




The evolution between Mycobacterium tuberculosis infection and active tuberculosis is multifactorial and involves different biological scales. The synthesis of ESAT-6 or the induction of alveolar macrophage necrosis are key, but to understand it, it is necessary to consider the dynamics of endogenous and exogenous reinfection, drainage of lung parenchyma and respiratory mechanics, local fibrosis processes and blood supply. Paradoxically, the immune response generated by the infection is highly protective (90%) against active tuberculosis, although as it is essentially based on the proliferation of Th1 lymphocytes, it cannot prevent reinfection. Severe immunosuppression can only explain 10% of active tuberculosis cases, while the remainder are attributable to comorbidities, a proinflammatory environment and an unknown genetic propensity. The pathogenic capacity of environmental mycobacteria is discrete, linked to deficits in the innate and acquired immune response. The ability to generate biofilms and the ability of M. ulcerans to generate the exotoxin mycolactone is remarkable.
La evolución entre la infección por Mycobacterium tuberculosis y la tuberculosis activa es multifactorial e implica diferentes escalas biológicas. La síntesis de ESAT-6 o la inducción de la necrosis de los macrófagos alveolares son claves, pero para entenderla se requiere tener en cuenta las dinámicas de reinfección endógena y exógena, el drenaje del parénquima pulmonar y la mecánica respiratoria, los procesos de fibrosis locales y la irrigación sanguínea. Paradójicamente, la respuesta inmune generada por la infección es altamente protectora (90%) contra la tuberculosis activa, aunque al basarse esencialmente en la proliferación de linfocitos Th1 no puede evitar la reinfección. La inmunosupresión severa tan solo puede explicar un 10% de los casos de tuberculosis activa, mientras que el resto es favorecido por comorbilidades, un ambiente proinflamatorio y una propensión genética desconocida. La capacidad patogénica de las micobacterias ambientales es discreta, ligada a déficits en la respuesta inmune innata y adquirida. Remarcable es la capacidad de generación de biofilms y la capacidad de M. ulcerans para generar la exotoxina micolactona.
Infected aerosols must be deposited on the pulmonary alveolus to be able to generate infection. In fact, this is one of keys of the success of Mycobacterium tuberculosis: its ability to infect the alveolar macrophage (AM). It is true that there are certain “protective” factors that can prevent its infective capacity. Firstly, the quality of the aerosol. Not all patients are able to generate a sufficient quantity of susceptible aerosol particles to be able to penetrate the alveolus.1 Secondly, the quality of the surfactant that prevents the collapse of the alveoli. The surfactant remains a surfactant and, as such, it has the ability to destroy the lipophilic wall of the mycobacterium, as it may be destroyed by the AM when it is phagocytised.2 In any case, it is not known to what extent this factor is important for evaluating the infectious dose. What is known is that there is close contact between patients that have not been infected who are constantly exposed to infectious aerosols and who have active pulmonary tuberculosis (APTB).3 It must be taken into account that the people most likely to suffer from active tuberculosis (ATB) are those that have been continuously in contact with a case of ATB, i.e. more than six hours a day for a period which depends on the diagnostic delay, and is somewhere between 60 and 90 days in countries with a good healthcare system.4 This means that to develop ATB, a single infection is not enough. A process of continuous reinfection is required.5
The alveolar space and alveolar macrophageThe physiological function of the pulmonary alveolus must always be taken into account to understand the essence of M. tuberculosis infection. The alveolus is a very delicate structure, made up of epithelial cells, type I pneumocytes, or flat alveolar cells, which make up 95% of the surface. They have a very low thickness to allow the diffusion of gases, which in turn have to cross the endothelial cells of the capillaries that cover the alveoli. In turn, these cells are firmly stuck to each other to prevent the entry of plasma. This fact is significant, since it allows a low surface tension to be maintained, thanks to the surfactant generated by the type II pneumocytes. However, there is a negative counterpart: it prevents the entry of antibodies. In the same way, each alveolus has its AM which is dedicated to constantly cleaning this space.6 It must be taken into account that the alveolus expands approximately every six seconds to allow the entry of external air, and with it all types of particles and pathogens. The function of the AM is to keep the alveolus clean to allow the exchange of gases and to prevent at all cost any inflammatory development that could break its delicate structure. The AM is a kind of “Mr Clean”, not “police”, dedicated to identifying pathogens in order to immediately generate an inflammatory response, as would be the case with Langerhans cells in the skin. This cleaning also includes the surfactant, converted into alveolar fluid, which not only serves to maintain surface tension but also cleans the alveolar space since it is constantly drained with the respiratory movement towards the bronchioles, the bronchial tree and the pharynx to be swallowed and directed towards the stomach. We drain approximately 500ml of alveolar fluid towards the stomach every day.
Necrosis of the alveolar macrophage as an essential virulence mechanismWhen the viable bacillus is phagocytised by the AM, it spreads its pathogenic capacity by secreting 6kDa early secretory antigenic target (ESAT-6). This peptide is essential to prevent the phagosome–lysosome union and apoptosis and eventually allows entry of the bacillus into the cytoplasm.7 In this way, the bacillus makes the most of its multiplication capacity in a single AM, between approximately 5–6 division cycles, to achieve a concentration of between 32 and 64 bacilli.8 This process develops over 5–6 days, considering that each division cycle in M. tuberculosis requires around 24hours, causing necrosis of the AM.9 Then, the bacilli become extracellular and are phagocytised by the AM from the interstitial space once more, which replaces the necrotised one, and by the AM of neighbouring alveoli, which are reached due to the constant drainage generated by the movement of inspiration/expiration. The process is repeated at least once more, generating up to 1000 bacilli, causing sufficient generation of chemokines by the infected AMs to produce an inflammatory response.
