





La leucemia mieloide aguda (LMA) es un grupo de trastornos hematológicos malignos de progresión rápida, fenotípica y genéticamente heterogéneos, los cuales se caracterizan por la proliferación clonal desregulada de células inmaduras que han perdido la capacidad de diferenciarse normalmente. El proceso de transformación leucémica, o leucemogénesis, es un proceso complejo en múltiples pasos, resultante de la acumulación de mutaciones que modifican en algún punto su sistema de señalización celular. Gracias al estudio de las bases genético-moleculares de las diferentes mutaciones que acompañan a la LMA, la humanidad ha comprendido, aunque no completamente, los efectos que desempeñan estas sobre procesos clave como proliferación, diferenciación y apoptosis celular. En el presente trabajo se hace una revisión de los diferentes mecanismos que intervienen en el proceso de carcinogénesis, haciendo uso de la LMA como modelo debido a su gran variabilidad genético-molecular, haciendo énfasis en las principales afectaciones citogenéticas y genéticas reportadas.
Acute myeloid leukemia (AML) is a group of haematological diseases, phenotipic an genetically heterogeneous, characterised by clonal expansion of myeloid precursors with a decreased capacity of differentiation. The process of leukaemic transformation or leukaemogenesis is a complex multi-step process arising from the accumulation of mutations that modify somewhere the cell signalling system. By studying the molecular basis in different mutations associated with AML, we have began to understand, but not completely, the effects that these play on key processes, such as proliferation, differentiation, and cell survival. In this paper a review is presented on the different mechanisms involved in the process of carcinogenesis, using AML as a model, because it has a large genetic and molecular variability, emphasising the main cytogenetic and genetic damage reported.
La producción normal de células sanguíneas —hematopoyesis— es un proceso complejo a través del cual células germinativas hematopoyéticas (CGH) proliferan y se diferencian dando lugar a los distintos tipos de células maduras circulantes (eritrocitos, plaquetas, neutrófilos, eosinófilos, basófilos, monocitos y linfocitos, entre otros)1. Las CGH tienen 2 propiedades esenciales que se requieren para el mantenimiento de la hematopoyesis: la pluripotencialidad y la capacidad de autorrenovación. La pluripotencialidad se refiere a la capacidad de una sola CGH para generar todas las células hematopoyéticas maduras, mientras que la capacidad de autorrenovación describe la característica de generar células hijas con las mismas propiedades que su progenitor. Cuando una CGH se divide, al menos una de las células hijas debe autorrenovarse para evitar la depleción de células germinativas2.
Los tumores de origen hematopoyético se asocian a menudo con mutaciones que bloquean la maduración de la célula progenitora o que anulan su dependencia de los factores de crecimiento3. El proceso de transformación leucémica o leucemogénesis es un proceso complejo en múltiples pasos, resultante de la acumulación de mutaciones que modifican en algún punto su sistema de señalización celular (receptor, segundo mensajero, proteína efectora o factor de transcripción) (fig. 1)4. El efecto neto de estas perturbaciones es una expansión clonal no regulada y alteraciones de los procesos de muerte celular y diferenciación, de tal manera que la CGH se transforma en lo que se ha denominado como célula madre leucémica5.
 interactúa con un receptor y lo activa; el receptor activado se relaciona con la maquinaria celular produciendo una segunda señal que desemboca en un cambio en los patrones de expresión genética o en la actividad funcional de otras proteínas. La modificación de la actividad metabólica es el principal blanco que sufre cambios en este sistema.")
Esquema general del sistema de señalización celular. Existen varios sistemas de traducción de señales dentro de la célula, pero las características generales son comunes en todos: una señal (ligando) interactúa con un receptor y lo activa; el receptor activado se relaciona con la maquinaria celular produciendo una segunda señal que desemboca en un cambio en los patrones de expresión genética o en la actividad funcional de otras proteínas. La modificación de la actividad metabólica es el principal blanco que sufre cambios en este sistema.
En el presente trabajo se hace una revisión de los diferentes mecanismos que intervienen en el proceso de carcinogénesis, haciendo uso de la leucemia mieloide aguda como modelo debido a su gran variabilidad genético-molecular y poniendo énfasis en las principales afectaciones citogenéticas y genéticas reportadas.
Generalidades de la leucemia mieloide agudaEl término «leucemia mieloide aguda» (LMA) se refiere a un grupo de trastornos hematológicos malignos de progresión rápida, fenotípica y genéticamente heterogéneos, los cuales se caracterizan por la proliferación clonal desregulada de células inmaduras que han perdido la capacidad de diferenciarse normalmente6.
