


La hemocromatosis hereditaria (HH) tipo 1 se debe a mutaciones del gen HFE, localizado en el cromosoma 6 (6p21.3). Se caracteriza por aumento de la absorción intestinal de hierro y su almacenamiento progresivo en el hígado, piel, páncreas, huesos, corazón y órganos endocrinos. El pronóstico de los pacientes depende del diagnóstico temprano de la enfermedad y del comienzo temprano del tratamiento.
Pacientes y métodoSe revisan las historias clínicas de los miembros de esta familia (8 varones y 5 mujeres) con HH tipo 1 estudiados y seguidos entre 1985 y 2008 en el Hospital Donostia de San Sebastián. Se realizó estudio molecular del gen HFE y de los parámetros de sobrecarga férrica en todos los familiares (17) de dos generaciones. En los familiares con la mutación C282Y/H63D y fenotipo incompleto, la cuantificación de hierro hepático se realizó por resonancia magnética (RM).
ResultadosLa mutación C282Y en homocigosis estaba presente en 3 familiares, en heterocigosis (asociada a la H63D) en otros 3 y en heterocigosis sola en 6. La biopsia hepática reveló fibrosis y/o cirrosis en 4 familiares (3 con C282Y/C282Y y otro con C282Y/-) mayores de 40 años con hepatomegalia, artralgias y, en algunos, pigmentación cutánea, astenia e hipogonadismo. La RM hepática en dos familiares con heterocigosis (C282Y/H63D) e índice de saturación de transferrina mayor del 50% no detectó sobrecarga férrica tras varios años de seguimiento, lo que ha evitado la realización de sangrías.
ConclusionesSe comentan las formas clínicas de la HH tipo 1 en esta familia y las alteraciones moleculares encontradas. La RM hepática se debería incorporar en el seguimiento de los individuos con alteraciones del metabolismo férrico y mutaciones en el gen HFE.
Hereditary hemochromatosis (HH) type I is due to mutations in the HFE gene, located on chromosome 6 (6p21.3). This disease is characterized by increased intestinal iron absorption and progressive storage in the liver, skin, pancreas, bones, heart and endocrine organs. Patient prognosis depends on early diagnosis and prompt treatment of the disease.
Patients and methodsWe reviewed the medical records of family members (eight men and five women) with HH type I, studied and followed-up between 1985 and 2008 in Hospital Donostia, San Sebastian (Spain). Molecular studies were performed of the HFE gene and of iron overload parameters in all the family members (N=17) belonging to two generations. In family members with the C282Y/H63D mutation and incomplete phenotype, liver iron quantification and monitoring was done by magnetic resonance imaging (MRI).
ResultsThe homozygous C282Y mutation was present in three family members, the heterozygous mutation (associated with H63D) in three, and the heterozygous mutation alone in six. Liver biopsy revealed fibrosis and / or cirrhosis in four family members (three with C282Y/C282Y and another with C282Y/-) aged over 40years with hepatomegaly, arthralgia, and in some cases, skin pigmentation, fatigue, and hypogonadism. Liver MRI in two family members with the heterozygous mutation (C282Y/H63D) and transferrin saturation greater than 50% showed no iron overload after several years of monitoring, avoiding the need for phlebotomy.
ConclusionsWe present the clinical manifestations of HH type 1 in this family and the molecular alterations found. Liver MRI should be incorporated into the monitoring of individuals with impaired iron metabolism and HFE gene mutations.
1 motako hemokromatosi hereditarioa (HH) 6 (6p21.3) kromosoman dagoen HFE genearen mutazioagatik gertatzen da. Burdinaren heste-xurgapena handitzen da, eta apurka, burdin hori gibelean, azalean, pankrean, hezurretan, bihotzean eta organo endokrinoetan pilatzen da. Gaixoen pronostikoa gaixotasunaren diagnosi goiztiarraren eta tratamendua goiz hastearen arabera aldatzen da
Gaixoak eta metodoa1 motako HH duen familia bateko kideei (8 gizon eta 5 emakume) 1985 eta 2008 artean Donostiako Ospitalean aztertutako historia klinikoak berraztertu dira. Bi belaunalditako familiartekoen (17) HFE genearen eta burdinaren gainkarga-parametroen azterketa molekularra egin zen. C282Y/H63D mutazioa eta fenotipo osatugabea zuten familiartekoen kasuan, burdin hepatikoaren kuantifikazioa erresonantzia magnetikoaren (EM) bidez egin zen.