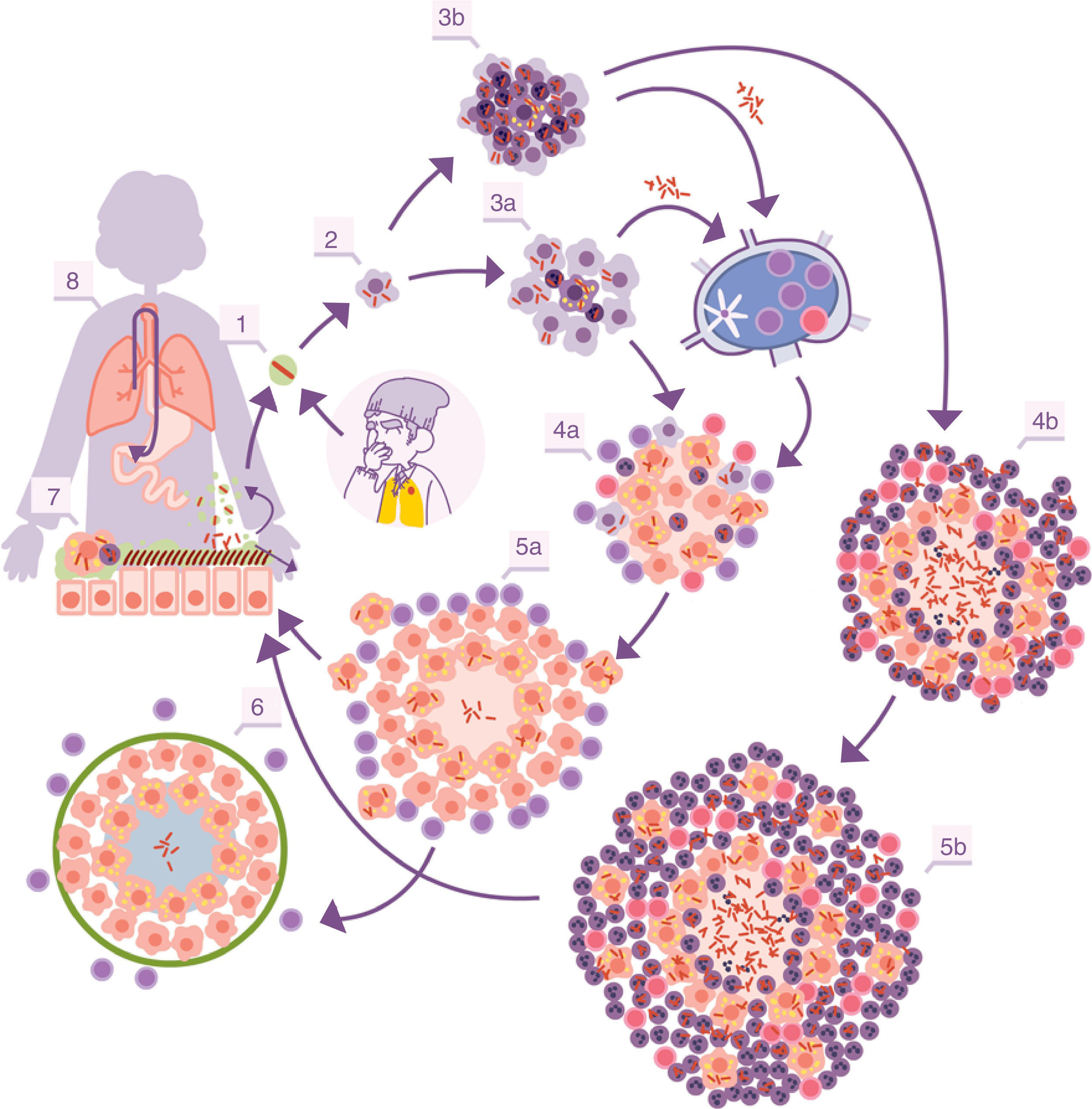
With the inflammation, the balance is broken when an exudate is generated in the capillaries which destroys the tightness of the alveolus and allows polymorphonuclear (PMN) cells to enter, normally neutrophils and monocytes, in proportions that will depend on the type of chemokines and cytokines secreted by the AMs. At the same time, it allows a more vigorous cleaning of the affected alveoli, draining into the lymph nodes through the afferent lymphatic capillaries. This is how M. tuberculosis first infects the macrophages of the nodules, generating lymphadenitis, and the dendritic cells (fig. 1).
 and subsequent multiplication inside. 3. Destruction of the AM, local dissemination of M. tuberculosis, phagocytosis by other AMs, and generation of a local inflammatory response dominated by monocytes (3a) or PMN cells (3b), thanks to which the bacilli can be drained into the regional lymph node, where Th1 or Th17 lymphocytes proliferate. 4. The lymphocytes are attracted by the inflammatory response of the lesions and activate the infected AMs or attract more PMN cells, depending on whether the immune response is Th1 (4a) or Th17 (4b), respectively. In the first case, there is a control of the bacillary population and the dormant bacilli are drained through the foam macrophages (5a) until it is controlled by encapsulation of the lesion (6). In the second, the lesions continue growing in size thanks to the entry of PMN cells and the extracellular bacillary growth in the NET, generating new peripheral lesions. In this case, the bacillary concentration is much higher and from here the drainage is much more significant, whether through the alveolar fluid or on a systemic level through neovascularisation of the granuloma (5b). In the lungs, the bacilli in the alveolar fluid (7) tend to be drained into the gastrointestinal tract (8), although they may form part of new aerosols, generating new lesions (1).")
