


¿ INTRODUCCIÓN
El cáncer cervicouterino (CaCu) es el segundo cáncer en frecuencia en mujeres de todo el mundo. La mayoría de los casos ocurre en países en vías de desarrollo.1-3 La infección por el virus del papiloma humano (HPV) ha sido reconocida como un factor etiológico para el desarrollo del CaCu.4-6 Se han descrito más de 120 tipos de HPV, de los cuales aproximadamente una tercera parte puede infectar el epitelio del tracto genital.7,8 Otros tumores relacionados con el HPV son los del canal anal, vagina, vulva, pene y orales, de tal manera que se ha estimado que el HPV es responsable del 5.2% de todos los cánceres en el mundo.9,10
Los HPV tienen tropismo por las células epiteliales. Así, pueden producir infecciones tanto de piel como de mucosas. Los virus capaces de infectar las mucosas se dividen en genotipos de alto y bajo riesgo, de acuerdo a si la infección que producen puede conducir o no al desarrollo de cáncer. Los HPV 6 y 11 producen verrugas benignas en el tracto genital y no son oncogénicos. La infección está caracterizada por lesiones verrugosas y el tratamiento para su eliminación es costoso. Por otro lado, los HPV de alto riesgo (HR-HPV), causan lesiones mucho menos evidentes y son clasificados como potencialmente oncogénicos, ya que están asociados con más del 99% de los cánceres del cérvix. El número de HR-HPV varía entre 13 y 19, pero los tipos 16, 18, 31, 33, 35, 45, 51, 52, 56 y 58 conllevan constantemente un alto riesgo.3,6 De estos, el HPV16 se encuentra en aproximadamente el 60% de todos los CaCu, mientras el HPV18 está involucrado en un 10-20%; los HPV 31, 33, 35, 39, 45, 51, 52, 56, 58,59, 68 y 73 juntos constituyen el 20-30% restante de los CaCu.3 El HPV ha sido implicado en el 85% de los casos de cáncer del canal anal, 50% de los cánceres de vulva, vagina y pene, 20% del cáncer orofaríngeo y 10% del cáncer laríngeo y esofágico.11,12 A pesar de que la tasa de progresión carcinogénica es relativamente baja, las infecciones por HR-HPV se encuentran en la mayoría de los CaCu humanos.5,6
Se requieren criterios claros para clasificar a los HPV en grupos de alto o bajo riesgo basados en estudios epidemiológicos y moleculares que provean el riesgo estimado sobre la evidencia funcional del potencial oncogénico de los diferentes tipos de HPV.