Emaitzak3 familiartekok zuten C282Y mutazioa homozigosian, beste hiruk heterozigosian (H63Drekin lotuta) eta beste seik heterozigosian bakarrik. Biopsia hepatikoaren arabera, 4 familiartekok fibrosi edota zirrosia zuten (3 familiartekok C282Y/C282Y eta besteak con C282Y/-). Familiako kide horiek 40 urtetik gorakoak ziren, hepatomegalia eta artralgia zituzten, eta batzuek azaleko pigmentazioa, astenia eta hipogonadismoa ere bai. Hainbat urtez monitorizatzen aritu ostean, erresonantzia magnetiko hepatikoak ez zuen hauteman burdinaren gainkarga heterozigosia eta siderofilina-saturazio indizea %50etik gorakoa zuten bi familiartekorengan (C282Y/H63D). Horrek odol-ateratzeak saihestu zituen.
OndorioakFamilia honetako kideengan 1 motako HH gaixotasunak dituen forma klinikoen eta asaldura molekularren gaineko iruzkinak egin dira. Erresonantzia magnetiko hepatikoa burdinaren metabolismoan asaldurak dituzten eta HFE genearen mutazioak dituzten pertsonei jarraipena egiteko erabili beharko litzateke.
La hemocromatosis genética se caracteriza por un aumento de la absorción intestinal de hierro y su almacenamiento progresivo en el hígado, piel, páncreas, huesos, corazón y órganos endocrinos. Esta sobrecarga férrica da lugar a lesiones celulares e insuficiencia funcional de los órganos afectados. Hasta hoy se conocen cinco tipos de hemocromatosis hereditaria (HH) producidos por mutaciones en diferentes genes. El tipo 1, de herencia autonómica recesiva, se relaciona con las mutaciones del denominado gen HFE (6p21.3). La HH tipo 1 (OMIM#235200) es la más frecuente en la población general. El pronóstico de los pacientes depende del diagnóstico temprano de la enfermedad y de la evaluación del contenido hepático de hierro, lo que permite iniciar el tratamiento de manera temprana y prevenir la aparición de manifestaciones tardías.
Se presentan las características clínicas de una familia con HH tipo 1 procedente de Guipúzcoa. En 13 de 17 familiares estudiados se han encontrado mutaciones del gen HFE (3 con la mutación C282Y/C282Y). Cuatro de ellos tienen lesiones hepáticas. Los familiares con doble heterocigosis (C282Y/H63D) y elevación del índice de saturación de transferrina (IST), tras varios años de seguimiento mediante resonancia magnética (RM) del hígado, hasta la fecha no han desarrollado sobrecarga férrica ni síntomas o signos de enfermedad.
Pacientes y métodoSe revisan las historias clínicas de los miembros de esta familia con HH tipo 1 procedente de Guipúzcoa, estudiados y seguidos entre 1985 y 2008 en el Hospital Donostia de San Sebastián. Los pacientes 1, 2 y 3 habían sido diagnosticados, en 1982 y en 1983, de hemocromatosis familiar por biopsia hepática en la Clínica Universitaria de Navarra, y el 4, en 1984, también por biopsia del hígado en el Hospital Txagorritxu de Vitoria. Los parámetros bioquímicos de sobrecarga férrica de los familiares 1, 2, 3 y 4 (tabla 1) corresponden a los realizados en 1985, comienzo del seguimiento en nuestro hospital, tras varios años de flebotomías.
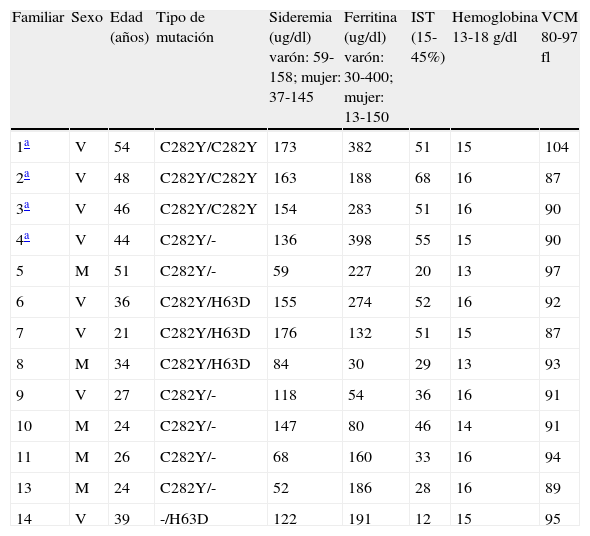
Datos bioquímicos del metabolismo férrico de los familiares con mutaciones del gen HFE
| Familiar | Sexo | Edad (años) | Tipo de mutación | Sideremia (ug/dl) varón: 59-158; mujer: 37-145 | Ferritina (ug/dl) varón: 30-400; mujer: 13-150 | IST (15-45%) | Hemoglobina 13-18 g/dl | VCM 80-97 fl |
| 1a | V | 54 | C282Y/C282Y | 173 | 382 | 51 | 15 | 104 |
| 2a | V | 48 | C282Y/C282Y | 163 | 188 | 68 | 16 | 87 |
| 3a | V | 46 | C282Y/C282Y | 154 | 283 | 51 | 16 | 90 |
| 4a | V | 44 | C282Y/- | 136 | 398 | 55 | 15 | 90 |
| 5 | M | 51 | C282Y/- | 59 | 227 | 20 | 13 | 97 |
| 6 | V | 36 | C282Y/H63D | 155 | 274 | 52 | 16 | 92 |
| 7 | V | 21 | C282Y/H63D | 176 | 132 | 51 | 15 | 87 |
| 8 | M | 34 | C282Y/H63D | 84 | 30 | 29 | 13 | 93 |
| 9 | V | 27 | C282Y/- | 118 | 54 | 36 | 16 | 91 |
| 10 | M | 24 | C282Y/- | 147 | 80 | 46 | 14 | 91 |
| 11 | M | 26 | C282Y/- | 68 | 160 | 33 | 16 | 94 |
| 13 | M | 24 | C282Y/- | 52 | 186 | 28 | 16 | 89 |
| 14 | V | 39 | -/H63D | 122 | 191 | 12 | 15 | 95 |
IST: índice de saturación de transferrina; M: mujer; V: varón; VCM: volumen corpuscular medio de hematíes.