Infectious cycle of M. tuberculosis. 1. Entry of bacilli into the pulmonary alveolus through an aerosol drop. 2. Phagocytosis by an alveolar macrophage (AM) and subsequent multiplication inside. 3. Destruction of the AM, local dissemination of M. tuberculosis, phagocytosis by other AMs, and generation of a local inflammatory response dominated by monocytes (3a) or PMN cells (3b), thanks to which the bacilli can be drained into the regional lymph node, where Th1 or Th17 lymphocytes proliferate. 4. The lymphocytes are attracted by the inflammatory response of the lesions and activate the infected AMs or attract more PMN cells, depending on whether the immune response is Th1 (4a) or Th17 (4b), respectively. In the first case, there is a control of the bacillary population and the dormant bacilli are drained through the foam macrophages (5a) until it is controlled by encapsulation of the lesion (6). In the second, the lesions continue growing in size thanks to the entry of PMN cells and the extracellular bacillary growth in the NET, generating new peripheral lesions. In this case, the bacillary concentration is much higher and from here the drainage is much more significant, whether through the alveolar fluid or on a systemic level through neovascularisation of the granuloma (5b). In the lungs, the bacilli in the alveolar fluid (7) tend to be drained into the gastrointestinal tract (8), although they may form part of new aerosols, generating new lesions (1).
The dendritic cells process M. tuberculosis and present epitopes that mostly correspond to the most abundant antigens secreted: ESAT-6 and the antigen 85 complex (Ag85 A, B or C). The latter is responsible for the construction of the cell wall, since it allows two essential molecules to be joined: arabinogalactan mycolate and trehalose dimycolate.11 The antigen presentation essentially stimulates the CD4 T cells, whose subtype depends on the type of chemokines and cytokines that transport the drained lymphatic fluid and which are essentially Th1, Th2, Th17 or Treg. CD8 T cells may also be generated on a smaller scale. Generally, the dominant subtype is Th1, responsible for generating interferon gamma, which allows the infected macrophages to be activated.
Although it is still not known for sure which immunological parameter is the determining one to evaluate the protective response against M. tuberculosis, what is clear is that it is protective and prevents the development of ATB in approximately 90% of cases. The Heimbeck studies in Ulleval Hospital in Oslo between 1924 and 1946 demonstrated that the nursing students that had a positive tuberculin test on admission had a very high protection against ATB in comparison with those that had a negative test. Specifically, in the first group there were 22 cases out of 668 (3.3%) and in the second group 97 out of 284 (34.2%), i.e. protection of more than 90%. In the case of students that had a negative tuberculin test and had been vaccinated with bacillus Calmette-Guérin, the incidence was 35 cases in 501 (6.9%), i.e. they were protected, but less than in the case of natural infection. The mortality also reflected this trend, with 0/668, 10/284 and 3/501 observed, respectively.12
The bacillus journey towards extrapulmonary disseminationNormally, the bacillus journey ends in the lymph node, but not necessarily. When lymphadenitis is caused, this may progress and release bacilli towards the efferent capillaries, which reach the vena cava and pass into the right atrium and ventricle to be once more transported towards the lungs. In this way, new infectious foci may be generated, especially if the bacilli are released en masse in the form of clumps, which obstruct capillaries, block circulation, destroy the tightness of the alveolar space and enter into it. They may also recolonise previously generated lesions which, as they are in a process of inflammation, have greater vascularisation and permeability. Finally, these bacilli may simply pass into the venous capillaries, reach the left atrium and ventricle, and disseminate systemically. The bacillus may potentially colonise any organ. This is linked to the characteristics of vascularisation. In some organs, the endothelial cells allow a greater permeability, such as bone tissue, especially in children when it is in the development phase, or the kidneys, and so these organs are common targets. In contrast, the meninges are much less commonly affected; mass entry of bacilli into the bloodstream is required, something which is associated with disseminated or active miliary TB, manifesting in the first few weeks after infection.13
An important point is that the lesions generated by M. tuberculosis develop new vessels which are more permeable and fragile than the structural ones, allowing both reinfection of the lesion and propagation of the bacillus towards the pulmonary venous capillaries.14
Another common dissemination route is the one generated by draining the bacillus itself through the alveolar fluid, which allows it to enter into the pharyngeal cavity and penetrate the mucous membrane, via small wounds, affecting the cervical lymph nodes; or causing intestinal ATB in the event that gastric acidification is not sufficient to destroy the bacillus. Furthermore, pleural ATB remains a variant of pulmonary TB. The mesenchymal cells of the pleural serosa are in charge of monitoring any small disturbance in the most superficial pulmonary parenchyma and if they detect any small lesion, they generate a mass influx of PMN cells and monocytes to isolate it and generate fibrous tissue around it.15
Extrapulmonary ATB represents approximately 30% of ATB cases and often indicates a delay in immune response, mostly affecting children below 5 years or people with immunodepression. However, there is a lot of geographical variability, something which may be interpreted as a possibility for there being a genetic factor that facilitates it.16
Control of lesions via immune response and local structuresThe “dynamic hypothesis”: process of endogenous reinfectionIn most cases, the proliferation of specific Th1 cells against the infection takes place in time to prevent the development of ATB. The Th1 cells are drained through the efferent vessels to be incorporated into pulmonary circulation and are mostly directed towards the infectious foci, given that the inflammation allows a greater probability of attraction. The Th1 cells come into contact with the infected macrophages and, through interferon gamma synthesis they activate them, allowing most of the bacilli to be destroyed. However, there is a small percentage that manage to enter into a metabolic slowing-down or dormancy phase, which allows them to survive by maintaining minimal metabolic activity. The activated macrophages, apart from maintaining these dormant bacilli inside, also phagocytise the remaining necrosed tissue to a point where it cannot metabolise the enormous load of fatty acids (especially cholesterol). This means that lipid bodies are produced, which accumulate within the cytoplasm and transform into foam cells.17 These cells end up being drained by the alveolar fluid and it is during this process that they may be destroyed. This releases the bacilli, which may form part of the aerosols that are constantly generated in the bronchioles and may reinfect the lungs (Fig. 1).