Los HPV son altamente transmisibles y la mayoría de los hombres y mujeres sexualmente activos adquiere la infección durante su vida. Las infecciones genitales por HPV son transmitidas principalmente por contacto sexual, pero no exclusivamente durante el coito. Mientras las infecciones son en su mayoría transitorias y benignas, la infección genital persistente con ciertos genotipos virales puede conducir al desarrollo de lesiones precancerosas y de cáncer en la región anogenital.
Muchos virus que originan infecciones persistentes debido a su capacidad de modular o evadir la respuesta inmune se caracterizan por una replicación continua a bajos o altos niveles (por ejemplo, el virus de la inmunodeficiencia humana y el virus de la hepatitis B) o por periodos de reactivación de una infección latente seguida por intervalos libres de la enfermedad (por ejemplo, el virus del herpes simple). Esto puede conducir a una variedad de problemas crónicos incluyendo neoplasias, inmunosupresión, enfermedades autoinmunes y falla orgánica selectiva.1,13
Las mujeres son infectadas por alguno de estos virus poco tiempo después del inicio de la vida sexual activa, ocurriendo la mayoría de las infecciones en menores de 25 años.14,15 Después de esa edad, la prevalencia disminuye rápidamente. En mujeres de mediana edad, las infecciones por HPV son transitorias, volviendo a observarse un incremento en las infecciones en mujeres de 30 años.16
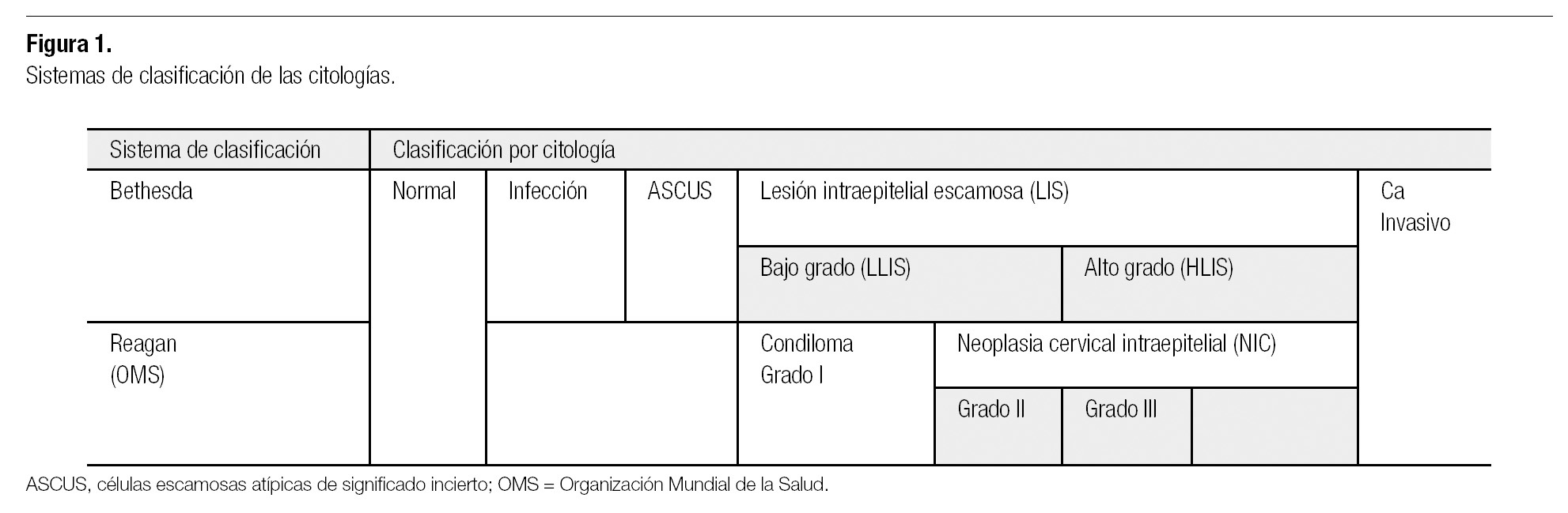
El CaCu está caracterizado por una fase premaligna bien definida, la cual puede ser detectada por examen citológico de células cervicales exfoliadas (prueba de Papanicolaou). Desde la implementación de la citología (Pap), la tasa del CaCu ha disminuido considerablemente,17 y a pesar de que la mayoría de las mujeres fallecidas por CaCu nunca se realizó una prueba de Pap, muchas que sí lo hicieron recibieron resultados negativos. Esto se debe a que la sensibilidad de la citología es limitada por error del muestreo, con colocación de pocas células en el frotis, agregándose el