Varón de 54 años de edad, sin antecedentes etílicos y con sinovectomía de la articulación metacarpofalángica derecha a los 45 años, prótesis de cadera izquierda a los 50 años y de la derecha a los 52 años, que acudió al hospital por astenia, dolor en manos, caderas y rodillas y discreto edema bimaleolar. A la exploración mostraba constantes normales, subictericia conjuntival, coloración morena de la piel, sobre todo en manos, escaso vello torácico, hepatomegalia de 3cm por debajo del reborde costal, de consistencia media y no dolorosa, atrofia testicular y edemas maleolares mínimos.
Por laparoscopia y biopsia se diagnosticó de cirrosis hemocromatósica (1982). Antígenos de histocompatibilidad (HLA): A2, 32; B7, -(Bw6). Se instauró tratamiento con flebotomías, al principio semanales y luego trimestrales, según los valores de hematocrito y de hemoglobina. En la analítica de 1985, transcurridos 30 meses del diagnóstico, destacaban: bilirrubina 1,2mg/dl, sideremia 173μg/dl, ferritina 382μg/dl e IST del 51% (tabla 1). Las enzimas hepáticas, el proteinograma y la coagulación fueron normales. Al año siguiente, la ecografía puso de manifiesto una colelitiasis. Dos años después presentó una hepatitis B aguda y, transcurridos 3 meses de ésta, tuvo un episodio de descompensación hidrópica. Se le han realizado varias intervenciones por hipertrofia prostática, estenosis uretral y recambios de ambas prótesis durante los últimos 22 años. Cuando estuvo disponible, se realizó test del gen HFE, siendo C282Y/C282Y. El paciente ha evolucionado favorablemente tanto desde el punto de vista clínico como analítico. Los datos actuales del metabolismo férrico (2008) son: sideremia 88μg/dl, ferritina 82,8μg/dl e IST del 26%.
Caso 2 (I-2): hermanoVarón de 48 años, con coloración morena de la piel y hepatomegalia de 3-4cm por debajo del reborde costal, de consistencia media. No presenta antecedentes etílicos. En 1983 una biopsia hepática ciega demostró cirrosis hepática con hemosiderosis. HLA: A2, 28; B7,-(Bw6). En 1985, 26 meses después del diagnóstico, se observa sideremia 163μg/dl, ferritina 188μg/dl, IST del 68%. Las pruebas hepáticas fueron normales. Durante los 25 años de seguimiento (ecografía abdominal y alfa-fetoproteína cada 6-12 meses) no se ha observado hipertensión portal ni descompensación de la hepatopatía. Los marcadores de hepatitis B y C fueron negativos. A los 62 años se observó litiasis de 6mm en el cuello de la vesícula y próstata adenomatosa I/IV; a los 67 años, pólipo hiperplásico en sigma, y a los 70 años, coxartrosis bilateral y artrosis de manos, cuello y rodillas. Test del gen HFE: C282Y/C282Y. Desde 1983 hasta la actualidad, ha tolerado bien las flebotomías. Continúa con sangrías de mantenimiento cada 3 o 4 meses. Por lo que respecta a la evolución de los síntomas, persiste la pigmentación cutánea y tiene astenia, artralgias de grandes y pequeñas articulaciones, junto a disfunción eréctil en las últimas 2 décadas.
