




Neisseria meningitidis is associated with invasive infections causing high mortality rates. The objective of this study was to describe the population structure of Colombian invasive isolates with ST-9493, a potentially emerging clonal group in the country.
MethodsThe complete genomes of 34 invasive isolates of serogroup B with ST-9493 and its variants at one or two loci were sequenced by Illumina to describe the phenotypic and genotypic characteristics of these isolates.
ResultsThe relationship of a clonal group associated with ST-136 CC41/44 was phylogenetically established, identifying two main clades composed of isolates from an outbreak or endemic. The most frequent alleles and peptides included porA 17, porB 44, fHbp 2.24, NHBA 10, and the FetA F5-17 variant. Most of the isolates were susceptible to the antibiotics evaluated.
ConclusionThis study shows that meningococcal isolates with ST-9493 are an autochthonous clonal group with population dynamics and the capacity to cause endemic and epidemic meningococcal disease in Colombia.
Neisseria meningitidis se asocia con infecciones invasivas que causan altas tasas de mortalidad. El objetivo de este estudio fue describir la estructura poblacional de aislamientos invasivos colombianos con ST-9493, un grupo clonal potencialmente emergente en el país.
MétodosSe secuenció por Illumina el genoma completo de 34 aislamientos invasivos, serogrupo B con ST-9493 y sus variantes en uno o doble locus para describir sus características fenotípicas y genotípicas.
ResultadosSe estableció filogenéticamente la relación como grupo clonal asociado con el ST-136 CC41/44, identificando 2 clados principales conformados por aislamientos de un brote o endémicos. Los alelos y péptidos más frecuentes fueron porA 17 y porB 44; fHbp 2.24, NHBA 10 y la variante FetA F5-17. La mayoría de los aislamientos fueron susceptibles a los antibióticos evaluados.
ConclusiónEste estudio muestra que los aislamientos meningocócicos con ST-9493 son un grupo clonal autóctono con dinámica poblacional y capacidad de causar enfermedad meningocócica endémica y epidémica en Colombia.
Neisseria meningitidis serogroup B is a cause of invasive meningococcal disease (IMD), such as meningitis and septicemia.1 The isolates are genetically diverse, and several genotypes have been associated with successful clonal complexes (CC11, CC41/44 and CC32) that have persisted in time and new clones can eventually emerge, become effective clones and lead to an increase in the incidence of IMD.1
After the introduction of conjugate vaccines, serogroup B of N. meningitidis has become the most frequent serogroup associated with invasive disease in European countries, North America and South America.2 Currently, two vaccines have been produced to prevent meningococcal disease caused by N. meningitidis serogroup B: 4CMenB (Bexsero) and Trumenba (Pfizer).
In Colombia, passive and voluntary surveillance of N. meningitidis has been performed since 1987. Until 2014, serogroup B isolates were the most frequent, but this serogroup was replaced by serogroup C isolates.3 Serogroup B ST-9493 was identified in isolates from patients in an outbreak of IMD in Cartagena city, Colombia and was related to ST-41/44 CC.4 The aim of this study was to describe the geographic distribution and the genomic characteristics of the N. meningitidis ST-9493 isolates, recovered during 2013–2016 that may constitute a potentially emerging clonal group that could be the cause of endemic IMD in the country.
MethodsIsolate collectionThirty-four N. meningitidis isolates recovered between 2013 and 2016 from meningococcal disease patients were analyzed. The isolates were received at the Colombian Instituto Nacional de Salud through the SIREVA II international surveillance network.5 Isolates were confirmed by standard microbiology techniques, and serogroup B was determined by slide agglutination (DIFCO, Beckton Dickinson). Agar dilution antimicrobial susceptibility testing was performed according to the Clinical and Laboratory Standards Institute guidelines.6
Whole genome sequencingDNA extraction was performed using a Qiagen DNA mini kit according to the manufacturer's instructions. DNA was sequenced by Illumina MiSeq technology (Universidad El Bosque-Colombia). Raw sequences were assembled using SPAdes program v 3.1. The quality control of the assemblies was run through the QUAST program and, the genomes were annotated using the Prokka program v1.12.