error de interpretación, donde pocas células anormales no son identificadas entre la multitud de células normales que también se encuentran en el frotis cervical debidamente tomado. El error de muestreo más común es la falta de células de la zona de transición cervical.18 Otro problema asociado al Pap, se reporta como resultado ASCUS (siglas en inglés de atypical squamous cells of undetermined significance, células escamosas atípicas de significado incierto; Figura 1), un hallazgo citológico sugestivo, pero no concluyente de lesiones escamosas intraepiteliales. Esto genera incertidumbre en el médico respecto a la conducta a seguir. ASCUS permanece como una entidad que se reporta en 5-10% de las pruebas de Pap.17
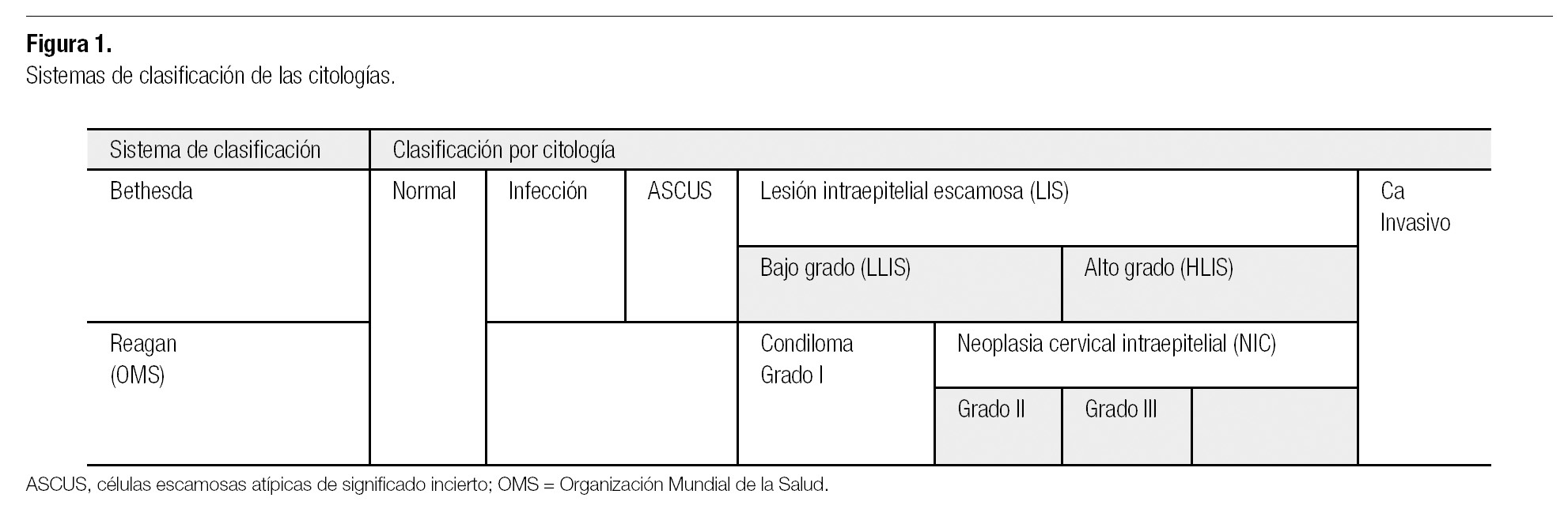
Figura 1. Sistemas de clasificación de las citologías.
Los cambios premalignos cervicales representan un espectro de anormalidades histológicas que van desde neoplasia intraepitelial cervical (NIC) 1 (displasia leve), NIC 2 (displasia moderada) y NIC 3 (displasia severa/carcinoma in situ) hasta cáncer invasivo (Figura 1).2 Aunque el tratamiento de los cambios cervicales premalignos es eficaz, también es un procedimiento ineficiente. Esto se debe a las incertidumbres que rodean a la historia natural de la neoplasia intraepitelial. Las pruebas citológicas e histológicas no logran distinguir a las pocas mujeres con frotis anormales que progresarán hacia un cáncer invasivo de la vasta mayoría que exhibe anormalidades que presentarán regresión espontánea.1
En los últimos años, con la intensa investigación realizada sobre la patogénesis del CaCu, se ha logrado un gran progreso en la comprensión de esta enfermedad.
La infección genital por HR-HPV es muy común y la mayoría de los individuos se cura de la infección con el tiempo, pero aproximadamente 15% de las pacientes no pueden eliminar el virus. La infección persistente por un virus HR-HPV es el principal factor de riesgo para desarrollar cáncer del tracto genital inferior.7,19,20