This process was the origin of the so-called “dynamic hypothesis” which involves latent tuberculosis infection (LTBI) as a constant endogenous reinfection process. Among other things, this hypothesis is able to explain why chemoprophylaxis with isoniazid works, taking into account that there is no action against dormant bacilli. The idea is that constant levels of isoniazid over 6–9 months allows the dormant bacilli to be drained into the gastrointestinal tract, preventing the risk of reinfection. The prolonged period is explained because it is the average time to be able to drain all the lesions before they fibrose and calcify, destroying the bacilli from the inside by generating multifactorial stress around them.18
Importance of interlobular septa in encapsulating lesionsThere is another local control mechanism that allows the lesion to be controlled: interlobular septa. These structures divide the pulmonary parenchyma in sections of approximately 1cm3 and serve to transmit the mechanical force generated by the diaphragm.19 In this way, the lungs can be inflated, allowing their expansion and preventing the delicate structure of the alveoli from being excessively strained due to the displacement required by respiratory movement. This structure is maintained by fibroblasts that have a special capacity for capturing surrounding mechanical variations, such as those generated with conformation of a lesion, even if it is very small such as that caused by initial infection due to M. tuberculosis (around 0.5mm).20 Therefore, the fibroblasts begin to weave a capsule around the lesion, which ends up isolating it in approximately 10 days.
It is interesting to highlight that infection due to M. tuberculosis generates a protective immune response.21 There are no mutation phenomena, nor are there big variations between the different lineages that have suffered different evolutionary contexts.22 The immune response is initially generated against several antigens but progressively focuses on vital antigens secreted for M. tuberculosis. This specialisation has a rationale. On the one hand, the bacillus in its active form is capable of causing damage to the host, thanks to its capacity to destroy macrophages. On the other hand, the ability of the bacillus to escape and cause reinfection after generating an immune response has already been described. Therefore, the focus against the bacilli in multiplication is a successful strategy, even more so taking into account that the dormant bacilli end up being drained into the gastrointestinal tract to eventually be expelled (Fig. 1).
Immunity based on lymphocytes explains the reactive nature of the immune responseThe other lesson to be learned is that due to the cellular nature of the immune response, it is always “after” the infection. This is completely the opposite of a humoral immune response, which in this case is not effective due to the lack of entry of antibodies into the alveolar space. Therefore, the host that has already generated a specific immune response gains a few days, due to memory cells which allow the bacillary population to be reduced. This is key for preventing excessive extrapulmonary dissemination and/or local bacillary concentration, needed for generating ATB.23 Even so, due to the fact that the multiplication of the bacillus is initially silent and does not generate an inflammatory response, these lymphocytes cannot be attracted in any way to the infectious focus. The conclusion from this information is that, in reality, infection and reinfection due to M. tuberculosis cannot be prevented.24 There is much desperation in the field of vaccinology in this area. In reality, it is impossible to achieve a prophylactic vaccine, as has been stated up to now.10 No vaccine is capable of preventing infection: the aim is to prevent ATB.