Caso 3 (I-3): hermanoVarón de 46 años, con pigmentación oscura de la piel, hepatomegalia de 3cm por debajo del reborde costal y dolores óseos en las articulaciones metacarpofalángicas (segundo, tercero y cuarto dedos de la mano derecha, y segundo y tercero de la izquierda), en las interfalángicas proximales y radiocarpianas, bilaterales, y en ambas rodillas. Sin hábitos tóxicos. En 1983 se realizó biopsia hepática ciega en la que se observó fibrosis portal con hemosiderosis. HLA: A2,28;B7,-(Bw6). En la analítica de 1985, a los 26 meses del diagnóstico, las pruebas hepáticas eran normales y los valores plasmáticos, compatibles con sobrecarga férrica (tabla 1). Test del gen HFE: C282Y/C282Y. En 2006, se practicó artoplastia total de rodilla izquierda por artrosis. Los marcadores de hepatitis B y C fueron negativos. Desde 1983 hasta 2008 sigue tratamiento con flebotomías. Durante el seguimiento, han persistido las artralgias de grandes y pequeñas articulaciones. En enero de 2008, la sideremia, IST y ferritina se mantienen dentro de la normalidad.
Caso 4 (I-4): hermanoVarón de 44 años, asintomático con hepatomegalia no dolorosa, de 2cm. No presenta antecedentes etílicos. En 1984, por hemocromatosis familiar, se practicó biopsia hepática que puso de manifiesto fibrosis con siderosis, grado III/IV. En 1985, a los 9 meses del diagnóstico, las pruebas de función hepática eran normales y los parámetros bioquímicos del metabolismo del hierro estaban alterados (tabla 1). Desde 1984ha seguido con sangrías periódicas semanales, mensuales o cada 45 días según el perfil férrico. En 1988 comenzó con dolor de cadera derecha y en 1993 se le implantó una prótesis por coxartrosis. En la actualidad se encuentra sin manifestaciones clínicas de la enfermedad, continúa con flebotomías y en mayo de 2008 la sideremia era de 83μg/dl, IST de 31% y ferritina de 57μg/dl. Test del gen HFE: C282Y/-. Los marcadores de hepatitis B y C fueron negativos.