Debido a la infección persistente del cérvix con genotipos carcinogénicos del HPV, se introdujo la prueba de detección del ADN del HPV para el tamizaje de CaCu.20 Después de ocho años de seguimiento, utilizando los dos tipos de test en la India, se advirtió que el diagnóstico por ADN reducía un 53% el número de casos con cáncer cervical avanzado y un 47% el número de muertes. Por su parte, el método Pap sólo redujo en un 25% el número de casos con cáncer avanzado y sólo un 11% el número de muertes.21-23
¿CARACTERÍSTICAS GENERALES DEL VIRUS DEl PAPILOMA HUMANO
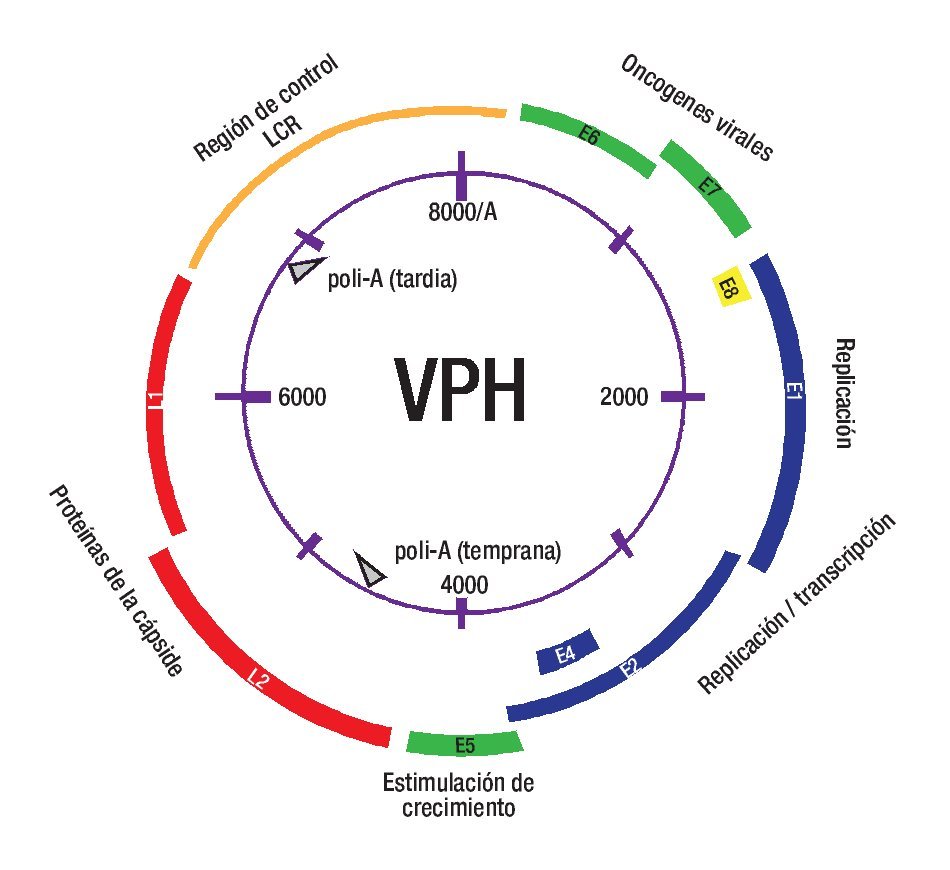
Los HPV son pequeños virus ADN de doble cadena, de la familia Papovaviridae. Aunque el genoma viral puede variar ligeramente entre los diferentes tipos de HPV, se acepta que característicamente contiene cerca de 8,000 pb y codifica en ocho o nueve marcos abiertos de lectura (Figura 2), los cuales son transcritos como ARNm policistrónicos.24,25 La cápside del virus está formada por dos proteínas. La proteína L1 es el elemento estructural primario, encontrándose 360 copias de ésta organizada en 72 capsómeros en los viriones infectantes.26 La proteína L2 es un componente menor del virión y se cree que puede estar presente en el centro de los capsómeros pentavalentes en los vértices del virión.27 Esta proteína L2 interviene en la entrada del virus a las células, en la localización de los componentes virales en el núcleo, en la unión al ADN, en la formación de la cápside y en la estabilidad.13 Ambas proteínas juegan un papel muy importante en mediar la eficiencia de la infectividad del virus.25,27 La infección por HPV requiere que las partículas virales accedan a la capa basal epitelial y penetren a las células basales en división. Es bien conocido que previo a que los HPV establezcan una infección, estos deben experimentar un complicado proceso para unirse y entrar a la célula huésped.28 Hay controversia sobre la naturaleza del receptor viral, pero se cree que los proteoglicanos de heparán sulfato (HSPG) son los receptores iniciales. Se ha observado que la L2 de todos los HPV secuenciados, contiene en su extremo amino terminal una secuencia consenso que es escindida por furina, una pro-proteína convertasa, y se ha supuesto que la escisión por furina29 es necesaria para la unión y entrada del virus a la célula, ya que esto