




La evolución entre la infección por Mycobacterium tuberculosis y la tuberculosis activa es multifactorial e implica diferentes escalas biológicas. La síntesis de ESAT-6 o la inducción de la necrosis de los macrófagos alveolares son claves, pero para entenderla se requiere tener en cuenta las dinámicas de reinfección endógena y exógena, el drenaje del parénquima pulmonar y la mecánica respiratoria, los procesos de fibrosis locales y la irrigación sanguínea. Paradójicamente, la respuesta inmune generada por la infección es altamente protectora (90%) contra la tuberculosis activa, aunque al basarse esencialmente en la proliferación de linfocitos Th1 no puede evitar la reinfección. La inmunosupresión severa tan solo puede explicar un 10% de los casos de tuberculosis activa, mientras que el resto es favorecido por comorbilidades, un ambiente proinflamatorio y una propensión genética desconocida. La capacidad patogénica de las micobacterias ambientales es discreta, ligada a déficits en la respuesta inmune innata y adquirida. Remarcable es la capacidad de generación de biofilms y la capacidad de M. ulcerans para generar la exotoxina micolactona.
The evolution between Mycobacterium tuberculosis infection and active tuberculosis is multifactorial and involves different biological scales. The synthesis of ESAT-6 or the induction of alveolar macrophage necrosis are key, but to understand it, it is necessary to consider the dynamics of endogenous and exogenous reinfection, drainage of lung parenchyma and respiratory mechanics, local fibrosis processes and blood supply. Paradoxically, the immune response generated by the infection is highly protective (90%) against active tuberculosis, although as it is essentially based on the proliferation of Th1 lymphocytes, it cannot prevent reinfection. Severe immunosuppression can only explain 10% of active tuberculosis cases, while the remainder are attributable to comorbidities, a proinflammatory environment and an unknown genetic propensity. The pathogenic capacity of environmental mycobacteria is discrete, linked to deficits in the innate and acquired immune response. The ability to generate biofilms and the ability of M. ulcerans to generate the exotoxin mycolactone is remarkable.
Los aerosoles infectados deben depositarse en el alvéolo pulmonar para poder generar la infección. De hecho, esta es una de las claves del éxito de M. tuberculosis: su capacidad para infectar el macrófago alveolar (MA). Es cierto que hay ciertos factores «protectores» que pueden evitar su capacidad infectiva. En primer lugar, la calidad del aerosol. No todos los enfermos son capaces de generar una cantidad suficiente de partículas aerosólicas susceptibles de poder internarse en el alvéolo1. En segundo lugar, la calidad del surfactante que permite evitar el colapso de los alvéolos. El surfactante no deja de ser un tensioactivo, y como tal, tiene la capacidad de destruir la pared lipofílica de la micobacteria de manera que puede ser destruida por el MA al ser fagocitada2. En todo caso, no sabemos hasta qué punto este factor es importante para evaluar la dosis infectiva. Lo que sí sabemos es que hay contactos próximos de enfermos con tuberculosis pulmonar activa (TBPA), expuestos constantemente a aerosoles infectivos, y que no han sido infectados3. Hay que tener en cuenta que las personas con más probabilidad de sufrir una tuberculosis activa (TBA) son aquellos que han estado en contacto con un caso de TBA de manera continuada, o sea más de 6h al día por un periodo que depende del retraso diagnóstico y que se sitúa entre los 60 y los 90 días en países con un buen sistema sanitario4. Esto quiere decir que para desarrollar una TBA no sirve simplemente una infección única. Se requiere de un proceso de reinfección continuo5.
El espacio alveolar y el macrófago alveolarSiempre hay que tener en cuenta la función fisiológica del alvéolo pulmonar para entender la esencia de la infección por M. tuberculosis. El alvéolo es una estructura muy delicada, configurada por unas células epiteliales, los neumocitos de tipo i, o células alveolares planas, que configuran el 95% de la superficie y que tienen un grosor ínfimo para permitir la difusión de gases, que a la vez han de atravesar las células endoteliales de los capilares que revisten los alvéolos. A la vez, estas células están adheridas firmemente entre ellas para evitar la entrada de plasma. Este hecho es trascendental, puesto que permite mantener una tensión superficial baja, gracias al surfactante generado por los neumocitos de tipo ii, pero tiene una contrapartida negativa: evita la entrada de anticuerpos. Igualmente, cada alvéolo tiene su MA que se dedica a limpiar constantemente este espacio6. Hemos de tener en cuenta que aproximadamente cada 6s el alvéolo se expande para permitir la entrada de aire del exterior, y con él todo tipo de partículas y de patógenos. La función del MA es mantener limpio el alvéolo para permitir el intercambio de gases y evitar a toda costa cualquier desarrollo inflamatorio que pueda romper su delicada estructura. El MA es, pues, una especie de «Don Limpio», no un «policía» dedicado a identificar patógenos para generar una respuesta inflamatoria inmediatamente, como sería el caso de las células de Langerhans de la piel. A este lavado también contribuye el surfactante, convertido en fluido alveolar, que sirve no solo para mantener la tensión superficial, sino que también lava el espacio alveolar, puesto que es constantemente drenado con el movimiento respiratorio hacia los bronquiolos, el árbol bronquial y la faringe, para ser deglutido y dirigido hacia el estómago. Diariamente drenamos aproximadamente 500mL de fluido alveolar hacia el estómago.
La necrosis del macrófago alveolar como mecanismo de virulencia esencialCuando el bacilo viable es fagocitado por el MA, despliega su capacidad patogénica secretando 6kDa early secretory antigenic target (ESAT-6). Este péptido es esencial para evitar la unión fagosoma-lisosoma y la apoptosis, y permite finalmente la entrada del bacilo en el citoplasma7. De esta manera, el bacilo aprovecha al máximo su capacidad de multiplicación en un único MA, aproximadamente entre 5-6 ciclos de división, para conseguir una concentración de entre 32 y 64 bacilos8. Este proceso se desarrolla durante unos 5-6 días, considerando que cada ciclo de división en M. tuberculosis requiere unas 24h, provocando la necrosis del MA9. Entonces, los bacilos vuelven a convertirse en extracelulares y vuelven a ser fagocitados por el MA proveniente del espacio intersticial que substituye al necrosado, y por los MA de los alvéolos vecinos, donde llegan a merced del constante drenaje generado por el movimiento de inspiración/espiración. El proceso se repite al menos una vez más, generándose hasta 1.000 bacilos, provocando la suficiente generación de quimiocinas por parte de los MA infectados como para generar una respuesta inflamatoria.
