





La diarrea por Clostridioides difficile es un importante problema de salud pública, cuyo tratamiento es complejo. La transferencia de microbiota fecal (TMF) se postula como una terapia útil para prevenir recidivas.
Material y métodosSe analizaron seis muestras fecales, una procedente del donante y cinco del paciente antes y después de la TMF. Se amplificó y secuenció el gen 16Sr mediante secuenciación masiva y se estudió la diversidad y composición taxonómica.
ResultadosLa diversidad aumentó en las muestras post-TMF, y se identificaron dos clústeres, uno formado por las muestras no patológicas (donante y paciente post-TMF), y otro por la muestra patológica. Los resultados obtenidos a través Qiime2 y Bioconductor fueron similares.
ConclusiónEl análisis realizado demostró un incremento en la diversidad taxonómica del paciente tras la TMF, sugiriendo su utilidad. Además, los resultados obtenidos con Qiime2 y Bioconductor reflejaron la importancia de unificar los análisis bioinformáticos.
Clostridioides difficile infection (CDI) has become a global healthcare challenge due to increases in its incidence and mortality rates. Faecal microbiota transfer (FMT) is postulated as a protocol to prevent CDI recurrence.
Material and methodsA donor faecal sample and patient faecal samples (pre-FMT and post-FMT) were analysed. The r16S gene was amplified and sequenced by NGS, and its diversity and taxonomy composition were examined.
ResultsMicrobial richness increased in post-FMT samples, and the β diversity studies grouped the samples into two clusters. One included the non-pathological samples (donor and pre-FMT samples), and the other included the pathological sample. The results obtained by Qiime2 and Bioconductor were similar.
ConclusionThe analysis showed an increase in taxonomic diversity after the FMT, which suggests its usefulness. Moreover, these results showed that standardisation of bioinformatics analysis is key.
La incidencia de infección por Clostridioides difficile (ICD) ha experimentado un gran incremento en las últimas dos décadas, y constituye un problema de salud global, debido tanto a su incidencia como a su morbimortalidad. La ICD es la causa principal de diarrea nosocomial de origen infeccioso asociada a la administración de antibióticos, conlleva un aumento de la mortalidad hospitalaria, y genera un importante gasto sanitario1.
El tratamiento de elección para la ICD es la terapia antibiótica, que es eficaz en aproximadamente el 85% de los casos. No obstante, la incidencia de recidivas es elevada, alrededor de un 30%, además este porcentaje se incrementa hasta el 45% tras el primer episodio, y en pacientes con múltiples recurrencias puede llegar a 75%2. Son varios los factores que se han asociado con un aumento del riesgo de recurrencias, entre ellos la necesidad de mantener un tratamiento antibiótico de amplio espectro, la edad avanzada o haber padecido un episodio previo de ICD3. Todos estos factores influyen sobre la microbiota propia del paciente, y por eso se está postulando que la causa subyacente de las recidivas es la disbiosis que se produce debido a la colonización por Clostridioides difficile (CD). Por ello, la TMF después del tratamiento antibiótico se presenta como un método eficaz frente a la prevención de las recidivas en pacientes con esta patología4.
Por otra parte, existen diferentes herramientas informáticas disponibles para llevar a cabo los análisis de microbiota, como Mothur5, Qiime26 o Bioconductor7. Se han realizado algunos estudios que comparan los resultados obtenidos con Mothur y Qiime2 sin encontrar diferencias significativas entre ellos8. No obstante, no se han publicado trabajos en los que se comparen estos métodos con los resultados obtenidos utilizando Bioconductor.
Material y métodosMuestras: Se analizó el excedente de seis muestras de heces, una del receptor pre-transferencia considerada patológica, y cinco no patológicas (del donante, y del receptor a los 7, 30, 90 y 180 días tras la intervención). El receptor era un paciente que había tenido seis recidivas de la ICD. Todos los procedimientos se han desarrollado siguiendo los estándares nacionales éticos y legales, y de acuerdo con las directrices establecidas en la declaración de Helsinki (2000).
Procedimiento de TMF: Se introdujo un concentrado de microbiota fecal de un donante sano mediante endoscopia. La selección del donante se realizó tras un estudio que incluyó un cuestionario clínico y estudios microbiológicos de muestras de suero, heces y exudado nasal. Se descartó la presencia de múltiples patógenos mediante cultivo y técnicas moleculares, según se propone en diversas guías clínicas9.