provoca un cambio conformacional de la cápside viral, seguido de la liberación de los HSPG para la posterior asociación con un receptor putativo secundario, que se cree es una integrina a6.4,27,28,30,31 Otros estudios evidencian que el rompimiento de furina puede tener lugar en la superficie celular o dentro de un compartimiento endosomal temprano,30 y las cápsides se liberan en un compartimiento endosomal tardío, llevando a liberar el genoma asociado del endosoma hacia el citoplasma por un mecanismo que involucra al extremo C-terminal de L2.32
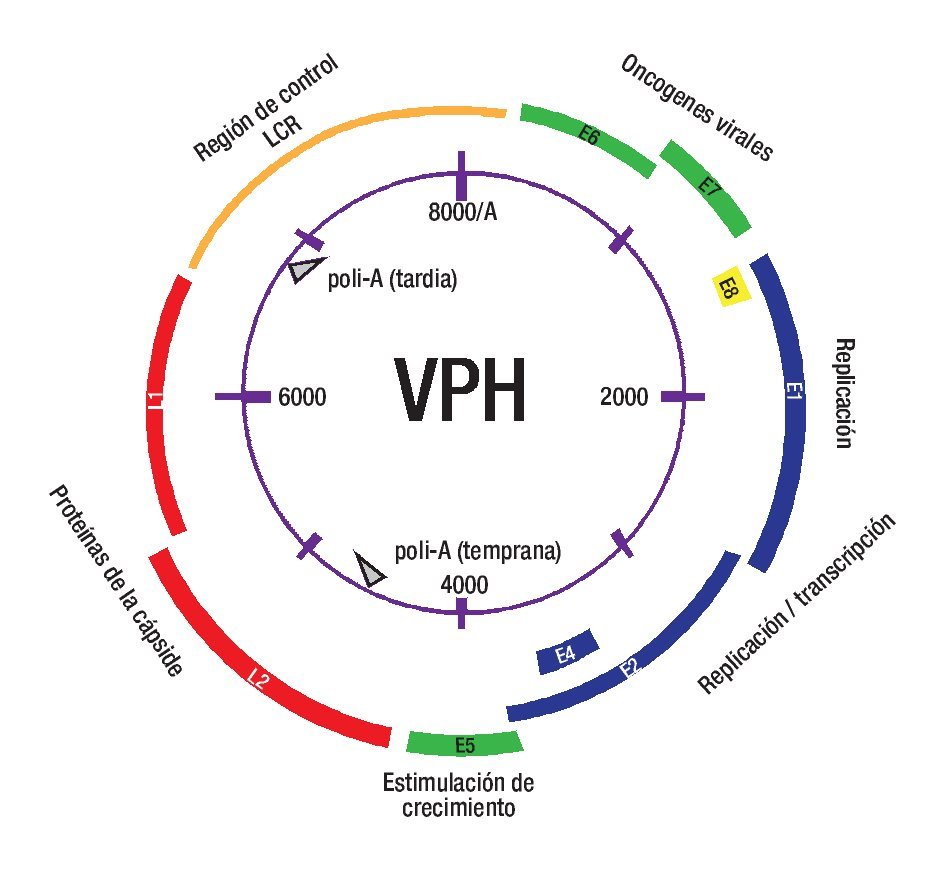
Figura 2. Virus del papiloma humano. LCR = regulación de la expresión génica y replicación viral. E6 y E7 considerados oncogenes ya que sus proteínas se unen a p53 y pRb, respectivamente. E1 y E2 son genes de expresión temprana necesarios para la replicación y transcripción del genoma viral. E4 es necesario para el ensamblaje y liberación viral. L1 y L2 codifican para las proteínas de la cápside.
¿ CICLO DE VIDA DEL VIRUS DEL PAPILOMA HUMANO
El ciclo de vida del HPV está ligado al programa de diferenciación de la célula huésped infectada, el queratinocito, pero la expresión de altos niveles de proteínas virales y el ensamblaje viral ocurren exclusivamente en las capas superiores, es decir, en los estratos espinoso y granuloso del epitelio escamoso.33 Las células en la capa basal consisten en células troncales y células en tránsito que se están dividiendo continuamente y proveen un reservorio de células para las regiones suprabasales.34 La infección de estas células por el HPV conduce a la activación de la expresión en cascada de los genes virales, que provoca la producción de aproximadamente 20 a 100 copias extracromosómicas de ADN viral por célula. Este promedio de número de copias permanece estable en las células basales indiferenciadas a través del curso de la infección.4,34 La integración viral ocurre más comúnmente en las células que contienen este número de episomas. En estos, la expresión de genes virales es mínima; en particular, la expresión de los oncogenes E6 y E7 está bajo un control muy estricto, y sus proteínas son discretamente detectables. Cuando el