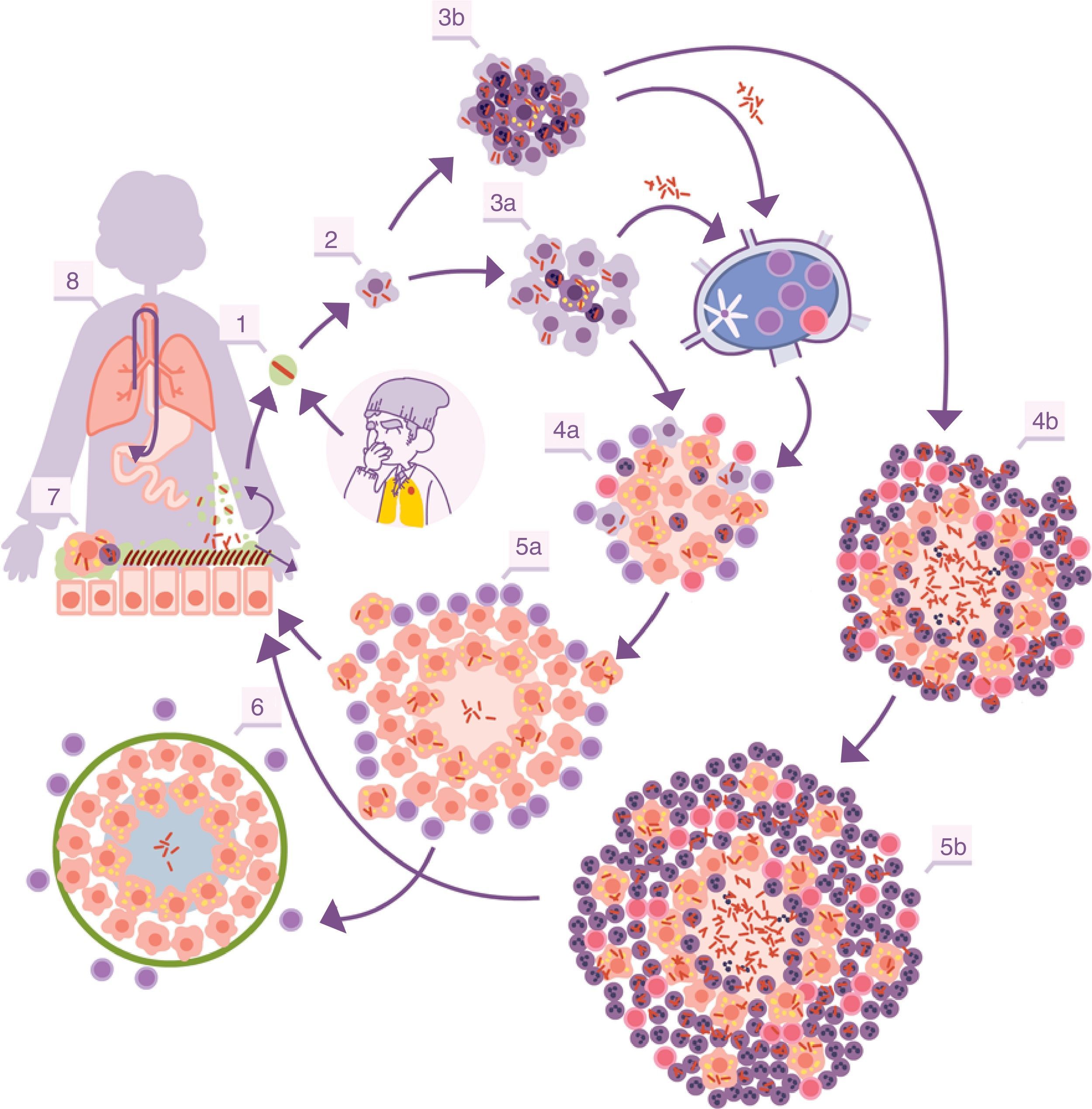
Con la inflamación se rompe el equilibrio, al generarse un exudado a nivel capilar que destruye la estanqueidad del alvéolo y permite la entrada de células polimorfonucleares (PMN) normalmente neutrófilos, y monocitos, en proporciones que dependerán del tipo de quimiocinas y citocinas secretadas por los MA. Al mismo tiempo, permite un lavado más enérgico de los alvéolos afectados, drenándose hacia los nódulos linfáticos a través de los capilares linfáticos aferentes. De esta manera es como M. tuberculosis infecta en primer lugar a los macrófagos de los nódulos, generando una linfadenitis, y a las células dendríticas (fig. 1).
 y posterior multiplicación en su interior. 3. Destrucción del MA, diseminación local de M. tuberculosis, fagocitosis por parte de otros MA y generación de una respuesta inflamatoria local dominada por monocitos (3a) o PMN (3b), merced a la cual los bacilos pueden ser drenados hacia el ganglio linfático regional, donde proliferan linfocitos Th1 o Th17. 4. Los linfocitos son atraídos por la respuesta inflamatoria de las lesiones y activan a los MA infectados o atraen más PMN, dependiendo de que la respuesta inmune se decante por una respuesta de tipo Th1 (4a) o Th17 (4b), respectivamente. En el primer caso hay un control de la población bacilar y hay un drenaje de bacilos adormecidos a través de los macrófagos espumosos (5a), hasta que se controla mediante la encapsulación de la lesión (6). En el segundo, las lesiones van creciendo de tamaño gracias a la entrada de PMN y el crecimiento bacilar extracelular en las NET, generando nuevas lesiones periféricas. En este caso, la concentración bacilar es mucho más alta, y de aquí que el drenaje sea mucho más importante, ya sea a través del fluido alveolar o a nivel sistémico mediante la neovascularización del granuloma (5b). A nivel pulmonar los bacilos del fluido alveolar (7) tienden a ser drenados hacia el tracto gastrointestinal (8), aunque pueden formar parte de nuevos aerosoles, generando nuevas lesiones (1). Adaptada de Cardona10.")
Ciclo infectivo de M. tuberculosis. 1. Entrada de bacilos al alvéolo pulmonar a través de una gota de aerosol. 2. Fagocitosis por parte de un macrófago alveolar (MA) y posterior multiplicación en su interior. 3. Destrucción del MA, diseminación local de M. tuberculosis, fagocitosis por parte de otros MA y generación de una respuesta inflamatoria local dominada por monocitos (3a) o PMN (3b), merced a la cual los bacilos pueden ser drenados hacia el ganglio linfático regional, donde proliferan linfocitos Th1 o Th17. 4. Los linfocitos son atraídos por la respuesta inflamatoria de las lesiones y activan a los MA infectados o atraen más PMN, dependiendo de que la respuesta inmune se decante por una respuesta de tipo Th1 (4a) o Th17 (4b), respectivamente. En el primer caso hay un control de la población bacilar y hay un drenaje de bacilos adormecidos a través de los macrófagos espumosos (5a), hasta que se controla mediante la encapsulación de la lesión (6). En el segundo, las lesiones van creciendo de tamaño gracias a la entrada de PMN y el crecimiento bacilar extracelular en las NET, generando nuevas lesiones periféricas. En este caso, la concentración bacilar es mucho más alta, y de aquí que el drenaje sea mucho más importante, ya sea a través del fluido alveolar o a nivel sistémico mediante la neovascularización del granuloma (5b). A nivel pulmonar los bacilos del fluido alveolar (7) tienden a ser drenados hacia el tracto gastrointestinal (8), aunque pueden formar parte de nuevos aerosoles, generando nuevas lesiones (1).