The origin of pulmonary tuberculosis. Size mattersClinical practice in diagnosing TBBearing in mind the reaction of the host, it is difficult to explain the development of APTB. Although infection seems inevitable, the consequences for the host are practically zero. Extraordinarily small lesions are generated, which do not cause any type of significant dysfunction. In fact, it is difficult to explain why a lesion that is 1mm in diameter can become a 10-mm lesion. Why 10mm? Because it is the size of the lesion that is required to be able to be detected via a chest X-ray scan.25
Years of combating tuberculosis have allowed effective clinical practice to be standardised. When ATB is suspected, a tuberculin test or interferon-gamma release assay should be performed to evaluate whether there has been an infection and if it has generated an immune response. If it is positive, a chest X-ray is performed. If a lesion is detected, it is considered that the patient has APTB and to be able to be detected, this lesion must be at least 10mm in size. Otherwise, it is considered that the patient has LTBI. In the event of a recent infection, it has been shown that the most significant risk of developing APTB is during the first two years, decreasing exponentially to zero after eight years.5
Importance of the size of the lesionThe question is: how can a 10-mm lesion develop from a 1-mm lesion, bearing in mind that the local control mechanisms work so well?
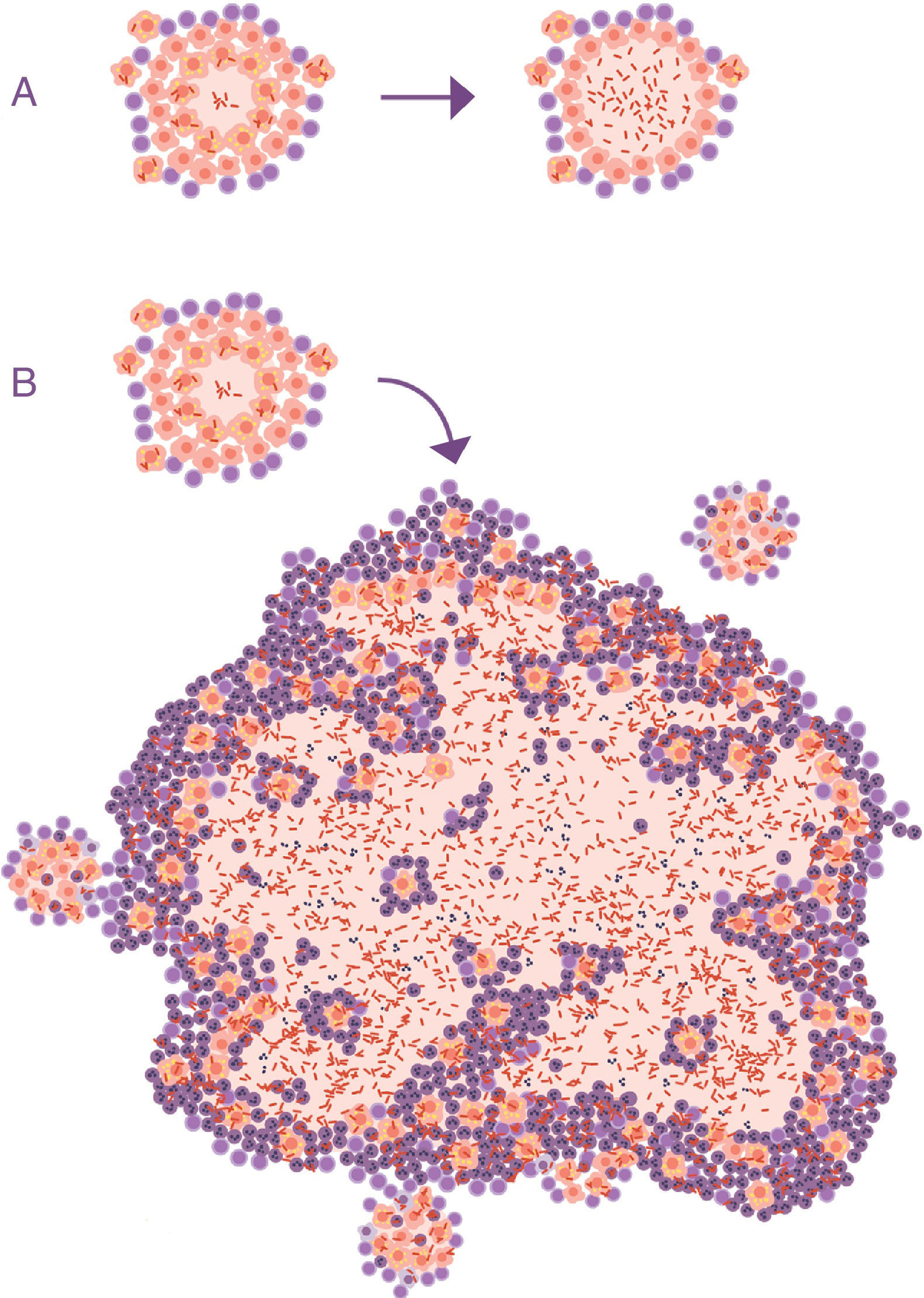
At this point, it is important to take into account the formulation of the question. In all the publications that study the progression of the tuberculosis infection to ATB, this issue is never raised. The questions focus on the reactivation capacity of the dormant bacilli or the development of some type of local immunodepression. No one considers what is actually seen in clinical practice: APTB is characterised by the presence of a visible lesion in the chest X-ray (fig. 2).
 to active tuberculosis (ATB). (A) Common representation; the difference is in the activity of the bacillus, omitting the fact that the main characteristic that clinically distinguishes LTBI from ATB is the size of the lesions (B).")
Progression of latent tuberculosis infection (LTBI) to active tuberculosis (ATB). (A) Common representation; the difference is in the activity of the bacillus, omitting the fact that the main characteristic that clinically distinguishes LTBI from ATB is the size of the lesions (B).
A characteristic aspect of the development of APTB in adults is the involvement of the upper pulmonary lobes. The theory with the most current success for explaining this phenomenon is that oxygen pressure is higher in this location, which allows M. tuberculosis to grow better. This theory, the “unitary theory”, is attractive due to its simple explanation, but in reality, it does not withstand systematic analysis.5 The reason for the accumulation of oxygen in this location is because the displacement of the pulmonary parenchyma due to respiratory movement is much less than at the base. This is due to the structure of the lung, which is held at its apex so that the expansion movement of the diaphragm can effectively generate the expansion of the parenchyma. Furthermore, the force of gravity causes the interlobular septa of the upper lung to be constantly tensed and have less reactivity than those situated at the base. The lack of movement also causes a deficiency in vascular drainage, which is why there is a lower exchange of gases leading to hyperoxygenation. However, despite this increase favouring an acceleration in the capacity of the bacillus to multiply, a fact that is doubtful from a microbiological standpoint, it does not explain the disproportionate growth of the lesions.26
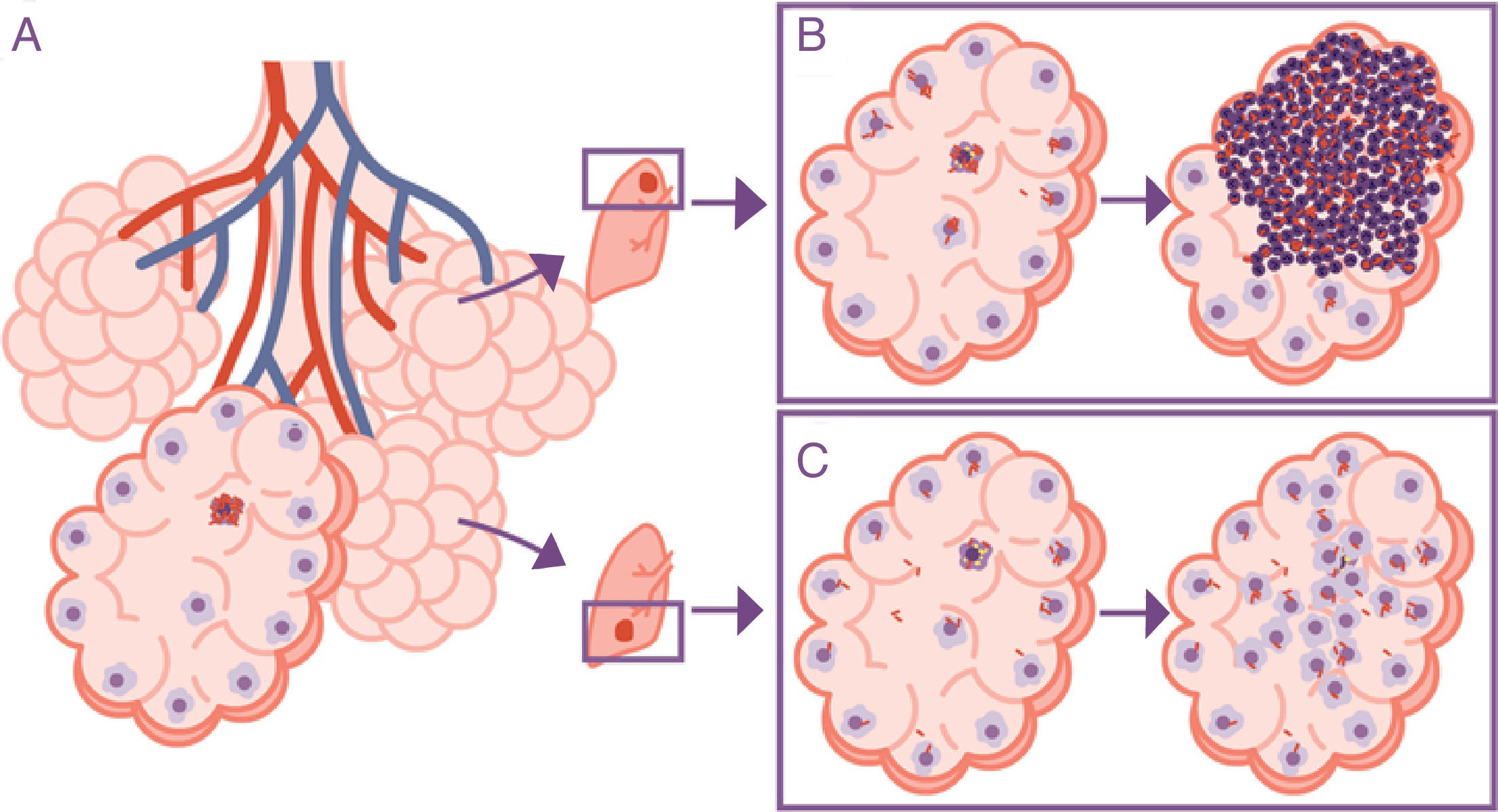
The response is in alveolar drainageWhat is really important is the reduction in draining the interior of the alveolus itself, since this causes an accumulation of extracellular bacilli locally. This means that the AM which has to phagocytise them faces a high bacillary concentration, causing more cellular destruction and a response from chemokines and cytokines that facilitates the intensive accumulation of PMN cells6 (Fig. 3).