queratinocito infectado entra al compartimento de diferenciación, sale del ciclo celular, ocurre una regulación positiva de la expresión de los genes virales, sobreviene la replicación del ADN viral y entonces el número de copias virales aumenta al menos a 1000 copias/célula, observándose abundante expresión de los genes tempranos E6 y E7 y de los genes tardíos (Figura 2).35
Las infecciones genitales por el HPV son transmitidas principalmente por contacto sexual, se considera que a través de microabrasiones del epitelio que expone a la infección viral a las células de la capa basal.4,36
¿ INTEGRACIÓN DEL VIRUS DEl PAPILOMA HUMANO
Los HPV pueden encontrarse en el material cervical en forma de episomas, en formas integradas o en forma mixta. La integración usualmente causa deleción o alteración del gen viral regulador E2, mientras retiene un segmento variable que incluye a los oncogenes E6 y E7 y la región reguladora corriente arriba. La sobreexpresión de E2 por los promotores heterólogos en las células huésped con el HR-HPV integrado, puede reprimir al promotor temprano del virus provocando una disminución drástica en la expresión de los genes E6 y E7. Así, la integración del HR-HPV y la deleción o alteración de E2 conduce a mayor expresión de los oncogenes virales.37 Las células que tienen integrado al HR-HPV adquieren una ventaja de crecimiento sobre aquellas que albergan episomas del HR-HPV (el estado natural del virus en infecciones productivas) y muestran un aumento de inestabilidad genómica.38,39
La integración viral al genoma de la célula huésped ocurre corriente abajo en la expresión de los genes tempranos E6 y E7, frecuentemente en la región E1 o E2; esta interrupción provoca la pérdida del control negativo de la expresión del oncogén por la expresión de la proteína del gen regulador viral E2. Los transcritos derivados del virus integrado son más estables que los derivados del ADN viral episomal y la integración del HPV 16 ha sido asociada con una ventaja de crecimiento selectiva en las células afectadas.38-40