Adaptada de Cardona10.
Las células dendríticas procesan M. tuberculosis y presentan unos epítopos que mayoritariamente corresponden a los antígenos secretados más abundantes: el ESAT-6 y el complejo antigénico 85 (Ag85 A, B o C). Este último es responsable de la construcción de la pared celular, ya que permite el ensamblaje de 2 moléculas esenciales: el arabinogalactano micolato y la trehalosa dimicolato11. La presentación antigénica estimulará esencialmente los linfocitos T CD4, cuyo subtipo dependerá del tipo de quimiocinas y citocinas que transporta el líquido linfático drenado, y que son esencialmente Th1, Th2, Th17 o Treg. También se pueden generar linfocitos T CD8, pero de manera minoritaria. Generalmente, el subtipo dominante es el Th1, responsable de generar interferón gamma que permite la activación de los macrófagos infectados.
Aunque todavía no se sabe con certeza qué parámetro inmunológico es el determinante para evaluar la respuesta protectora contra M. tuberculosis, lo que sí está claro es que es protectora y evita el desarrollo de la TBA en un 90% de los casos aproximadamente. Los estudios de Heimbeck en el Hospital Ulleval de Oslo durante los años 1924-1946 demostraron que las estudiantes de enfermería que al ingresar tenían un test de tuberculina positivo tenían una protección altísima contra la TBA en comparación con las que tenían un test negativo. Concretamente, en el primer grupo hubo 22 casos de 668 (3,3%) y en el segundo 97 de 284 (34,2%), o sea, una protección de más del 90%. En el caso de aquellas estudiantes que tenían un test de la tuberculina negativo y se vacunaban con bacilo de Calmette-Guérin, la incidencia fue de 35 casos en 501 (6,9%), es decir, que se protegían, pero menos que en el caso de la infección natural. La mortalidad también reflejaba esta tendencia, observándose 0/668, 10/284 y 3/501, respectivamente12.
El periplo del bacilo hacia la diseminación extrapulmonarNormalmente el periplo del bacilo finaliza en el ganglio, pero no necesariamente. Al generarse una linfadenitis, esta puede progresar y liberar bacilos hacia los capilares eferentes, los cuales llegan hasta la vena cava y pasan a la aurícula y el ventrículo derechos para ser trasladados de nuevo hacia el pulmón. De esta manera se pueden generar nuevos focos infecciosos, sobre todo si los bacilos son liberados masivamente, en forma de agregados o «clumps», que permitan obstaculizar un capilar, generar un estancamiento de la circulación, destruir la estanqueidad del espacio alveolar y entrar en el mismo. También pueden recolonizar las lesiones generadas previamente que, al estar en un proceso de inflamación, tienen una mayor vascularización y permeabilidad. Finalmente, estos bacilos pueden pasar simplemente a los capilares venosos, llegar a la aurícula y el ventrículo izquierdos y diseminarse sistémicamente. Potencialmente el bacilo puede colonizar cualquier órgano. Es un tema ligado a las características de la vascularización. De aquí que unos de los órganos en que las células endoteliales permiten una mayor permeabilidad, como el tejido óseo, especialmente en los niños, cuando está en fase de desarrollo, o el riñón, sean órganos diana habituales. En cambio, la afectación de las meninges es mucho menos habitual y requiere de una entrada masiva de bacilos en el torrente circulatorio, hecho que se asocia a las TBA diseminadas o miliares, manifestándose en las primeras semanas posteriores a la infección13.
Un tema importante es que las lesiones generadas por M. tuberculosis desarrollan nuevos vasos que son más permeables y frágiles que los estructurales, permitiendo tanto el fenómeno de reinfección de la lesión como el de propagación del bacilo hacia los capilares venosos pulmonares14.
Otra vía de diseminación habitual es la generada por el mismo drenaje del bacilo a través del fluido alveolar, que permite que entre en la cavidad faríngea, pudiendo penetrar en la mucosa a través de pequeñas heridas, afectando a los ganglios cervicales; o provocando una TBA intestinal, en el caso de que la acidificación gástrica no sea suficiente como para destruir el bacilo. Por otra parte, la TBA pleural no deja de ser una variante de la TB pulmonar. Las células mesenquimales de la serosa pleural se encargan de vigilar cualquier mínima alteración del parénquima pulmonar más superficial y ante cualquier mínima lesión generan un masivo influjo de PMN y monocitos para aislarla y generar tejido fibroso a su alrededor15.
La TBA extrapulmonar representa aproximadamente un 30% de los casos de TBA y suele denotar un retraso en la respuesta inmune, afectando mayoritariamente a niños menores de 5 años o a personas que sufren una inmunodepresión, aunque hay mucha variabilidad geográfica, hecho que puede interpretarse como que puede haber algún factor genético que la favorece16.
El control de las lesiones mediante la respuesta inmune y las estructuras localesLa «hipótesis dinámica»: el proceso de reinfección endógenoEn la mayoría de los casos, la proliferación de linfocitos de tipo Th1 específicos contra la infección tiene lugar a tiempo para evitar el desarrollo de la TBA. Los Th1 son drenados por los vasos eferentes, para incorporarse a la circulación pulmonar y dirigirse mayoritariamente hacia los focos infecciosos, dado que la inflamación permite una mayor probabilidad de atracción. Los Th1 entran en contacto con los macrófagos infectados, y mediante la síntesis de interferón gamma los activan, permitiendo la destrucción de la mayoría de los bacilos. No obstante, hay un pequeño porcentaje que consigue entrar en una fase de enlentecimiento metabólico o adormecimiento («dormancy») que le permite sobrevivir manteniendo una actividad metabólica mínima. Los macrófagos activados, aparte de mantener estos bacilos adormecidos en su interior, también fagocitan restos de los tejidos necrosados hasta un punto en que no pueden metabolizar la enorme carga de ácidos grasos (y sobre todo de colesterol) que supone, fabricando cuerpos lipídicos que van acumulando dentro del citoplasma, transformándose en macrófagos espumosos17. Estas células acaban siendo drenadas por el fluido alveolar, y es durante este proceso cuando pueden destruirse, liberando así bacilos que pueden formar parte de los aerosoles que se generan constantemente en los bronquiolos y volver a reinfectar el pulmón (fig. 1).
Este proceso fue el origen de la denominada «hipótesis dinámica», que entiende la infección tuberculosa latente (ITBL) como un proceso de reinfección endógena constante. Entre otras cosas, esta hipótesis permite explicar por qué funciona la quimioprofilaxis con isoniacida, teniendo en cuenta que no tiene actividad contra los bacilos adormecidos. El concepto es que los niveles constantes de isoniacida durante 6-9 meses permiten el drenaje de los bacilos adormecidos hacia el tracto gastrointestinal, evitando el riesgo de reinfección. El periodo tan prolongado se explica porque sería el tiempo medio para poder drenar todas las lesiones antes de que se fibrosen y calcifiquen, destruyendo los bacilos de su interior al generar un estrés multifactorial a su alrededor18.
