a anaerobios. Las de tercera generación (ciprofloxacino, levofloxacino) tienen mejor actividad frente a grampositivos y organismos fastidiosos y por sus propiedades farmacocinéticas permiten su empleo sistémico. Finalmente, las de cuarta generación (moxifloxacino, gemifloxacino) son muy activas frente a grampositivos y tienen una buena actividad antianaerobia.
NitroimidazolesAmplio grupo de compuestos de los cuales metronidazol, tinidazol y ornidazol son los más conocidos. Estos antibióticos penetran fácilmente en el citoplasma por difusión pasiva y allí el grupo NO2 del anillo imidazólico, que se comporta como aceptor de electrones, se reduce por nitroreductasas bacterianas del metabolismo anaerobio, liberándose radicales nitritos que dañan el ADN por oxidación. Tienen actividad frente a Clostridium spp., microorganismos gramnegativos anaerobios y microorganismos microaerofílicos (Helicobacter pylori, Campylobacter spp., Gardnerella vaginalis) y protozoos (tricomonas, giardias, amebas, Balantidium coli).
NitrofuranosLa nitrofurantoína es el antibiótico representativo de este grupo y se usa como antiséptico urinario. Al igual que los nitroimidazoles, estos compuestos se reducen en el citoplasma bacteriano para generar derivados tóxicos que dañan el ADN por un mecanismo no bien conocido. También parecen interferir con la síntesis proteica bacteriana al unirse al ribosoma 30S bloqueando el reconocimiento del codón-anticodón.
Bloqueo de la síntesis de factores metabólicosPara obtener determinados elementos esenciales como los aminoácidos o las bases púricas y pirimidínicas de los nucleótidos, se requiere la síntesis de folatos, que algunas bacterias son incapaces de obtener del medio, a diferencia de las células eucariotas. La síntesis de ácido tetrahidrofólico se obtiene a partir de una molécula de pteridina y de ácido paraaminobenzoico (PABA), y mediante la enzima dihidropteroatosintetasa se forma el ácido dihidropteroico. Posteriormente, por adición de ácido glutámico se forma el ácido dihidrofólico (ácido fólico), que reducido por la dihidrofolato reductasa forma el ácido tetrahidrofólico (ácido folínico).
Sulfamidas, diaminopirimidinasLas sulfamidas son análogos del ácido paraaminobenzoico, y por tanto, compiten por la enzima dihidropteroatosintetasa, impidiendo así la formación de ácido dihidropteroico, precursor del ácido fólico. Estos antibióticos no afectan a las células humanas, que obtienen ácido fólico de la dieta. De este grupo se usa en clínica, sulfametoxazol (asociado a trimetoprima), sulfisoxazol, suladiazina, sulfacetamida, etc.
Las diaminopirimidinas, como la trimetoprima y la pirimetamina, compiten por la enzima dihidrofolatoreductasa que cataliza la conversión de ácido dihidrofólico en ácido tetrahidrofólico. El trimetoprima tiene mucha menos afinidad por la dihidrofolatoreductasa humana, que, sin embargo, puede llegar a afectarse con dosis altas o en pacientes con alteraciones hemáticas preexistentes.
El cotrimoxazol31 es la combinación de trimetoprima y sulfametoxazol en proporción 1:5, y por tanto, actúa en dos etapas de la síntesis de ácido folínico, pudiendo llegar a tener efecto bactericida por la sinergia ente sus 2 componentes.
Otras combinaciones utilizadas son las formadas por pirimetamina y sulfadiazina para el tratamiento de toxoplasmosis, o pirimetamina y sulfadoxina para el paludismo.
Note: Sectión acreditada por el SEAFORMEC. Consultar preguntas de cada artículo en: http://www.elsevier.es/eimc/formaction