Estudio del microbioma mediante secuenciación masiva (NGS): El estudio de la microbiota se llevó a cabo siguiendo el protocolo recomendado por Illumina10. En el análisis bioinformático se utilizaron los programas Qiime26 y Bioconductor7.
Se estudió la diversidad α calculando los índices de Shannon y Chao1, y la diversidad β a través de la estimación de la distancia Unifrac unweighted. La taxonomía se asignó utilizando la base de datos SILVA (Release 132)11. Los taxones diferenciales se identificaron a través de ANCOM-Composition y Gneiss en Qiime2, DESeq2 y EdgeR en Bioconductor, y LefSe mediante el servidor Galaxy.
El análisis comparativo de los resultados con Qiime2 y Bioconductor se llevó a cabo comparando el número total de detección de taxones (OTU's) detectados por ambos programas, y el número de secuencias no identificadas. Se utilizó la prueba estadística t de Student para las variables paramétricas, y la prueba U de Mann-Withney para las variables no paramétricas.
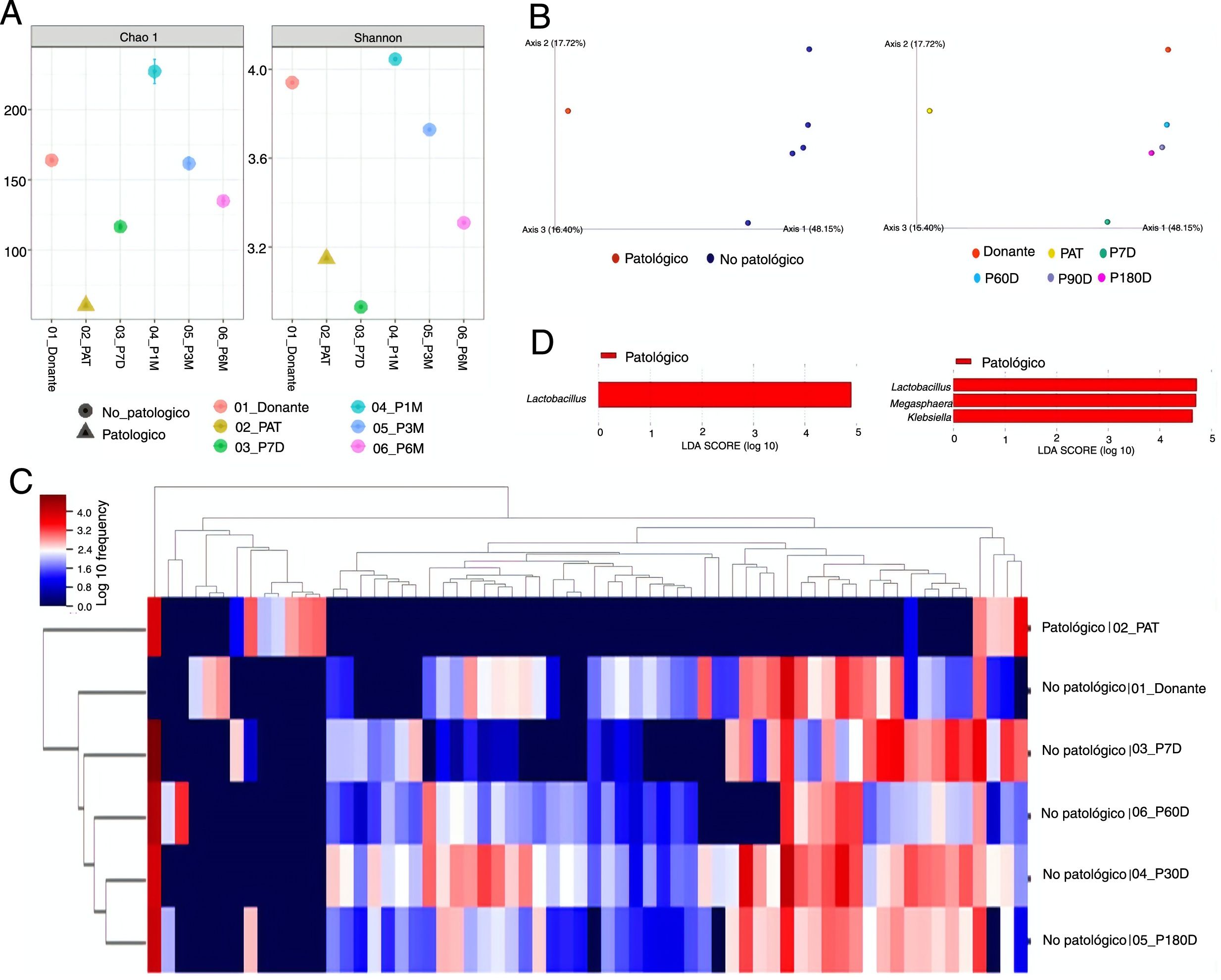
ResultadosAnálisis de la diversidadLos índices de Chao1 y Shannon más bajos fueron los obtenidos para las muestras pre-TMF, y a los 7 días post-TMF tanto con Qiime2 como con Bioconductor, por lo que se pudo afirmar que fueron las muestras que presentaron una menor diversidad α. La muestra obtenida a los 30 días post-TMF fue la que presentó una mayor diversidad α (fig. 1 A).