Importancia de los septos interlobulares en la encapsulación de las lesionesHay otro mecanismo de control local que permite controlar la lesión: se trata de los septos interlobulares. Estas estructuras dividen el parénquima pulmonar en porciones de aproximadamente 1cm3 y sirven para transmitir la fuerza mecánica generada por el diafragma19. De esta manera se puede insuflar el pulmón, permitiendo su expansión y evitando que la delicada estructura de los alvéolos sea tensionada excesivamente por el desplazamiento que requiere el movimiento respiratorio. Esta estructura es mantenida por fibroblastos que tienen una especial capacidad para captar variaciones mecánicas a su alrededor, como las generadas con la conformación de una lesión, incluso si es tan pequeña como la que ocasiona la infección inicial por M. tuberculosis (de unos 0,5mm)20. Así pues, los fibroblastos empiezan a tejer una cápsula alrededor de la lesión que acaba aislándola en aproximadamente unos 10 días.
Es interesante reseñar que la infección por M. tuberculosis genera una respuesta inmune protectora21, que no hay fenómenos de mutación ni existen grandes variaciones entre los diferentes linajes que han sufrido contextos evolutivos diferentes22. La respuesta inmune se genera inicialmente contra varios antígenos, pero progresivamente se va focalizando contra antígenos secretados vitales para M. tuberculosis. Esta especialización tiene una lógica. Por un lado, el bacilo en su forma activa es el que es capaz de generar daño al hospedador, gracias a su capacidad para destruir macrófagos. Por otra parte, ya hemos reseñado la capacidad de escape y de reinfección del bacilo incluso después de haberse generado la respuesta inmune, por lo que la focalización contra los bacilos en multiplicación supone una estrategia exitosa, más aún teniendo en cuenta que los bacilos adormecidos acaban siendo drenados hacia el tracto gastrointestinal, para ser finalmente expulsados (fig. 1).
La inmunidad basada en linfocitos explica la naturaleza reactiva de la respuesta inmuneLa otra lección que aprendemos es que, debido a la naturaleza celular de la respuesta inmune, esta siempre va constantemente «después» de la infección. Todo lo contrario a lo que supone una respuesta inmune humoral, que en este caso no es eficaz debido a la falta de entrada de anticuerpos al espacio alveolar. Por tanto, el hospedador que ya ha generado una respuesta inmune específica previa tiene una ganancia de unos pocos días, al tener disposición de células de memoria, que permite reducir la población bacilar, hecho que sería clave para evitar una excesiva diseminación extrapulmonar y/o concentración local bacilar, necesarias para la generación de la TBA23. Aun así, debido a que inicialmente la multiplicación del bacilo es silente y no genera respuesta inflamatoria alguna, estos linfocitos no pueden ser atraídos de ninguna manera al foco infeccioso. La traducción de esta información es que, en realidad, la infección y la reinfección por M. tuberculosis no pueden evitarse24. De aquí la enorme desesperación en el campo de la vacunología. En realidad, es imposible conseguir una vacuna profiláctica, tal como hemos venido constatando hasta la fecha10. Ninguna vacuna es capaz de evitar la infección: el objetivo es evitar la TBA.
El origen de la tuberculosis pulmonar. El tamaño importaLa praxis clínica en el diagnósticoTeniendo en cuenta la reacción del huésped, se hace difícil explicar el desarrollo de la TBPA. Si bien la infección parece inevitable, las consecuencias para el huésped son prácticamente nulas. Se generan lesiones extraordinariamente pequeñas, que no ocasionan ningún tipo de disfunción significativa. De hecho, se hace difícil explicar por qué una lesión de 1mm de diámetro puede convertirse en una lesión de 10mm. ¿Por qué 10mm? Porque es el tamaño de lesión que se requiere para poder ser detectada a través de una exploración radiográfica de tórax25.
Años de lucha contra la tuberculosis han permitido estandarizar una praxis clínica eficaz. Ante una sospecha de TBA, lo que debe hacerse es un test de tuberculina o de interferon-gamma release assay para valorar si ha habido infección y se ha generado una respuesta inmune. En caso de ser positivo, se procede a realizar una radiografía de tórax. Si se detecta una lesión se considera que se sufre una TBPA y para poder ser detectada esta lesión debe tener un tamaño mínimo de 10mm. En caso contrario, se considera que la persona sufre una ITBL y en caso de infección reciente se ha demostrado que el riesgo más importante de desarrollar una TBPA es durante los 2 primeros años, decreciendo exponencialmente hasta ser nulo a los 8 años5.
Importancia del tamaño de la lesiónLa pregunta es: ¿cómo puede desarrollarse una lesión de 10mm a partir de una lesión de 1mm teniendo en cuenta que los mecanismos de control local funcionan tan bien?
En este punto es importante tener en cuenta la misma formulación de la pregunta. En la totalidad de las publicaciones que estudian el fenómeno de la evolución de la infección tuberculosa a TBA, esta cuestión ni se plantea. Las preguntas se focalizan en la capacidad de reactivación de los bacilos durmientes o en el desarrollo de algún tipo de inmunodepresión local. Nadie plantea lo que realmente se ve en la clínica: la TBPA se caracteriza por la presencia de una lesión visible en la radiografía de tórax (fig. 2).
 a tuberculosis activa (TBA). A. Representación habitual, la diferencia está en la actividad del bacilo, obviando el hecho de que la característica principal que clínicamente distingue la ITBL de la TBA es el tamaño de las lesiones (B). Extraída de Cardona10.")
Evolución de infección tuberculosa latente (ITBL) a tuberculosis activa (TBA). A. Representación habitual, la diferencia está en la actividad del bacilo, obviando el hecho de que la característica principal que clínicamente distingue la ITBL de la TBA es el tamaño de las lesiones (B).
Extraída de Cardona10.
Un aspecto característico del desarrollo de la TBPA en adultos es la afectación de los lóbulos pulmonares superiores. La teoría que actualmente tiene más éxito para explicar este fenómeno es que en esta localización la presión de oxígeno es más elevada y permite un mejor crecimiento de M. tuberculosis. Esta teoría, la «teoría unitaria», es atractiva debido a su fácil enunciación, pero realmente no resiste un análisis sistemático5. El origen de la acumulación de oxígeno en esta localización se debe a que el desplazamiento del parénquima pulmonar con el movimiento respiratorio es mucho menor que en la base. Ello es debido a la estructura del pulmón, que se sujeta en el vértice para que el movimiento de expansión del diafragma pueda efectivamente generar la expansión del parénquima. Por otra parte, la fuerza de la gravedad condiciona el hecho de que los septos interlobulares del pulmón superior estén constantemente tensados y tengan una menor reactividad que los situados en la base. La falta de movimiento también provoca un déficit en el drenaje vascular, por lo que hay un menor intercambio de gases que se traduce en la hiperoxigenación. Sin embargo, aunque este incremento favoreciese una aceleración de la capacidad de multiplicación del bacilo, hecho dudoso desde el punto de vista microbiológico, no explicaría el crecimiento desmesurado de las lesiones26.