 but the progression is different in the upper lobe, where the accumulation of bacilli means that the alveolar macrophages (AMs) must deal with a higher quantity of bacilli, generating a response preferentially based on the accumulation of PMN cells (B). In contrast, in the lower lobe, more significant drainage allows a greater distribution of bacilli and an inflammatory response based on the accumulation of AM and monocytes (C).")
Importance of the location of the infection in the upper lobes. The infection takes place in a pulmonary alveolus (A) but the progression is different in the upper lobe, where the accumulation of bacilli means that the alveolar macrophages (AMs) must deal with a higher quantity of bacilli, generating a response preferentially based on the accumulation of PMN cells (B). In contrast, in the lower lobe, more significant drainage allows a greater distribution of bacilli and an inflammatory response based on the accumulation of AM and monocytes (C).
During the modern phase of TB study, which starts with the declaration of a global emergency in 1993, PMN cells were considered secondary actors in pathogenesis, when they were not simply ignored. However, after reviewing the experiences of pathologists that studied APTB in the pre-antibiotic era, it is found that two types of lesions were described: proliferative (or “tubercle”), which contains a small bacillary population composed of epithelioid cells and fibroblasts that progress towards fibrosis and calcification; and exudative tuberculosis or local neutrophilic condensation, which contains a high bacillary population and is related to the development of APTB.27
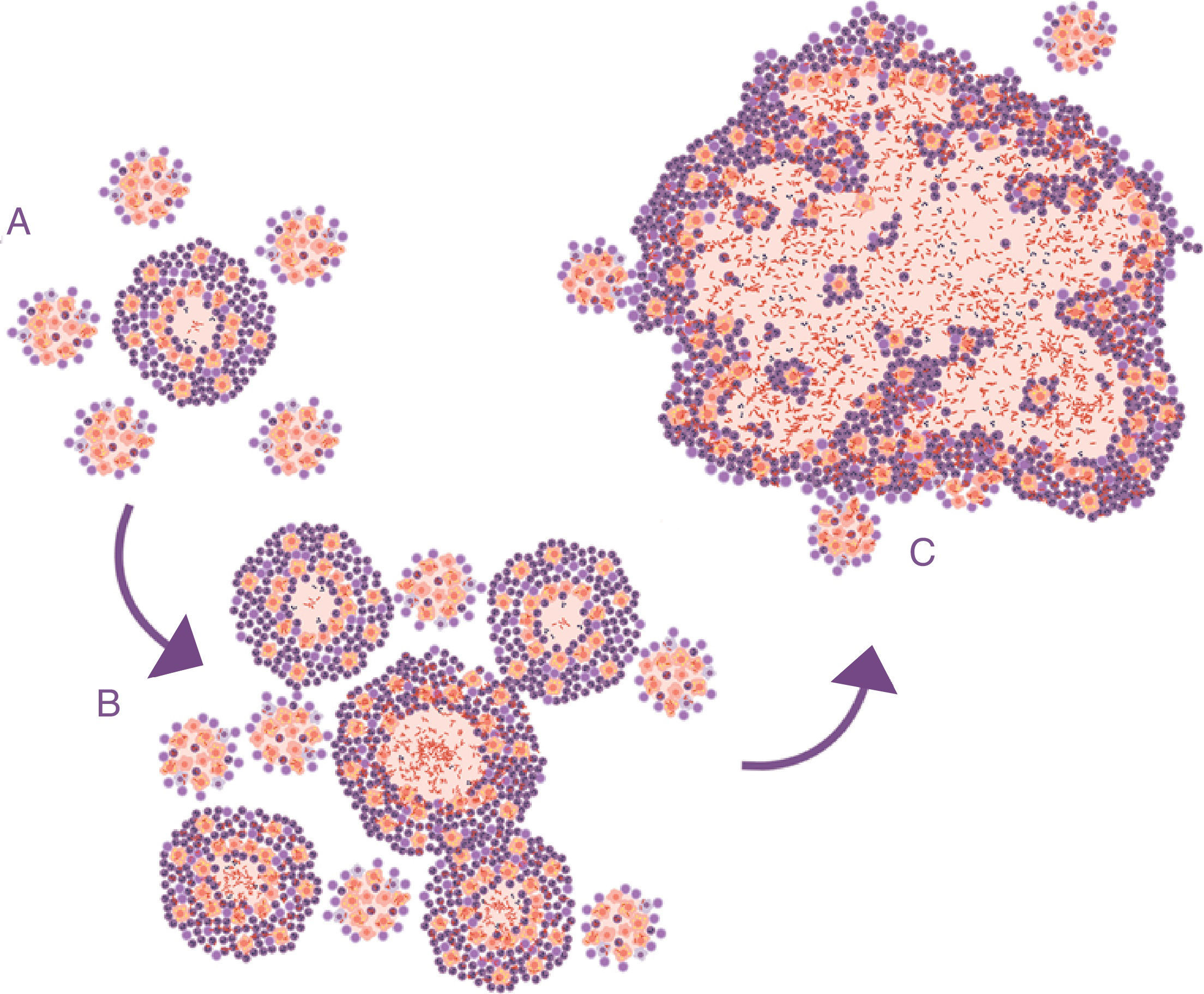
Lessons learned from the ATB model in mice. The soap bubbles modelThe development of the experimental model using mice with the C3HeB/FeJ strain has allowed a very significant leap forward in understanding progression from LTBI to APTB. In this model, it has been possible to demonstrate that M. tuberculosis strengthens the formation of neutrophil extracellular traps, a mechanism developed by the PMN cells to deal with infections caused by extracellular pathogens. It consists of cell lysis to release enzymes contained in the lysosomes, which in turn traps the pathogens via tissue formation with nuclear DNA. However, in the case of M. tuberculosis, they do not manage to destroy it. In contrast, the neutrophil extracellular traps make a platform for extracellular growth of the bacillus.28 In this way, it causes exponential growth in the size of the lesion, which involves the generation of surrounding satellite (or “daughter”) lesions and a final fusion process between all of them over a time period of between 10 and 15 days. This prevents the protective encapsulation mechanism and it is capable of generating APTB in the form of alveolar infiltration by PMN cells that finally liquefies and cavitates. The previously mentioned constant reinfection from contact with cases of APTB clearly helps this phenomenon. In the case of reinfection, it is located adjacently, the same as constant endogenous reinfection, as a consequence of draining dormant bacilli from previous lesions (Fig. 1) and those that come from haematogenous dissemination that manage to pass the filter of the lymph nodes. The evolution of this process is very similar to the dynamic of the formation of soap bubbles. That is why a parallel has been made and it has been possible to show that the induction of this type of lesion depends on three factors: rapid growth of the initial lesion (as with PMN infiltration), the formation of surrounding “daughter lesions” and the fusion of all of them29 (Fig. 4).
, minimal lesions are generated which grow rapidly thanks to the accumulation of PMN cells and the extracellular growth of the bacilli. Subsequently, “daughter” lesions are generated which undergo the same process (B) and eventually they all fuse into a big lesion, evading the encapsulation process (C).")