 Análisis de diversidad α. Índice de Chao1 (izquierda) y Shannon (derecha) obtenidos con el programa Bioconductor. (B) Análisis de diversidad β. Representación del resultado del cálculo de la distancia Unifrac unweighted, y el posterior análisis de componentes principales con Qiime2. (C) Heatmap obtenido con Qiime2. Horizontalmente se encuentran los datos de las muestras. Verticalmente la información referente a los géneros. (D) Géneros diferencialmente encontrados utilizando el algoritmo LefSe. La imagen de la izquierda corresponde al realizado con datos de Qiime2, y la de la derecha al realizado con datos de Bioconductor. Donante: muestra obtenida del donante; AT: muestra obtenida del paciente pre-TMF; P7D, P30D, P90D y P180D: muestras obtenidas del paciente post-TMF a los 7,30,90 y 180 días.")
(A) Análisis de diversidad α. Índice de Chao1 (izquierda) y Shannon (derecha) obtenidos con el programa Bioconductor. (B) Análisis de diversidad β. Representación del resultado del cálculo de la distancia Unifrac unweighted, y el posterior análisis de componentes principales con Qiime2. (C) Heatmap obtenido con Qiime2. Horizontalmente se encuentran los datos de las muestras. Verticalmente la información referente a los géneros. (D) Géneros diferencialmente encontrados utilizando el algoritmo LefSe. La imagen de la izquierda corresponde al realizado con datos de Qiime2, y la de la derecha al realizado con datos de Bioconductor. Donante: muestra obtenida del donante; AT: muestra obtenida del paciente pre-TMF; P7D, P30D, P90D y P180D: muestras obtenidas del paciente post-TMF a los 7,30,90 y 180 días.
Los resultados de diversidad β agruparon las muestras en dos grandes clústeres, separando las muestras no patológicas (donante y muestras post-TMF) de la muestra patológica (pre-TMF). Las muestras que más similitud presentaron fueron las del donante, y del paciente a los tiempos largos tras la transferencia, estando muy separadas de la muestra pre-TMF y de la muestra obtenida siete días post-TMF (fig. 1 B).
Análisis de composición taxonómicaEl análisis de composición taxonómica reveló que las muestras no patológicas fueron más parecidas entre sí, y diferentes de la muestra del paciente pre-TMF. Se observó la existencia de grupos de microorganismos que caracterizaron tanto las muestras patológicas como las no patológicas (fig. 1 C).
Los géneros Lachnospira, Butyricimonas, Paraprevotella, Odoribacter y Anaerostipes fueron descritos como taxones asociados al estado no patológico por el algoritmo DESeq. Asimismo, los algoritmos ANCOM y Gneiss identificaron algunos de estos géneros como asociados a dicho estado. Del resto de taxones señalados como diferencialmente encontrados entre las muestras estudiadas, siete de ellos fueron detectados por al menos tres algoritmos: Blautia, Bacteroides y Alistipes asociados a las muestras no patológicas, y Klebsiella, Veillonella, Megasphaera y Lactobacillus al estado patológico (tabla 1).