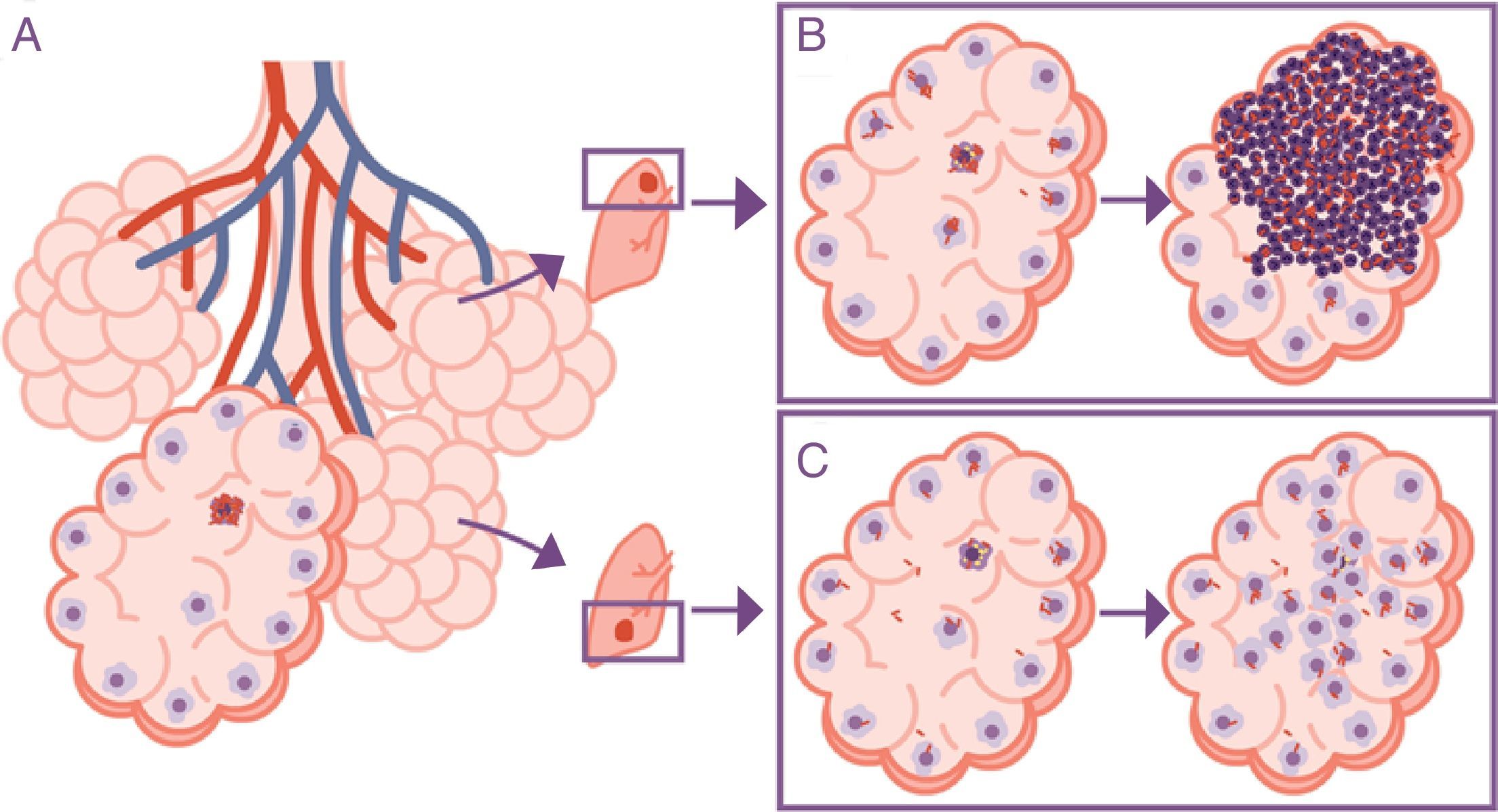
La respuesta está en el drenaje alveolarLo que es relevante en realidad es la disminución del drenaje del interior del propio alvéolo, puesto que se traduce en una acumulación de bacilos extracelulares localmente. Ello provoca que el MA que debe fagocitarlos se enfrente a una gran concentración bacilar, provocando una mayor destrucción celular y la generación de una respuesta quimiocínica y citocínica que favorece la acumulación intensiva de PMN6 (fig. 3).
, pero la evolución es diferente en el lóbulo superior, donde la acumulación de bacilos hace que los macrófagos alveolares (MA) deban hacer frente a una cantidad más elevada de bacilos, generando una respuesta basada preferentemente en la acumulación de PMN (B). Contrariamente, en el lóbulo inferior el drenaje más importante permite una mejor distribución de los bacilos y una respuesta inflamatoria basada en la acumulación de MA y monocitos (C). Adaptada de Cardona10.")
Importancia de la localización de la infección en los lóbulos superiores. La infección tiene lugar en un alvéolo pulmonar (A), pero la evolución es diferente en el lóbulo superior, donde la acumulación de bacilos hace que los macrófagos alveolares (MA) deban hacer frente a una cantidad más elevada de bacilos, generando una respuesta basada preferentemente en la acumulación de PMN (B). Contrariamente, en el lóbulo inferior el drenaje más importante permite una mejor distribución de los bacilos y una respuesta inflamatoria basada en la acumulación de MA y monocitos (C).
Adaptada de Cardona10.
Durante la fase moderna del estudio de la TB, la que empieza con la declaración de la Emergencia Mundial en 1993, los PMN eran considerados unos actores secundarios en la patogenia, cuando no eran simplemente ignorados. Sin embargo, al revisar las experiencias de los patólogos que estudiaron la TBPA en la era preantibiótica, se observa que describían 2 tipos de lesiones: la proliferativa (o «tubérculo»), que contiene una población bacilar pequeña compuesta por células epitelioides y fibroblastos que progresa hacia la fibrosis y la calcificación, y la exudativa, o condensación neutrofílica local, que contiene una elevada población bacilar y se relaciona con el desarrollo de TBPA27.