Allelic profiles of the multilocus sequence typing (MLST) genes and ribosomal multilocus sequence typing (rMLST) genes were determined by PubMLST and rMLST databases. Additionally, clonal complex (CC), allelic and peptide variants of porA, porB, NHBA, fHbp, nadA, and fetA genes were established (www.pubmlst.org/neisseria/). The sequencing data were deposited in the GenBank database (BioProject: PRJNA400226).
The clonal relationship of N. meningitidis isolates was performed using the goeBURST program with previously reported genomes belonging to CC 41/44. The variant calling process included bwa_mem (github.com/lh3/bwa), samtools-BcfTools (github.com/samtools/) and snp-sites (github.com/sanger-pathogens/snp-sites) software. The phylogenic tree was generated using IQ_TREE software, including the reference genomes CP002421.1 (M01-240149-ST-41) and CP021520 (Alpha710-ST-136). Microreact program was used to visualize data.
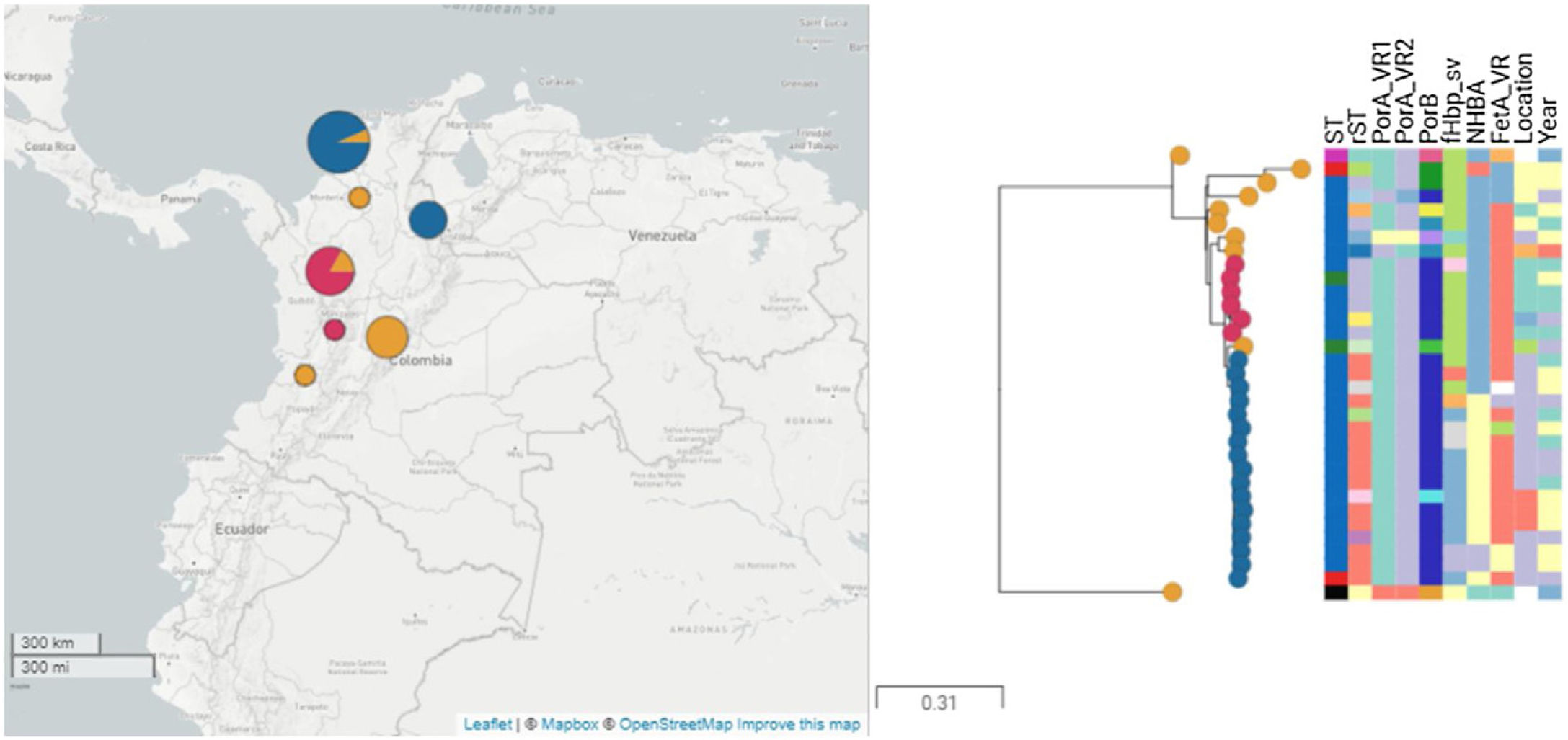
ResultsThirty-four isolates related to ST-9493 serogroup B were identified during the study period. The isolates came from seven political divisions of the country, mainly Bolívar (47.05%), Antioquia (20.5%) and Bogotá (14.7%). The reported diagnoses were acute bacterial meningitis (76.4%) and meningococcemia (17.6%). The ages of the patients ranged from 1 month to 60 years (average of 20 years). The most frequent age groups were 20–49 years (26.4%) and 1–5 years (26.4%).