Caso 5 (I-5): hermanaMujer de 51 años, con antecedentes familiares de hemocromatosis, que presentaba molestias ocasionales en hipocondrio derecho y artralgias en hombros, cuello y caderas. Sin hábitos tóxicos. La exploración física era normal, salvo hepatomegalia de 1cm por debajo del reborde costal, de consistencia blanda. Los datos analíticos revelaron pruebas hepáticas y valores bioquímicos del metabolismo férrico (tabla 1) sin alteraciones y HLA: A2,26; B7,38 (Bw4,6). Test del gen HFE: C282Y/-. Marcadores de hepatitis B y C, negativos. Después de la menopausia, los parámetros de sobrecarga férrica siempre fueron normales en los sucesivos controles efectuados. A los 54 años se le colocó una prótesis de cadera por coxartrosis derecha que se recambió transcurridos 10 años y a los 69 años se diagnosticó cáncer de mama izquierda. Tras cirugía y quimioterapia, falleció a los 3 años de la intervención.
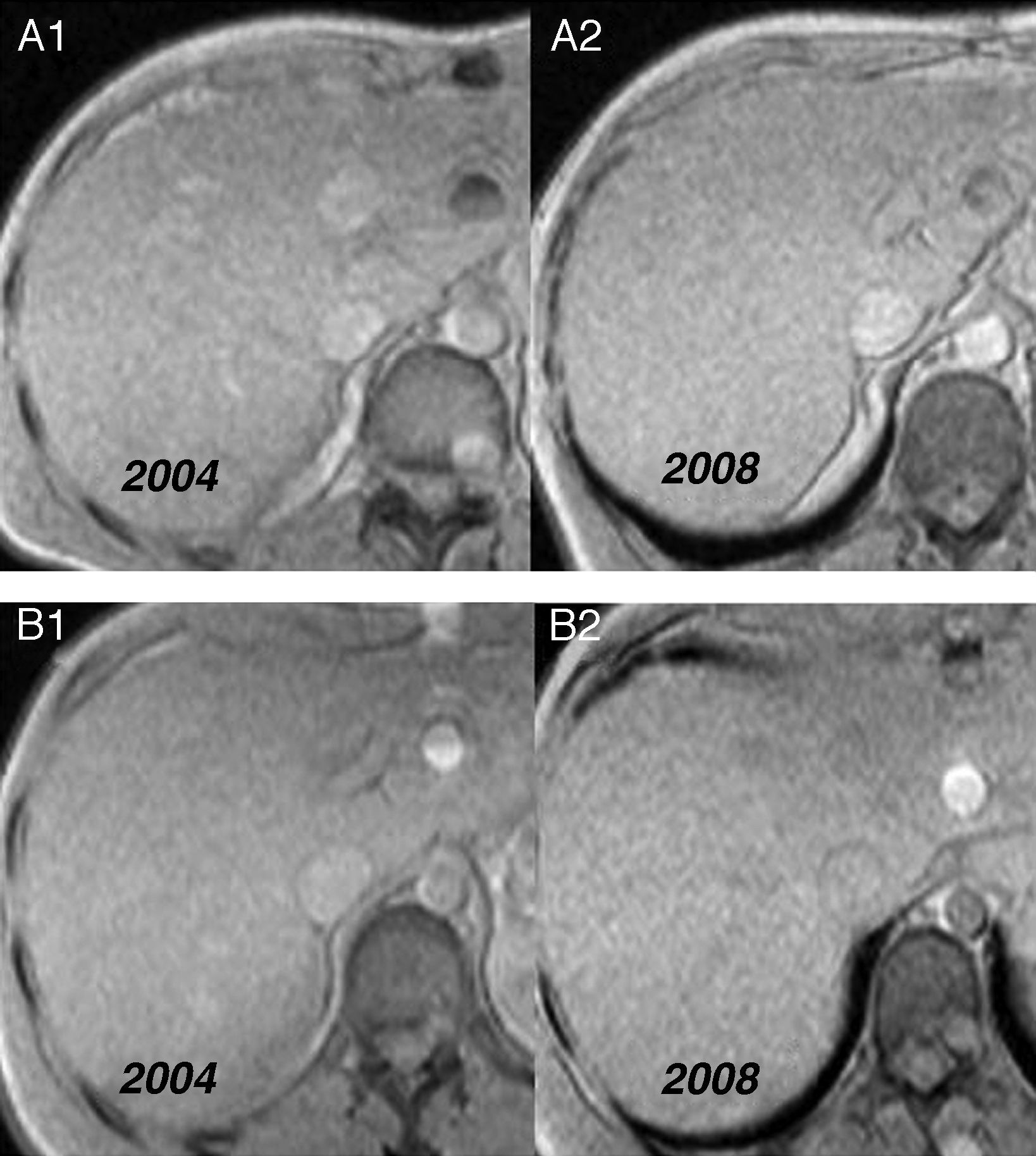
Otros familiaresEl genotipo y el fenotipo de los familiares de segundo grado del caso índice se determinaron a partir de la adolescencia (tabla 1). Hasta ahora no se ha encontrado ningún tipo de síntoma ni signo de la enfermedad. Los familiares 6 y 7 (heterocigotos dobles y con HLA: A2,-;B7,44;Cw4,-;DR2,6), a los 35 y 21 años presentaron un IST superior al 50% que persiste hasta hoy, por lo que se ha realizado un seguimiento mediante RM hepática para evaluar los depósitos de hierro. En el caso 6, a los 36, 38 y 41 años la concentración de hierro hepático (CHH) ha sido de 23,5, 23 y 24μmol de hierro (Fe)/g, respectivamente, y en el caso 7, a los 22, 24 y 28 años, de 15,6, 14 y 10, respectivamente, valores que según nuestra experiencia indican ausencia de sobrecarga férrica (fig. 1).
Estudio de la cuantificación de hierro hepático por resonancia magnética.")
En los familiares (casos 6 y 7) la cuantificación de hierro hepático se llevó a cabo mediante una máquina de 1,5 teslas (Philips Intera, Osatek, San Sebastián) según el modelo de Alustiza et al1, con dos secuencias en eco de gradiente (DP=120/4/20° [TR/TE/Flip angle] y T2=120/14/20°) aplicando el algoritmo CHH=e (5,808 – [0,877×T2] – [1.518×DP]) en el que DP y T2 corresponde a la ratio hígado/músculo en cada una de las dos secuencias.