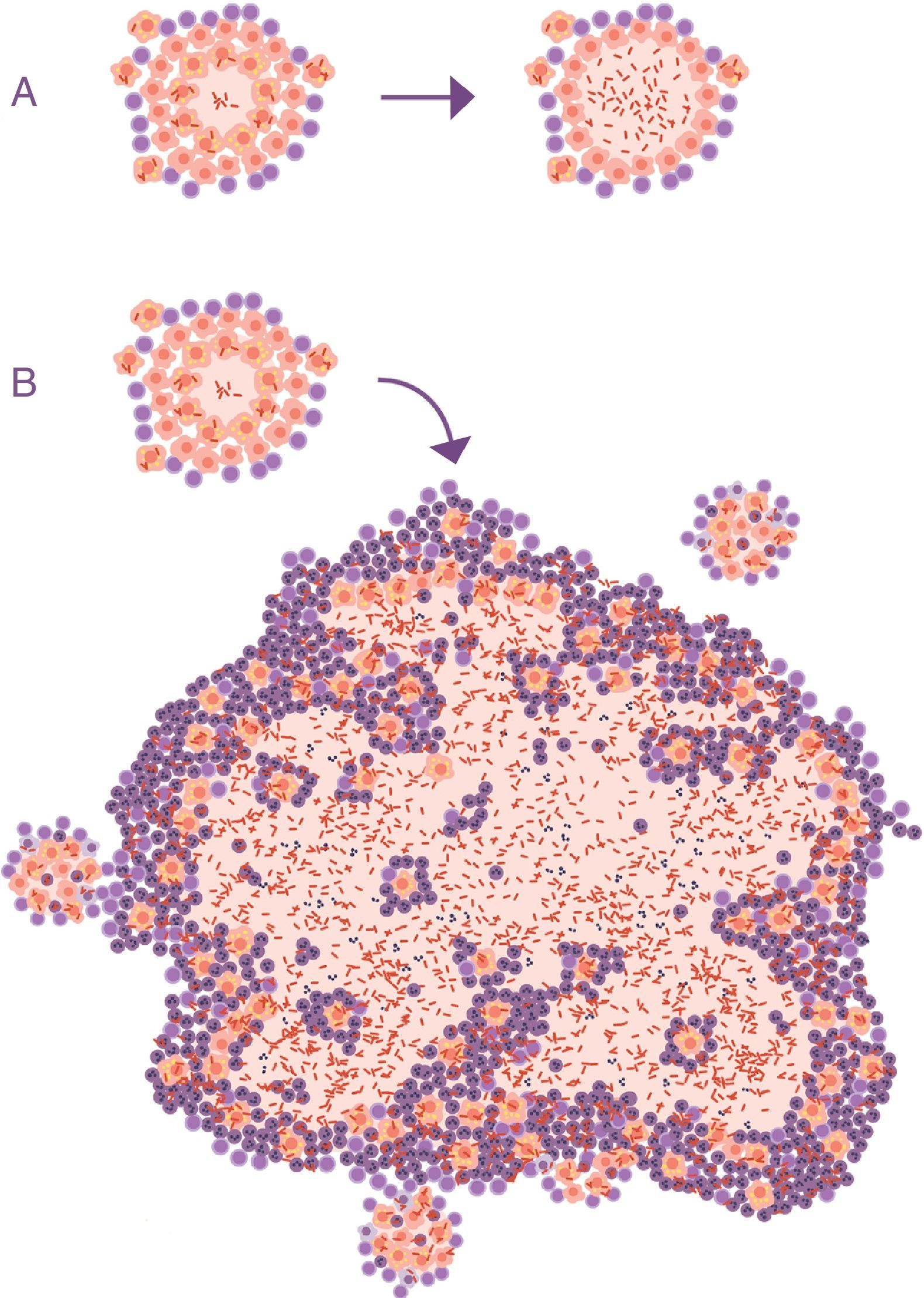
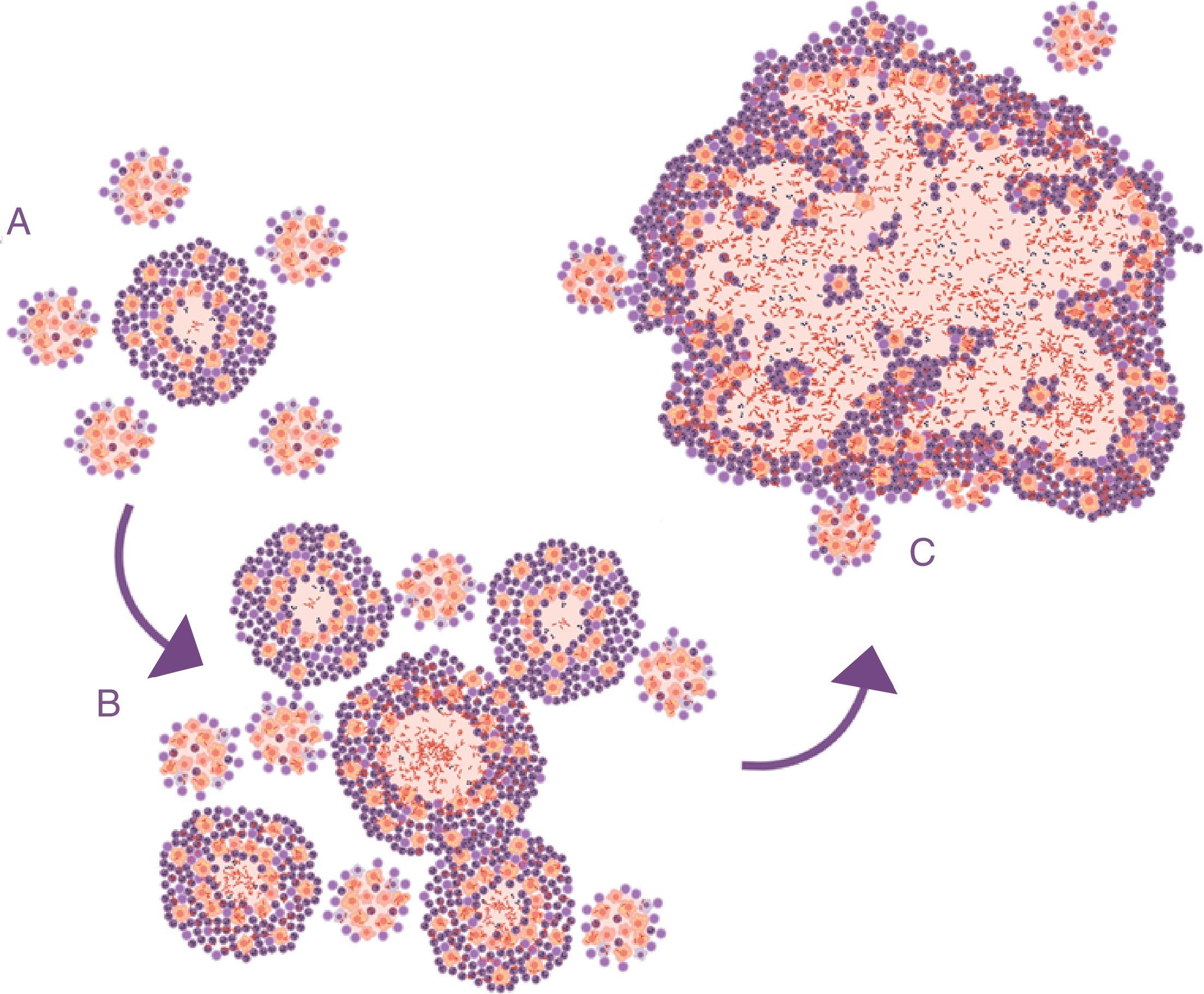
Lecciones aprendidas del modelo de tuberculosis activa en ratón. El modelo de pompas de jabónEl desarrollo del modelo experimental utilizando ratones de la cepa C3HeB/FeJ ha permitido un impulso muy importante en la comprensión de la evolución entre la ITBL y la TBPA. En este modelo se ha podido demostrar que M. tuberculosis potencia la formación de trampas extracelulares de neutrófilos, un mecanismo desarrollado por los PMN para hacer frente a infecciones ocasionadas por patógenos extracelulares. Consiste en la lisis de la célula para liberar las enzimas contenidas en los lisosomas, atrapando a la vez a los patógenos a través de la formación de un tejido mediante el ADN nuclear. Pero en el caso de M. tuberculosis no consiguen destruirlo; al contrario, las trampas extracelulares de neutrófilos constituyen una plataforma para el crecimiento extracelular del bacilo28. De esta manera se produce un crecimiento exponencial del tamaño de la lesión, que implica la generación de lesiones satélites (o «hijas») alrededor y un proceso de fusión final entre todas ellas, en un periodo temporal de entre 10 y 15 días. Esto permite evitar el mecanismo protector de la encapsulación y es capaz de generar una TBPA en forma de infiltración alveolar por PMN que finalmente licuefacta y cavita. La reinfección constante que sufren los contactos de los casos de TBPA citada anteriormente ayuda claramente a este fenómeno, en el caso de que la reinfección se localice de manera adyacente, al igual que la reinfección endógena constante como consecuencia del drenaje de los bacilos adormecidos a partir de lesiones previas (fig. 1) y las que provienen de la diseminación hematógena que consigue burlar el filtro de los ganglios linfáticos.