All isolates were sensitive to ceftriaxone, rifampicin, chloramphenicol and ciprofloxacin. Ten (32.3%) isolates presented intermediate sensitivity to penicillin (minimum inhibitory concentration (MIC)=0.125–0.25μg/ml).
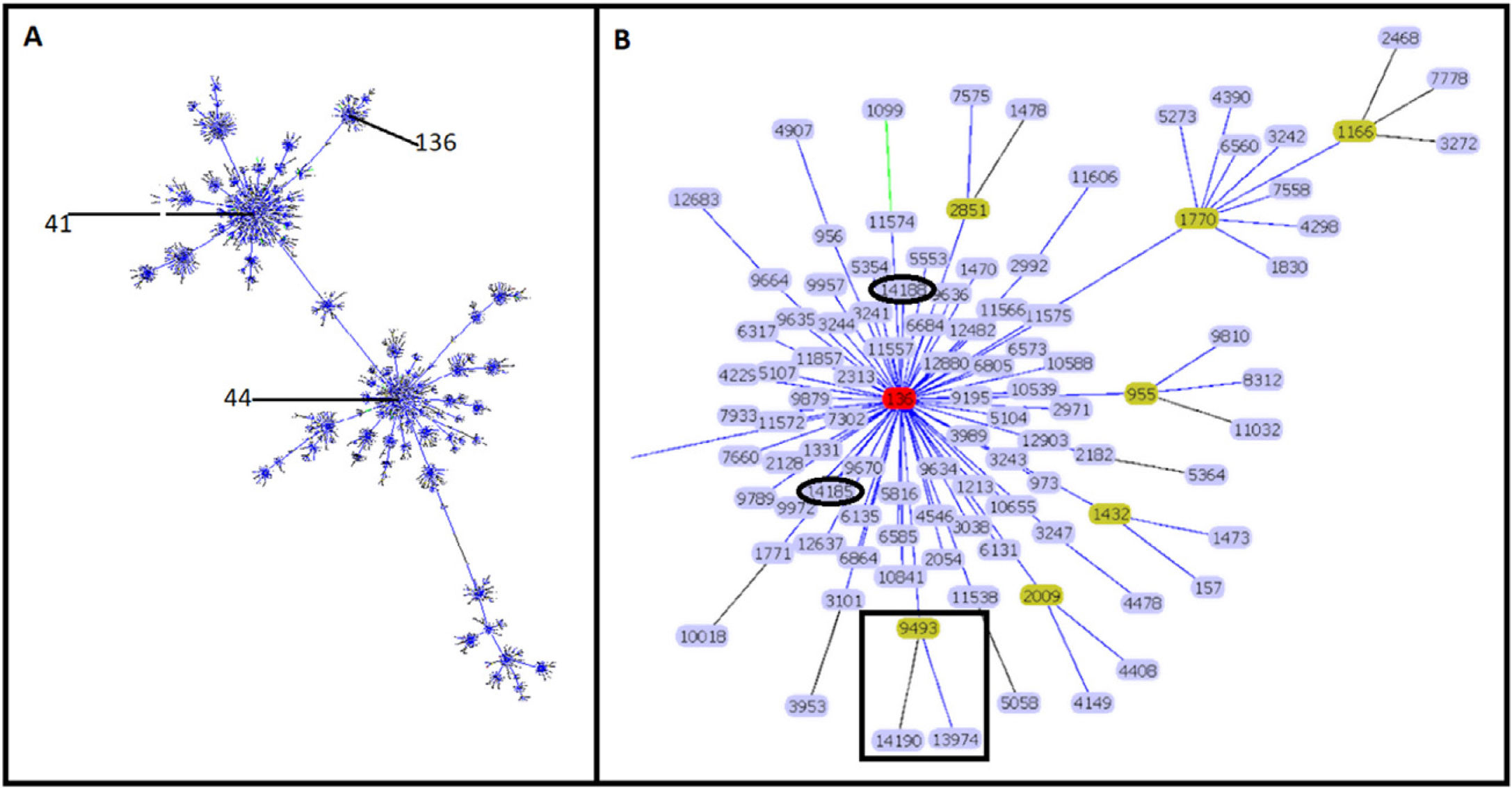
Molecular characterizationEight STs were identified in the 34 isolates. ST-9493 was identified in 27 isolates, and the other seven isolates corresponded to new STs. Four isolates (ST-13974, ST-14185, ST-14188 and ST-14190) had a single locus variant (SLV), the other three isolates were double locus variant (DLV) (ST-13976, ST-13977 and ST-14191) compared to ST-9493. Sixteen new rSTs were found, which were subsequently recorded in the pubMLST database. rST-98571 was the most prevalent with 13 isolates followed by rST-98570 with seven isolates. GoeBURST showed that ST-9493, ST-14185 and ST-14188 correspond to SLVs, and ST-13974 and ST-14190 correspond to DLV from ST-136. The ST-13976, ST-13977 and ST-14191 were assigned to singletons (Fig. 1) and were not included in additional analyses.
. The image shows the relationship of Colombian STs and ST-136 (highlighted in red). ST-9493, ST-13974 and ST-14190 are identified as black squares. ST-14185 and ST-14188 are represented by black ovals (B).")
Clonal complex 41/44. Analysis by goeBURST shows CC 41/44 with all its STs represented by dots and the founder ST-41. ST-136 is also identified in the image (A). The image shows the relationship of Colombian STs and ST-136 (highlighted in red). ST-9493, ST-13974 and ST-14190 are identified as black squares. ST-14185 and ST-14188 are represented by black ovals (B).