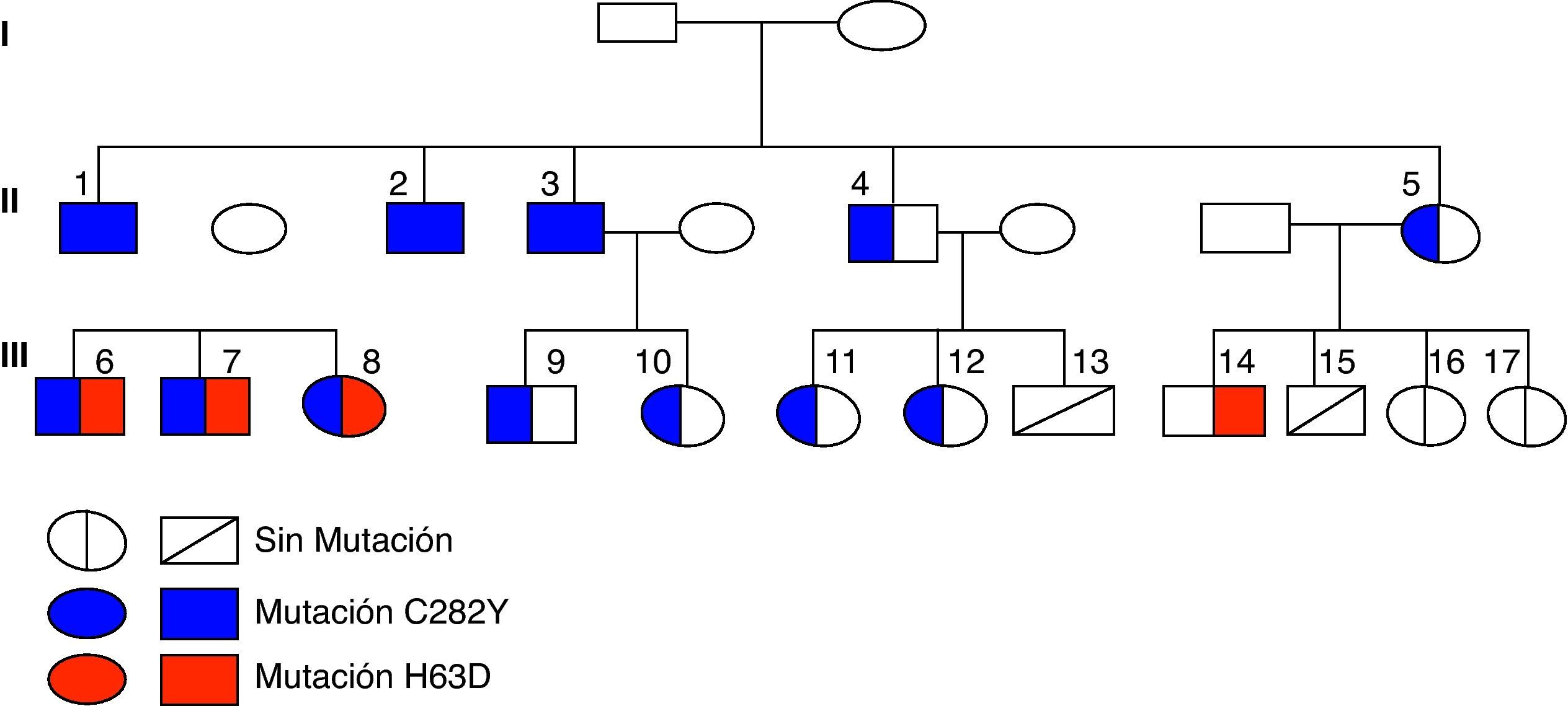
ResultadosEn la figura 2 se muestra el árbol genealógico, donde se detallan las mutaciones detectadas en los distintos miembros de la familia. La mutación C282Y en homocigosis estaba presente en el caso índice y en 2 hermanos (casos 2 y 3), y en heterocigosis, asociada a la mutación H63D, en sus hijos (casos 6, 7 y 8). En 6 miembros de la familia (casos 4, 5, 9, 10, 11 y 12) también se detectó la mutación C282Y sola en heterocigosis (tabla 1).

El IST elevado con ferritina sérica normal (tabla 1) se comprobó en 6 integrantes de la familia, 3 homocigotos (casos 1, 2 y 3, tras varios años de flebotomías), 2 heterocigotos dobles (casos 6 y 7) y otro con heterocigosis sola (caso 4, después de algunos meses de sangrías). La biopsia hepática confirmó la presencia de fibrosis y/o cirrosis en 4 familiares (casos 1, 2, 3 y 4) mayores de 40 años con hepatomegalia, dolor en pequeñas y grandes articulaciones y, en algunos, pigmentación cutánea, astenia e hipogonadismo. De los 4 casos con daño hepático, 3 son homocigotos para la mutación C282Y.
La RM en 2 familiares heterocigotos (C282Y/H63D) que tienen un IST mayor del 50%, tras varios años de seguimiento, ha permitido cuantificar el hierro hepático y, por el momento, corroborar la ausencia de sobrecarga férrica. En ninguno de los familiares de la tercera generación con o sin mutación en el gen HFE han aparecido síntomas o signos de la enfermedad.
DiscusiónLa hemocromatosis genética es el trastorno hereditario más frecuente en la población blanca y se transmite de forma autosómica recesiva, con penetrancia incompleta y expresividad baja. En 1996, Feder et al2 identificaron el gen de la enfermedad, que está situado en el brazo corto del cromosoma 6 y se relaciona estrechamente con el locus HLA-A3. Se han descrito tres mutaciones principales en el gen HFE de la HH tipo 1: a) C282Y, sustitución del aminoácido tirosina por cisteína en la posición 282 del cromosoma 6; b) H63D, sustitución de histidina por ácido aspártico en la posición 63, y c) S65C, sustitución de serina por cisteína en la posición 65.