Progression of latent tuberculosis infection lesions to pulmonary tuberculosis. The soap bubbles model. In the initial infection (A), minimal lesions are generated which grow rapidly thanks to the accumulation of PMN cells and the extracellular growth of the bacilli. Subsequently, “daughter” lesions are generated which undergo the same process (B) and eventually they all fuse into a big lesion, evading the encapsulation process (C).
What would be the difference between a pulmonary abscess and APTB? The answer is time. Pulmonary abscesses have a very rapid progression, generated by pathogens with a much quicker multiplication capacity (one division every 20min) than that of M. tuberculosis (one division every 24h). This means that the destruction of the alveolar walls takes place quickly in the case of an abscess but not in the case of APTB, so that the detritus of the PMN cells remain confined in each alveolus. When the pathologist divides the lesion, the necrotic area has a caseous texture and, therefore, it is called “caseum”.30 However, over several days the alveolar walls finally degenerate which allows all the affected alveoli to converge, generating the so-called liquefaction. It is interesting to take into account this paradigm change. M. tuberculosis becomes an extracellular pathogen before previously thought. Traditionally, it was believed that the extracellular phase started with liquefaction and was strengthened by incoming air in the case of causing erosion of the bronchus and subsequent drainage.30 Nowadays, we can state that this is not what happens and that extracellular growth constitutes the mechanism through which there is a progression from LTBI to APTB.
Primary or post-primary ATB?Generally, primary APTB is considered to affect the basal lobes and there is visible involvement of the hilar lymph nodes, whereas post-primary or adult APTB affects the upper lobes. This concept comes from APTB detection campaigns via chest X-ray that were performed after the Second World War. At that time, when the incidence of APTB and LTBI were very significant, they began to observe calcified lesions in the lower lobe and lymph nodes in adults, whereas APTB manifested in the upper lobes. In 1967, the “unitary theory” was announced, which gave a global sense to those observations, taking into account that it was believed that once a person was infected, they could not be reinfected.31 The interpretation was that lesions in the upper lungs that had remained dormant and had subsequently been reactivated had arisen from old lesions in the pre-immune phase. It has now been shown that this is not true. Firstly, although infection gives good protection against ATB, it does not prevent reinfection. Secondly, molecular epidemiological studies have shown that the location of APTB depends on the host's immune response.32 In the case of children under five years or immunosuppressed adults, the proliferation of the bacillus does not require the advantage given by location in the upper lung, and so it may develop in both areas. In the case of immunocompetent adults, it generally develops in the upper lung due to the advantage from the lack of drainage and the induction of an inflammatory response dominated by PMN cells. One of the elements that is often not taken into account in ATB developed in patients that contracted ATB in the pre-antibiotic era, i.e. before the 1950s, is that very significant lesions were often resolved via classic methods, either with “rest cures”, pneumothorax or thoracoplasty, which aimed to control lesions through fibrosis. In all these cases, eradication of the bacillary population was very different but generally ineffective. Hence, in this case the possibility of generating ATB, especially when reaching old age, was much higher than in the case where LTBI was detected and, therefore, no lesion was observed in the chest X-ray.