The most frequent alleles of the porA variable region were 17 (n=28, 90.3%) and 16-3 (n=25, 80.6%) in variants 1 and 2, respectively. Allele 44 (n=23, 74.2%) class 3 of porB was the most frequent. The fHbp gene was present in all genomes with eight distinct peptides. Twenty-six isolates (83.8%) coded for the variant 2 peptide, and the most common fHbp peptides were 2.24 (n=14, 53.8%) and 2.1187 (n=9, 34.6%). The nadA gene was not identified in any of the isolates. These results were confirmed by a local alignment with the M07162 genome. Three different NHBA peptides were identified; 10 (n=16; 51.6%), 1358 (n=2, 6.4%) and 1359 (n=1, 3.2%). Twelve isolates contained a frameshift mutation. The fetA gene was present in 30 genomes, encoding four different FetA variable regions, of which F5-17 (n=23, 74.2%) was the most frequent, and one isolate harboured a frameshifted protein. Among all isolates, ten (32.2%) had fHbp 2.24, NHBA 10 and FetA 15-17, and nine (29.0%) carried fHbp 21.187, NHBA 1510 and FetA 15-17 profiles (Fig. 2). According to Meningococcal Deduced Vaccine Antigen Reactivity (MenDeVAR, https://pubmlst.org/organisms/neisseria-spp/mendevar), cross-reactive to vaccine variants NHBA-10 was identified in 16 (51.6%) isolates, fHbp-12 in one isolate (3.2%) and fHbp-25 in other isolate (3.2%).
 and Alpha (ST-136), are presented. Isolates belonging to the major clade are represented in blue, isolates of minor clade are presented in red, and others are presented in orange.")