En el norte y centro de Europa, el 80-90% de los casos se atribuye a la mutación C282Y en homocigosis, mientras que las formas heterocigotas y homocigotas (H63D o S65C) son menos frecuentes2,3. La prevalencia de la mutación C282Y varía según un gradiente norte (heterocigosis 5-10% y homocigosis 0,4-0,7%4,5)-sur (2-4 y 0,5%, respectivamente6) y oeste-este europeos, y según las diferencias regionales observadas en algunos países7. En 2001 De Juan et al8 estudiaron la prevalencia de las mutaciones del gen HFE en la población de origen vasco con hemocromatosis de Guipúzcoa y la cotejaron con la de la población general. El 57% tuvo homocigosis para la mutación C282Y y el 11%, doble heterocigosis C282Y/H63D. En el resto hubo otras mutaciones o no encontraron mutaciones en el gen HFE. En 2005 Bander et al9 corroboraron estos hallazgos en el País Vasco francés, donde encontraron un 53% de homocigosis para la mutación C282Y en la población con hemocromatosis. La frecuencia de la mutación H63D en Europa oscila del 15-40% (20-25% en heterocigosis y 2-4% en homocigosis si la población es de origen céltico10) y la de la mutación S65C, del 2-3%.
Los sujetos con predisposición genética hasta los 20 años están asintomáticos, no tienen sobrecarga férrica y el contenido en hierro del hígado no llega a los 5g. De los 20 a los 40 años, la mayoría no suelen presentar manifestaciones clínicas, excepto artralgias y/o debilidad, el IST y la ferritina sérica superan los valores normales y el contenido hepático de hierro es de 10-20g. El daño hepático ocurre en los varones de más de 40 años y en las mujeres de 50 años, que tienen fibrosis o cirrosis con su sintomatología habitual y una cantidad de hierro en hígado mayor de 20g. Los pacientes que expresan completamente el fenotipo se caracterizan por el IST y ferritina elevados. La sobrecarga férrica se descubre de manera temprana por aumento del IST y más tardíamente por el de la ferritina.
No se conoce la evolución natural de los individuos con hemocromatosis hereditaria sin tratar. En una serie de varones homocigotos C282Y no tratados y seguidos durante un período de 12 años, la ferritina sérica aumentó en el 58% y descendió en el 42%, mientras que en las mujeres homocigotos bajó en el 30%11. El seguimiento de los adultos homocigotos C282Y indica que, si no existe aumento progresivo de la ferritina cuando se hacen los controles, es poco probable que se desarrolle hepatopatía. Por otro lado, la supervivencia de los homocigotos C282Y asintomáticos sin tratamiento es similar a la de una población control12. La mutación C282Y/C282Y tiene una penetrancia incompleta, por lo que alrededor de la cuarta parte de los portadores de ésta no tienen sobrecarga férrica ni síntomas o signos de la enfermedad4,13,14.
Los sujetos heterocigotos C282Y/H63D tienen menor grado de sobrecarga de hierro que los homocigotos C282Y/C282Y. El 2-5% cursan con un IST mayor del 45% y pueden desarrollar síntomas semejantes a los de los homocigotos15,16. En los individuos heterocigotos para la mutación C282Y sola (C282Y+/-), la sobrecarga férrica es infrecuente, y en menos del 10% hay elevación del IST y ferritina séricos16. La mutación H63D, homocigota o heterocigota tiene escasa penetrancia clínica y sólo en el 5% de los portadores de esta mutación hay evidencia de sobrecarga férrica10. En los parientes estudiados de esta familia, se ha encontrado la mutación C282Y en homocigosis en 3. Cuatro de ellos (3 con la mutación C282Y/C282Y y 1 con la C282Y/-) tienen daño hepático y manifestaciones clínicas.