Taxones diferencialmente encontrados utilizando los algoritmos ANCOM, Gneiss, DESeq, EdgeR y LefSe. En verde se marcan los asociados al estado no patológico y en rojo los asociados al estado patológico
| Taxones | ANCOM | Gneiss | DESeq | EdgeR | LEfSe |
|---|---|---|---|---|---|
| Blautia | x | x | x | ||
| Bacteroides | x | x | x | ||
| Alistipes | x | x | x | ||
| Klebsiella | x | x | x | x | x |
| Veillonella | x | x | x | x | |
| Megasphaera | x | x | x | x | x |
| Lactobacillus | x | x | x |
Los resultados obtenidos en el análisis de diversidad y taxonomía tanto con Qiime2 y Bioconductor fueron similares. No obstante, para comprobar si existían diferencias en los OTU's, o en la asignación taxonómica se llevó a cabo una comparación evaluando la diferencia entre los OTU's totales observados, y la cantidad de secuencias no identificadas por cada método. La media de OTU's totales observados en Qiime2 fue de 140, y en Bioconductor de 141 (valor p = 0,97), por lo que se pudo afirmar que no existían diferencias significativas.
Con relación al porcentaje de secuencias no identificadas, la media para el programa Qiime2 fue de 9,96%, y para Bioconductor de 7,91%. En este caso, se observó que el porcentaje obtenido fue ligeramente superior con el programa Qiime2, aunque se obtuvo un valor p = 0,197, por lo que se estableció que no existían diferencias en la asignación taxonómica.
En cuanto a la comparativa de los algoritmos utilizados para la identificación de los taxones diferencialmente encontrados, el más restrictivo fue LefSe, que sólo detectó un taxón diferencial (Lactobacillus) con los datos del Qiime2, y tres (Lactobacillus, Megasphaera y Klebsiella) con los datos de Bioconductor (fig. 1 D). El menos restrictivo fue DESeq que identificó un total de 34 géneros asociados a uno u otro estado. El algoritmo Gneiss fue el que detectó un número mayor de taxones, que a su vez fueron detectados también por otros algoritmos (7/11, 63,6%). Contrariamente, el algoritmo DESeq fue el que señaló un mayor número de taxones no coincidentes con otros algoritmos, un total de 28 de 34 (82,3%) (tabla 1).
DiscusiónEl mayor factor de riesgo en el desarrollo de ICD es la administración de antibióticos, lo que provoca una alteración de la composición de la comunidad bacteriana intestinal aumentando la susceptibilidad del huésped al patógeno12. Independientemente del sistema de análisis, nuestros resultados mostraron que la microbiota del paciente antes del proceso de transferencia de microbiota es diferente a la del donante, considerada como una microbiota sana, tanto en su diversidad como en su composición taxonómica.
La microbiota sana contiene trillones de bacterias, principalmente Bacteroidetes, Firmicutes, Actinobacteria y Proteobacteria13, y es una barrera natural que previene la ICD. Los cambios en la composición bacteriana de la microbiota tras el TFM engloban la aparición de diferentes géneros, como Lachnospira, Butyricimonas, Paraprevotella, Odoribacter y Anaerostipes, que no se encuentran en el microbioma del paciente pre-transferencia debido a la disbiosis que genera la colonización masiva del colon por CD14. Estos cambios también se observaron en las muestras estudiadas en este trabajo, donde, de forma progresiva, fueron identificándose estos géneros, que estaban ausentes en la muestra tomada del paciente antes de la TMF.
Por otra parte, los resultados obtenidos tanto con Qiime2 como con Bionconductor son similares, y por lo tanto ambos pueden ser usados sin esperar variaciones en la detección de taxones asociada al análisis bioinformático. Este tipo de estudios es esencial para establecer un flujo de trabajo común en el análisis de estos datos, para que los resultados obtenidos sean equiparables independientemente del lugar donde se obtengan. Tras la utilización de ambos programas, siendo Qiime2 una herramienta muy potente y completa, Bioconductor permitió una mayor adaptabilidad de los «pipelines» a diferentes situaciones, así como una mayor versatilidad debido a la existencia de multitud de paquetes. Por ello, aunque ambas opciones son igualmente válidas, se podría sugerir el uso de Bioconductor.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesTodos los autores declaran no tener conflictos de intereses.