La evolución de este proceso se asemeja mucho a la dinámica de formación de pompas de jabón. De ahí que se hiciera un paralelismo y se consiguiera demostrar que la inducción de este tipo de lesiones depende necesariamente de los 3 factores: un crecimiento rápido de la lesión inicial (como el experimentado con la infiltración de PMN), la formación de «lesiones hijas» alrededor y la fusión de todas ellas29 (fig. 4).
 se generan unas lesiones mínimas que crecen rápidamente gracias a la acumulación de los PMN y al crecimiento extracelular de los bacilos. Posteriormente, se generan lesiones «hijas», que sufren el mismo proceso (B), y finalmente todas ellas se fusionan en una gran lesión, burlando el proceso de encapsulación (C). Adaptada de Cardona10.")
Evolución de las lesiones de infección tuberculosa latente a tuberculosis pulmonar. El modelo de las pompas de jabón. En la infección inicial (A) se generan unas lesiones mínimas que crecen rápidamente gracias a la acumulación de los PMN y al crecimiento extracelular de los bacilos. Posteriormente, se generan lesiones «hijas», que sufren el mismo proceso (B), y finalmente todas ellas se fusionan en una gran lesión, burlando el proceso de encapsulación (C).
Adaptada de Cardona10.
¿Cuál sería la diferencia entre un absceso pulmonar y una TBPA? La respuesta es el tiempo. Los abscesos pulmonares tienen una progresión muy rápida, generados por patógenos con una capacidad de multiplicación mucho más rápida (una división cada 20min) que la que tiene M. tuberculosis (una división cada 24»h). Ello se traduce en que la destrucción de las paredes alveolares tiene lugar rápidamente en el caso del absceso, mientras que en el caso de la TBPA no, de manera que los detritus de los PMN quedan circunscritos en cada alvéolo. Cuando el patólogo secciona la lesión, la zona necrótica tiene una textura caseosa y, por ello, la denomina «caseum»30. Sin embargo, con el paso de los días, las paredes alveolares finalmente degeneran y permiten confluir el contenido de todos los alvéolos afectados, generándose la denominada licuefacción.
Es interesante tener en cuenta este cambio de paradigma. M. tuberculosis se convierte en un patógeno extracelular antes de lo pensado. Tradicionalmente se pensaba que la fase extracelular se iniciaba con la licuefacción, y se potenciaba con el aire entrante en el caso de causar una erosión del bronquio y proceder al drenaje30. Hoy podemos sostener que no es así, y que el crecimiento extracelular constituye el mecanismo por el cual hay una evolución entre ITBL y TBPA.
¿Tuberculosis primaria o posprimaria?En general, la TBA primaria se considera aquella que afecta a los lóbulos basales y hay una implicación visible de los ganglios hiliares, mientras que la TBA postprimaria o del adulto es aquella que afecta a los lóbulos superiores. Este concepto surge de las campañas de detección de la TBA mediante radioscopia de tórax que se realizaron posteriormente a la Segunda Guerra Mundial. En aquel momento, cuando la incidencia de la TBA y la ITBL era tan importante, se empezaron a observar en los adultos lesiones calcificadas en el lóbulo inferior y en los ganglios, mientras que la TBA se manifestaba en los lóbulos superiores. En 1967 se enunció la «unitary theory», que daba sentido global a aquellas observaciones, teniendo en cuenta que se creía que una vez infectada una persona, no podía volver a infectarse31. La interpretación era que de aquellas lesiones antiguas habían surgido en la fase preinmune unas lesiones en los pulmones superiores que habían quedado dormidas y que posteriormente se habían reactivado. Actualmente se ha demostrado que esto no es cierto. En primer lugar, si bien la infección da una buena protección contra la TBA, no evita la reinfección. En segundo lugar, los estudios epidemiológicos moleculares han demostrado que la localización de la TBA depende de la respuesta inmune del hospedador32. En el caso de los niños menores de 5 años o en adultos inmunodeprimidos, la proliferación del bacilo no necesita la ventaja que le da la localización en el pulmón superior, por tanto, se puede desarrollar en ambas zonas. En el caso de los adultos inmunocompetentes, se desarrolla generalmente en el pulmón superior debido a la ventaja que le proporciona la falta de drenaje y la inducción de una respuesta inflamatoria dominada por PMN.
Uno de los puntos que habitualmente no se tienen en cuenta en las TBA desarrolladas en pacientes que contrajeron una TBA en la era preantibiótica, es decir, antes de los años 50, es que a menudo lesiones muy importantes se solucionaron mediante métodos clásicos, ya sea con «curas de reposo», neumotórax o toracoplastias, que buscaban el control de las lesiones mediante su fibrosis. En todos estos casos, la erradicación de la población bacilar fue muy diverso, pero en general inefectivo. De aquí que en este caso la posibilidad de generar una TBA, especialmente al llegar a la vejez, fuera mucho más elevada que en el caso de detectarse una ITBL y, por tanto, de no observarse ninguna lesión con la radiografía de tórax.