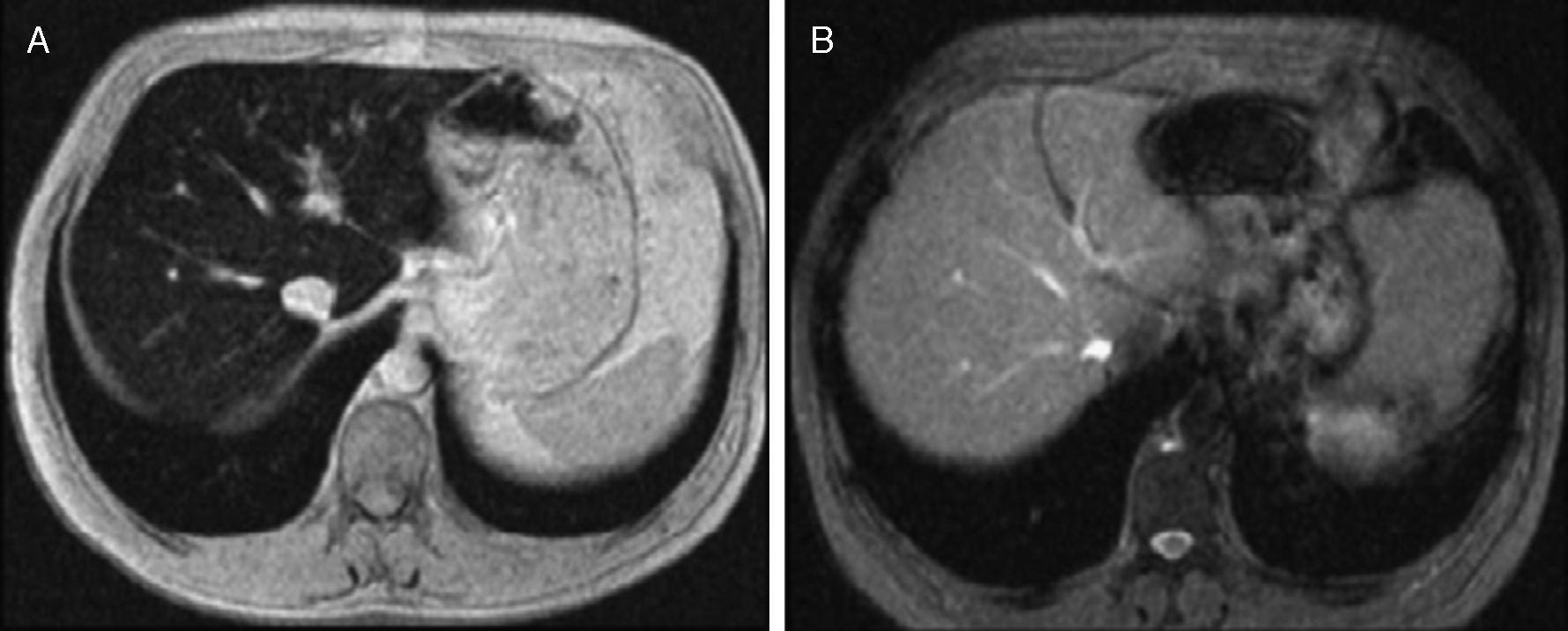
Hace algunos años la determinación de la CHH requería la realización de una biopsia hepática, por lo que era importante disponer de un método no invasivo para la cuantificación de la CHH. La RM se considera en la actualidad la técnica de elección para la cuantificación no invasiva de la CHH17. La sobrecarga férrica genera un acortamiento de T2 que se traduce en una disminución de la señal del hígado proporcional a la intensidad de la sobrecarga de hierro18–24. En diversos trabajos se ha demostrado la existencia de una correlación alta entre la CHH medida sobre la biopsia hepática y las mediciones de la intensidad de la señal de la RM, de manera que pueden hacerse estudios fiables de cuantificación19,22–24. Durante el período 1999-2001, nuestro grupo elaboró un método de cuantificación por RM. La correlación entre la CHH calculada mediante dicho algoritmo y la medida sobre la biopsia hepática fue buena (r=0,937)1. De acuerdo con este modelo diseñado, la cuantificación de 80μmol de hierro/g por RM tiene un valor predictivo positivo del 100% para el diagnóstico de hemocromatosis, con una sensibilidad del 86% y una especificidad del 100%. Una cuantificación<40μmol Fe/g tiene un valor predictivo negativo del 100% para el diagnóstico de hemocromatosis, con una sensibilidad del 100% y una especificidad del 81%1. La RM junto a otros parámetros (mutación C282Y y ferritina) ha facilitado la predicción no invasiva de lesión hepática y la cuantificación de los depósitos de hierro tras flebotomías25–28. En nuestro hospital se utiliza desde 2002 la RM para evaluar la CHH en individuos con mutaciones indicativas (C282Y/C282Y o C282Y/H63D) de HH tipo 1 y fenotipo incompleto, en sujetos con mutaciones no puntuales y en los tratados mediante flebotomías (fig. 3). En los casos 6 y 7 de esta familia, heterocigotos dobles y con IST mayor del 50%, la CHH no ha superado los valores de 24 y 15,6μmol de hierro/g, respectivamente, tras 6 años de seguimiento por RM.
 y a los 3 años, tras flebotomías (B: cuantificación de hierro en hígado, normal).")
El estudio del genotipo HFE es ineludible para el cribado de los pacientes asintomáticos de primer grado de un paciente con hemocromatosis y mutaciones en este gen. Sólo la mutación C282Y con carácter homocigoto, y a veces la H63D en estado heterocigoto combinada con C282Y, se consideran indicativas de HH ligada al gen HFE, siempre asociadas a un fenotipo compatible. En los últimos años se han descrito otras mutaciones, como en el gen SLC40A1 y C326S29,30. La RM del hígado se debe incluir en el algoritmo diagnóstico y en el seguimiento de los individuos con alteraciones del metabolismo de hierro y mutaciones en el gen HFE.
Conflictos de interésLos autores declaran no tener ningún conflicto de intereses.