La prevalencia en células exfoliadas de cérvix o de tejido cervical de episomas o formas integradas del HPV o ambas, varía según el grado de severidad de la enfermedad, del tipo de HPV que se encuentre presente y del método utilizado para determinar el estado físico del virus.31,32,41 Se ha propuesto que la identificación de formas integradas del HPV podría ser un biomarcador muy útil para la enfermedad progresiva. Sin embargo, hay varios problemas con esta propuesta. Primero, la identificación del reducido número de formas integradas sobre una base de formas episomales es un reto técnico cuando sólo se dispone de células exfoliadas para el análisis. Segundo, cuando los genomas integrados están transcripcionalmente silentes o se obtienen poco tiempo después de la integración, su detección puede tener una utilidad pronóstica limitada.4 Aunque las formas integradas se detectan en más del 40% de las mujeres con NIC 3, su transcripción activa se ha reportado solamente en el 15% de las pacientes.4,40 La detección de transcritos derivados de virus integrados nos proporcionaría información pronóstica más útil. Sin embargo, se ha demostrado que en los queratinocitos cervicales a los cuales se integrará el virus, solamente puede haber transcritos después de que ocurra una disminución del número de episomas que expresen E2.34 Esta pérdida del gen E2 en los episomas se asocia con la activación endógena de los genes antivirales aumentando la expresión de los oncogenes virales en las células que poseen las formas integradas.34,42
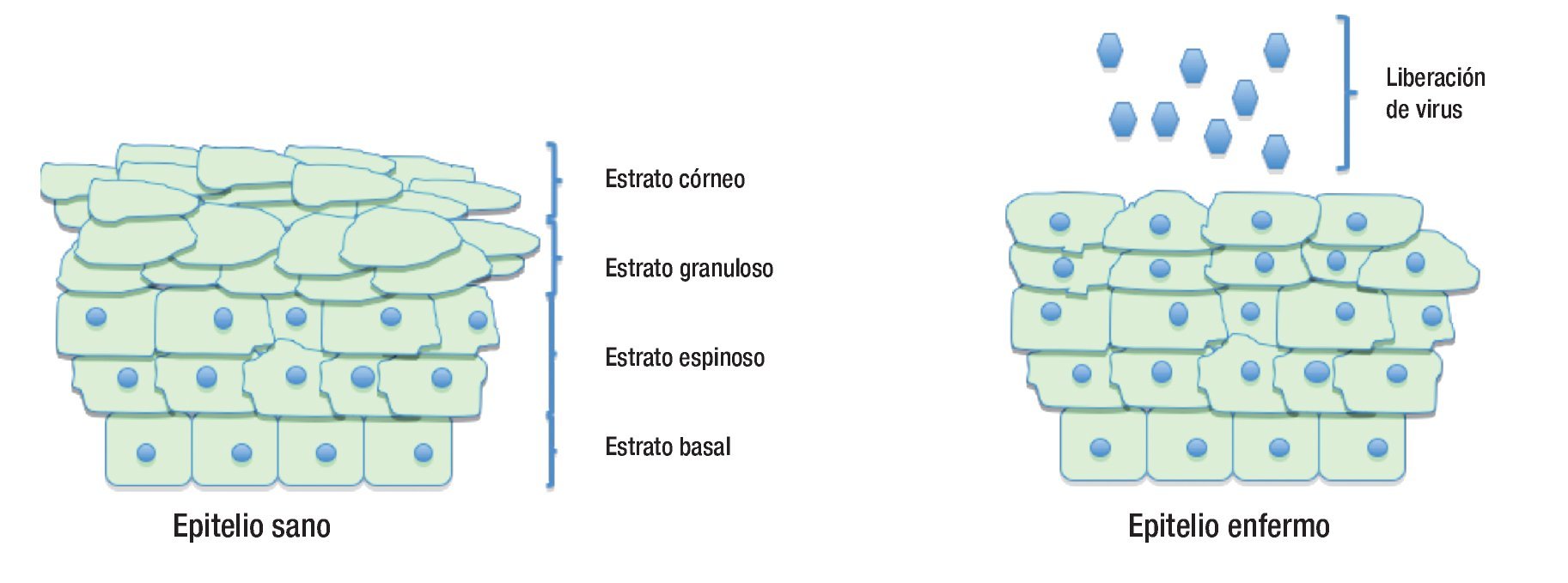
Una vez que el virus logra penetrar a la célula huésped inicia la expresión de sus genes. Los productos de los genes pueden dividirse en tempranos (E) y tardíos (L), dependiendo del momento en que se expresen durante el ciclo de vida viral. Las moléculas críticas en la replicación viral son E6 y E7, las cuales inactivan funcionalmente los productos de dos genes supresores de tumores muy importantes, el gen p53 y Rb respectivamente. Ambos oncogenes inducen la proliferación, inmortalización y transformación maligna de las células infectadas (Figura 3).
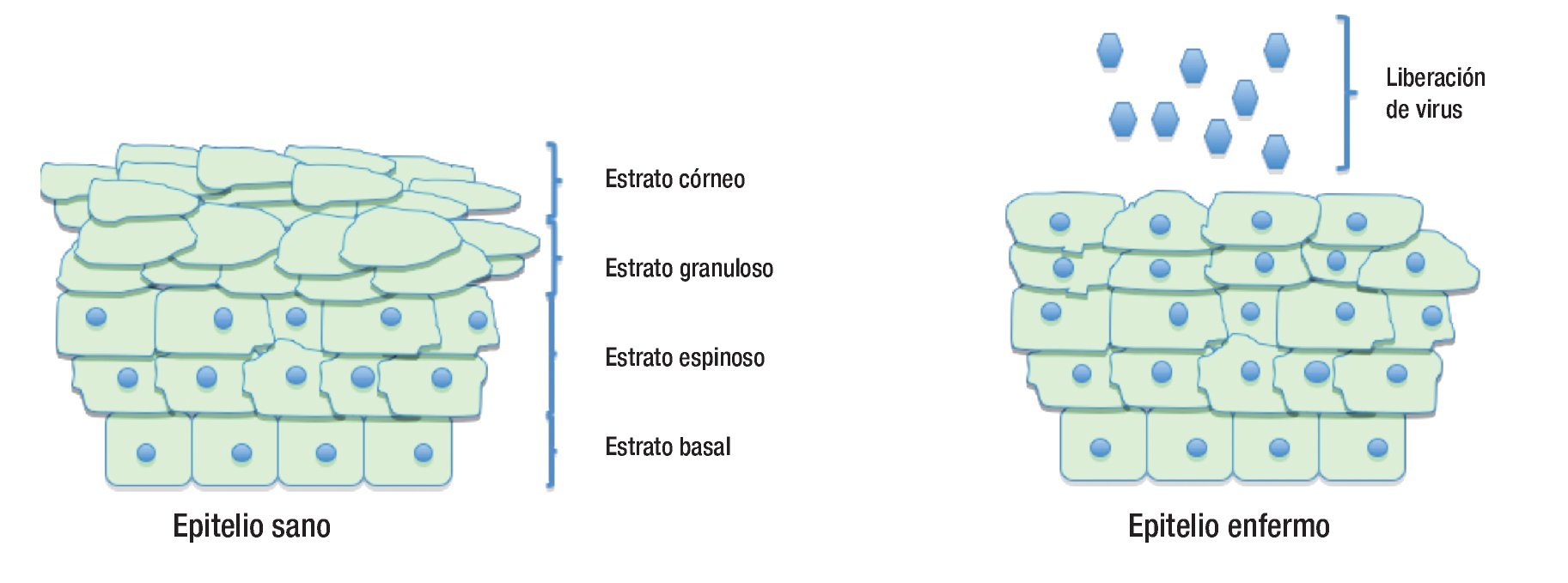
Figura 3. Diferencias en las capas del epitelio cervical cuando está sano y cuando está infectado por el virus del papiloma humano.