Conclusión: ¿por qué un 10% de los infectados desarrollan tuberculosis activa?¡Esta es la gran pregunta! Por un lado, se han identificado los antígenos más importantes, cuya expresión es muy estable; por otro lado, la naturaleza de la respuesta inmune de tipo celular no evita la infección, pero sí que tiene una gran eficacia para evitar la TBA. En este sentido, la aparición del sida ha ayudado a entender la importancia de los linfocitos CD4. Sin embargo, solo explica un 10% de los casos de TBA33. La gran complejidad en las circulaciones del bacilo a través del drenaje pulmonar o el torrente sanguíneo, los movimientos respiratorios, la capacidad de encapsulación por parte de los septos interlobulares, conjuntamente con la capacidad que tienen los aerosoles externos para generar infecciones efectivamente, generan una gran complejidad que impide encontrar marcadores con un valor predictivo suficiente. Lo que está claro es que hay factores de comorbilidad que favorecen el desarrollo de la TBA. El consumo de tabaco34 o la polución doméstica por el uso de combustibles sólidos35 se pueden relacionar claramente con una capacidad deficiente en el drenaje pulmonar. Sin embargo, hay un factor que parece tener un gran valor, como es el de aquellas personas que tienen un ambiente proinflamatorio y que, por tanto, favorece la atracción de PMN a las lesiones. Este aspecto puede explicar por qué la TBA afecta más al género masculino36 y a los pacientes con diabetes mellitus tipo 237. De hecho, en la mayoría de los casos, la inducción de TBA se incluiría dentro del marco conceptual de la «teoría del daño»38, por la cual la inducción de TBA puede ser debida a una inmunodepresión, pero mayoritariamente es debida a una excesiva respuesta por parte del hospedador, en este caso en forma de infiltrados de PMN.
Históricamente hay una gran evidencia de la susceptibilidad familiar a sufrir TBA, sin embargo, hasta la fecha no se ha reconocido todavía una asociación concreta replicable39. Finalmente, siempre se tiene que tener en cuenta que los pacientes que han sufrido una TBPA y se han curado con el tratamiento estándar, y no han sufrido una recaída, tienen una mayor probabilidad de sufrir otro episodio de TBPA ante una nueva reinfección exógena. Ello puede ser debido a 2 factores: a una susceptibilidad debido al mismo factor o factores de tipo genético que ya favorecieron la inducción de la primera TBPA y/o a las secuelas locales generadas por el primer episodio40.
Micobacteriosis ambientalesLas micobacterias ambientales tienen mucho interés para poder contrastar la patogenia de la TB. Se trata de unos microorganismos que viven en el medio natural y que comparten muchos aspectos con M. tuberculosis, no tan solo en cuanto a las características de la pared celular, rica en ácidos micólicos, sino compartiendo diversos antígenos capaces de generar respuestas protectoras41. Sin embargo, a nivel patogénico han sido poco estudiadas y es difícil extraer conclusiones. Su interés clínico surge, en primer lugar, con la enfermedad pulmonar generada en poblaciones envejecidas42, produciendo en pacientes negativos para el VIH una dolencia muy similar a la TBPA; en segundo lugar, en pacientes positivos para el VIH, demostrando el valor de la respuesta inmune de tipo Th1 para evitar infecciones diseminadas43. Igualmente, estas micobacterias son capaces de generar linfadenopatías, infecciones cutáneas, óseas o de articulaciones42. Cabe reseñar que una de sus particularidades es que, en general, su temperatura óptima de crecimiento se sitúa alrededor de los 30°C. Esta desventaja competitiva limita su actividad patogénica a epitelios superficiales donde la temperatura puede acercarse a este valor.
Puesto que la secreción de ESAT-6 parece ser uno de los factores clave en la patogenia de M. tuberculosis, podemos clasificar las micobacterias ambientales en relación con este aspecto.
Especies secretoras de ESAT-6 o ESAT-6-likeM. kansasii es capaz de generar lesiones similares a la TBPA en individuos expuestos a aerosoles de aguas con una gran concentración de este bacilo. La incidencia de la enfermedad pulmonar por M. kansasii es muy baja y probablemente la explicación se encuentra en que en realidad se necesita una dosis infectiva muy elevada. Este hecho significa que en realidad M. tuberculosis tiene otros mecanismos patogénicos que no hemos sabido valorar, seguramente relacionados con la capacidad de generar una respuesta necrótica. Recientemente se ha podido demostrar que las micobacterias ambientales inducen una producción rápida de apoptosis, hecho vinculado con diferencias moleculares en uno de los principales componentes de la pared celular: el lipoarabinomanano, distinto del de M. tuberculosis, que sí es capaz de provocar necrosis celular44.
Mycobacterium marinum también genera lesiones muy similares a las inducidas por M. tuberculosis, tal como reflejan los extensos estudios realizados en el modelo de zebrafish45. Sin embargo, debido a que su temperatura de crecimiento óptimo está alrededor de los 30°C, la capacidad de generar patogénesis humana es limitada, centrándose en localizaciones con una temperatura similar, como la superficie cutánea, y siempre ligado a la disrupción del epitelio por causas físicas, que permite al bacilo la introducción en la dermis.
No secretoras de ESAT-6En este caso debemos citar el grupo extensísimo de M. avium-intracellulare, que surgió como un problema médico importante al ocasionar infecciones diseminadas en pacientes con sida42. Este grupo ya se conocía por su capacidad de generar linfadenopatías en niños de países nórdicos, como en el caso de M. malmoense46, o infecciones respiratorias en pacientes con bronquiectasias47, aspecto ligado seguramente a su capacidad para generar biofilms48. Este aspecto ha sido también especialmente importante tanto en micobacterias de rápido crecimiento como de lento para poder mantenerse en las conducciones de agua potable, cañerías, duchas, etc.49, y poder genera dosis infectivas suficientemente importantes como para poder contactar con diferentes epitelios humanos, sea cutáneo o respiratorio, y poder generar procesos infecciosos en personas con alguna deficiencia en la inmunidad innata o adquirida. O colonizar prótesis o dispositivos médicos.
Mención aparte merece M. ulcerans, un microorganismo cuyo hábitat natural son los ríos o los lagos, y que requiere una incubación a una temperatura menor de 32°C42. Pues bien, este bacilo es capaz de sintetizar una exotoxina, la micolactona, con efectos devastadores en el epitelio cutáneo. Esta toxina es capaz de inducir un efecto citotóxico en todas las estirpes celulares de la piel y suprimir la producción de citocinas, evitando entre otras la capacidad microbicida inducida por el interferón gamma en los macrófagos; la capacidad de maduración y de migración de las células dendríticas, así como su capacidad para estimular inmunidad celular. En los linfocitos inhibe la capacidad de generar IL-2, y la capacidad dependiente de antígeno de producir citocinas en linfocitos Th1, Th2 y Th17. De hecho, la micolactona también afecta en el proceso de atracción de los linfocitos, causando depleción de linfocitos T a nivel de los nódulos linfáticos periféricos50. Se trata pues del factor patogénico más importante detectado en una micobacteria ambiental.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.
A Júlia Gallardo Andrés, por las magníficas ilustraciones. A Paula Cardona, por repasar el manuscrito.