Phylogenetic tree of isolates under study. Thirty-one isolates and two reference genomes, M01-240149 (ST-41) and Alpha (ST-136), are presented. Isolates belonging to the major clade are represented in blue, isolates of minor clade are presented in red, and others are presented in orange.
The clonal relationship of CC 41/44 is represented in Fig. 1; ST-41 is represented as the founder of CC 41/44 and is the major group. All Colombian ST isolates were related to the subfounder ST-136. Phylogenetic analysis (Fig. 2: https://microreact.org/project/ohdzZuF2g6Yyx483ZorNKS) showed that the nearest isolate to the reference strain ST-136 was 1021 with a 1734 SNP distance, whereas the closest isolate to the reference strain ST-41 was 1043 with 8552 SNP sites. The major clade grouped 17 isolates (54.8%) with an average of 2440 SNPs related to ST-136. Most isolates showed rST-98571 (76.5%) and 11 (84.6%) were recovered from patients of the Bolivar political division. The minor clade was composed of six isolates (19.35%) with an average of 2319 SNPs to ST-136, of which most (83.3%) were rST-98570 and obtained from the Antioquia political division. The other isolates were genetically diverse.
DiscussionThis study provides a genotypic analysis of ST-9493 N. meningitidis isolates from IMD cases in Colombia. Phylogenetic analysis shows that ST-9493 isolates were related to ST-136 and divided into two sublineages. One sublineage is associated with an epidemic outbreak, and the other is associated with sporadic cases of the disease. The autochthonous emergence of this ST has epidemiological significance that justifies monitoring of future spread.
In Colombia, the incidence of bacterial meningitis and meningococcal disease caused by N. meningitidis was 0.23 cases per 100,000 inhabitants in 2019.7 Serogroup B was predominant until 2014 and was displaced in frequency by isolates of serogroup C3; these serogroups are responsible for most of the cases reported in Latin America.2 In Colombia, serogroup B isolates are genetically more diverse, consisting of seven clonal groups. One of the most frequent clonal groups is ST-9493, which is related to CC 41/444 and described in isolates recovered in an outbreak of meningococcal disease8 and in healthy carriers.9
ST-9493 shows different genotypic profiles with respect to the genes encoding protein antigens to developed vaccines against serogroup B N. meningitidis. Previous studies have shown that the clonal complex cannot be used to predict vaccine antigen genotypes given the dynamic antigenic expression within clonal complexes with different antigenic profiles and strains with the same peptide variant in several clonal complexes.10,11 According to Meningococcal Deduced Vaccine Antigen Reactivity (MenDeVAR),12 predictions based on the antigenic peptide sequence indicated that isolates of the ST-9493 clonal group might be partially covered (51.6%) by the protein-based meningococcal vaccines in accordance with the isolates analyzed in this study. A vaccine that contains NHBA-10 peptide may be more effective against this clonal group.
The Colombian isolates evaluated were more closely related to ST-136, a subfounder of CC 41/44 witch can be mainly recovered from asymptomatic carriers.13 Two important clades were identified. The first clade was composed of isolates related to an endemic outbreak,8 indicating clonality and persistence over time. A second minor paraphyletic clade was identified in a different geographic region, and this clade could have endemic behaviour. The other isolates presented greater genomic diversity. These findings show that ST-9493 is highly dynamic.
In Colombia, the molecular characterization of meningococcal isolates is not included in the surveillance program; therefore, it is necessary to perform genomic surveillance to establish the population dynamics of this clonal group as well as to detect the emergence of new autochthonous clones.
Authors’ contributionsStudy conception and design: ZA, JM, DP, CD, JMG.
Acquisition of data: ZA, JM, DP.
Analysis and interpretation of data: ZA, JM, DP.
Drafting of manuscript: Critical revision: all authors.
FundingThe study was supported by the Instituto Nacional de Salud, Colombia and the Pan American Health Organization/World Health Organization (PAHO/WHO).
Conflict of interestThe authors have no potential conflicts of interest to disclose.
The authors would like to thank to the national network of integrated by 33 public health laboratories in Colombia, which regularly send isolates to the National Institute of Health.