Conclusion: why do 10% of those infected develop ATB?This is the big question! On the one hand, the most important antigens, whose expression is very stable, have been identified; on the other hand, the nature of the cell immune response does not prevent infection but it does have good efficacy at preventing ATB. In this sense, the onset of AIDS has helped to understand the importance of CD4 lymphocytes. However, it only explains 10% of ATB cases.33 Bacillus circulations through pulmonary drainage or the bloodstream, respiratory movements, the encapsulation capacity of the interlobular septa, together with the capacity of external aerosols to effectively cause infections cause great complexity that inhibits finding markers with a sufficient predictive value. What is clear is that there are comorbidity factors that facilitate the development of ATB. Tobacco use34 or domestic pollution through the use of solid fuels35 can be clearly related to a deficient capacity in pulmonary drainage. However, there is a factor that appears to be very important, which is those people that have a proinflammatory environment and therefore facilitate PMN cell attraction to lesions. This factor may explain why ATB affects more males36 and patients with type 2 diabetes mellitus.37 In fact, in most cases, the induction of ATB is included within the conceptual framework of the “damage theory”,38 by which the induction of ATB may be due to immunodepression but is mostly due to an excessive response from the host, in this case in the form of PMN infiltrates.
Historically, there is a lot of evidence for familial susceptibility to ATB. However, up to now no replicable specific association has been recognised.39 Finally, it must always be taken into account that patients that have had APTB and have been cured with standard treatment without suffering a relapse have a greater probability of having another episode of APTB through exogenous reinfection. This may be due to two factors: a susceptibility due to the same genetic factor or factors that already facilitated the induction of the first APTB and/or local sequelae caused by the first episode.40
Environmental mycobacteriosisEnvironmental mycobacteria have a lot of interest to be able to verify TB pathogenesis. They are microorganisms that live in the natural world and that share many aspects with M. tuberculosis, not only in terms of the characteristics of the cell wall, rich in mycolic acids, but also by sharing different antigens capable of generating protective responses.41 However, on a pathogenic level they have barely been studied and it is difficult to draw conclusions. Clinical interest has arisen, firstly, with pulmonary disease generated in elderly populations,42 causing a disease very similar to APTB in patients that are negative for HIV; secondly, in patients that are positive for HIV, demonstrating the value of the Th1-type immune response for preventing disseminated infections.43 In the same way, these mycobacteria are capable of generating lymphadenopathy and infections in the skin, bone or joints.42 It should be underlined that one of its peculiarities is that, in general, its optimum growth temperature is around 30°C. This competitive disadvantage limits its pathogenic activity to superficial epithelia where the temperature may be close to this value.
Since the secretion of ESAT-6 seems to be one of the key factors in the pathogenesis of M. tuberculosis, the environmental mycobacteria may be classified in relation to this aspect.
ESAT-6 or ESAT-6-like secreting speciesMycobacterium kansasii is capable of generating lesions similar to APTB in individuals exposed to aerosols in water with a high concentration of this bacillus. The incidence of pulmonary disease due to M. kansasii is very low. The explanation for this is probably due to the fact that in reality, a very high infectious dose is required. This means that in reality M. tuberculosis has other pathogenic mechanisms that have not been known to be evaluated, surely related to the capacity to generate a necrotic response. It has recently been possible to demonstrate that environmental mycobacteria induce a rapid production of apoptosis, a fact that is linked to molecular differences in one of the main components of the cell wall: lipoarabinomannan, different from M. tuberculosis, which is capable of causing cell necrosis.44
Mycobacterium marinum also generates very similar lesions to those induced by M. tuberculosis, as reflected in extensive studies performed on the zebrafish model.45 However, due to the fact that its optimum growth temperature is around 30°C, its capacity to generate human pathogenesis is limited and is focused on locations with a similar temperature, such as the surface of the skin, and is always linked to epithelium disruption due to physical causes that allow the bacillus to be introduced into the skin.
Non-ESAT-6-secretingIn this case, the very extensive group of M. avium-intracellulare must be mentioned, which emerged as a significant medical problem by causing disseminated infections in patients with AIDS.42 This group was already known for its capacity to generate lymphadenopathy in children in Nordic countries, such as in the case of Mycobacterium malmoense,46 or respiratory infections in patients with bronchiectasis,47 an aspect surely linked to its capacity to generate biofilms.48 This aspect has also been especially important in both fast- and slow-growth mycobacteria in order to be able to support itself in drinking water pipes, plumbing, showers, etc.,49 and to be able to generate sufficiently significant infectious doses to enter into contact with different human epithelia, whether cutaneous or respiratory, and to be able to generate infectious processes in people with an innate or acquired immunity deficiency. Or to colonise prosthesis or medical devices.
M. ulcerans also deserves to be mentioned, a microorganism whose natural habitat is rivers or lakes and which requires incubation at a temperature below 32°C.42 This bacillus is also capable of synthesising an exotoxin, mycolactone, with devastating effects on the cutaneous epithelium. This toxin is capable of inducing a cytotoxic effect on all the skin's cell lines and suppressing the production of cytokines, preventing among other things the microbicidal capacity induced by the interferon gamma in the macrophages; the capacity for maturation and migration of dendritic cells, as well as its capacity to stimulate cell immunity. In lymphocytes, it inhibits the capacity to generate IL-2 and the antigen-dependent capacity to produce cytokines in Th1, Th2 and Th17 lymphocytes. In fact, mycolactone also affects the process of attracting lymphocytes, causing a depletion of T-cells in the peripheral lymph nodes.50 It is therefore the most important pathogenic factor detected in a mycobacteria environment.
Conflicts of interestThe author declares that there are no conflicts of interest.
To Júlia Gallardo Andrés, for her magnificent illustrations. To Paula Cardona, for reviewing the manuscript.
Please cite this article as: Cardona P-J. Patogénesis de la tuberculosis y otras micobacteriosis. Enferm Infecc Microbiol Clin. 2018;36:38–46.