¿ DE LA INFECCIÓN AL CÁNCER
Los virus infectan a los queratinocitos basales primitivos, pero los niveles de expresión elevada de proteínas virales y el ensamblaje viral, ocurren exclusivamente en las capas de los estratos espinoso y granuloso del epitelio escamoso.33 La expresión de genes virales está confinada al queratinocito, y no hay evidencia de que tales genes se expresen en otra célula que no sea queratinocito. Poco después de la infección, la replicación de los episomas virales parece ser independiente del ciclo celular, produciéndose aproximadamente de 50 a 100 copias por célula.8 Se cree que la célula deja este estado primitivo para transformarse en una célula proliferativa del epitelio. En esta etapa la expresión viral es mínima, la expresión de los oncogenes virales E6 y E7 está bajo un control muy estricto, por lo que sus transcritos son escasamente detectables. Cuando el queratinocito entra al estatus de diferenciación, sale del ciclo celular e inicia un aumento masivo en la expresión de los genes virales, formándose al menos 1000 copias de virus por célula, con abundante expresión de los genes tempranos E6 y E7 y expresión de genes tardíos.35
El HPV codifica sólo una proteína para la replicación del ADN, la enzima E1 y además de ésta la proteína viral E2; fuera de ello, la replicación viral es totalmente dependiente de la maquinaria de síntesis del ADN celular. El problema para los virus es que las ADN polimerasas celulares y los factores de replicación sólo se producen en células con mitosis activa. Para resolver este problema, los virus codifican proteínas del ciclo de vida viral, que reactivan la síntesis de ADN celular en células sin ciclo celular, inhiben la apoptosis y retardan el programa de diferenciación del queratinocito infectado, creando un ambiente permisivo para la replicación del ADN viral.43 Los detalles a fondo no son bien conocidos, pero los genes virales centrales para estas funciones son el E6 y el E7.8
En esta estrategia de replicación el ADN viral se replica y el virus se ensambla en una célula que estaba destinada a morir por causas naturales; no hay citólisis inducida por el virus, tampoco hay necrosis y de ahí que no induzca inflamación, lo cual dificulta la activación de las células dendríticas y del inicio de una respuesta inmune efectiva. Este virus no produce viremia, por lo que pasa desapercibido para el sistema inmune. Además, al igual que todos los virus ADN, tiene mecanismos para inhibir la síntesis de interferón por la célula huésped. Estas estrategias virales generan infecciones crónicas por largos periodos de tiempo sin que el huésped se entere.
Durante la fase inicial de infección, el HPV existe como un episoma nuclear, pero la integración del HR-HPV al ADN del genoma huésped es un paso importante en la progresión neoplásica del cérvix.44 La integración causa deleción o alteración del gen regulador viral E2, mientras retiene un segmento variable que incluye a los genes E6 y E7, lo cual origina el incremento de la expresión de los oncogenes virales.37,38 Las células que contienen al virus integrado, adquieren ventajas de crecimiento sobre las que contienen episomas virales, lo cual provoca su expansión clonal. Estas células tienen inestabilidad genómica lo que conduce a la progresión de la malignización.37,45
Correspondencia: Dra. Guadalupe Zaldívar.
Clavel #200, Colonia Prados de la Capilla, CP 76170, Querétaro, Qro. México.
Teléfono: 1-92-12-00 Ext. 6234. Teléfono celular: 442-300-17-10. Fax. 52+ (442) 215-0928.
Correo electrónico:apizl@yahoo.com.mx