La LMA representa del 15 al 20% de las leucemias agudas en niños y el 80% en adultos. La LMA es la forma predominante de leucemia en el periodo neonatal y adulto, pero representa una pequeña proporción de casos durante la infancia y la adolescencia. El rango de incidencia es aproximadamente 1.5 por 100,000 en infantes menores de un año de edad, decrece a 0.4 por 100,000 en niños de edades entre 5 y 9 años, posteriormente se incrementa gradualmente a aproximadamente 1 por cada 100,000 personas hacia los 25 años de edad, y finalmente comienza a incrementarse exponencialmente hasta aproximadamente a 25 por 100,000 personas en octogenarios7.
Esta leucemia surge como resultado de la transformación de precursores hematopoyéticos a través de la adquisición de rearreglos cromosómicos y múltiples mutaciones genéticas que bloquean la diferenciación celular y confieren ventajas proliferativas y de supervivencia4,8. Estos eventos oncogénicos claves son a menudo clasificados de acuerdo al modelo de los 2 hits propuesto por Gilliland en el 2001. Este modelo supone que para el desarrollo de una LMA se deben asociar al menos 2 tipos de mutaciones: las mutaciones de clasei, que activan vías que confieren ventajas proliferativas o de supervivencia, y las mutaciones de claseii, que afectan los procesos de diferenciación celular y apoptosis. Sin embargo, recientemente, con los estudios de secuenciación masiva, se ha identificado otro grupo de mutaciones que no caen dentro de estas categorías, por lo que se encuentran sin clasificar, pero principalmente incluyen genes implicados en modificaciones epigenéticas (fig. 2)9–11.

Modelo de cooperación entre mutaciones asociadas a la aparición de LMA. El modelo de los 2 hits hipotetiza que la LMA es consecuencia de la colaboración entre al menos 2 tipos de mutaciones. Las mutaciones de clasei confieren ventajas proliferativas o de supervivencia, mientras que las mutaciones de claseii alteran los procesos de diferenciación celular y apoptosis. Actualmente se conocen diversas mutaciones que por no adecuarse a ninguna de las 2 clases se encuentran sin clasificar; sin embargo, estas principalmente promueven modificaciones epigenéticas.
La base de la leucemogénesis subyace en el daño genético no letal, y en el caso de LMA existen una gran variedad de factores que contribuyen a su desarrollo; sin embargo, los más importantes son la exposición a radiaciones ionizantes, altas concentraciones de benceno, agentes quimioterapéuticos e inhalación crónica de humo de cigarro. Estos agentes exógenos tienen la capacidad de producir daños en el ADN por diferentes mecanismos, pero principalmente mediante daño oxidativo12–16. Además, la obesidad es un factor endógeno que incrementa el riesgo; el mecanismo preciso aún es incierto, sin embargo se sugiere que la hiperinsulinemia, la resistencia a la insulina, los elevados niveles de leptina, bajos niveles de adeponectina y el acortamiento de los telómeros encontrados en estos pacientes están relacionados17,18. Por otro lado, la LMA puede desarrollarse como progresión de otro trastorno clonal de las CGH, resultado de la inestabilidad genómica y la adquisición de mutaciones adicionales7. Los principales ejemplos subyacen en las neoplasias mieloproliferativas (NMP), en las cuales aumenta la producción de uno o más tipos de células sanguíneas, y los síndromes mielodisplásicos (SMD), que se destacan por presentar defectos en la maduración que se asocian a una hematopoyesis ineficaz2. En el primer caso, las NMP se caracterizan por la presencia de proteínas tirosina cinasa mutadas cuantitativamente activadas, o bien afectaciones en la señalización por los efectores río abajo, lo cual ejemplifica perfectamente mutaciones de clasei. Por su parte, los SMD muestran defectos en factores de transcripción claves para la diferenciación hematopoyética normal y en moduladores de la apoptosis, que asemejan mutaciones de claseii9. De esta manera, ambas patologías presentan un primer hit, lo que las hace susceptibles de desarrollar LMA si son sometidas a una segunda mutación.
Caracterización genético-molecularLa clasificación de la LMA fue por mucho tiempo basada en la morfología (clasificación Francesa-Americana-Británica [FAB]) (tabla 1A) y el inmunofenotipo. Las primeras pruebas de la base genética de la LMA provienen del análisis citogenético, en el cual se detectaron cambios a nivel cromosómico como translocaciones, deleciones, inserciones, inversiones, monosomías, trisomías, poliploidías y otras aberraciones. Generalmente una o más anormalidades citogenéticas son encontradas en aproximadamente el 55% de los pacientes con LMA, y debido a esto configuran un fuerte factor pronóstico dentro de la clasificación de la Organización Mundial de la Salud (OMS) (tabla 1B)19.
Sistemas de clasificación de Francesa-Americana-Británica (FAB) y de la Organización Mundial de la Salud (OMS)
| A. Clasificación de la leucemia mieloide aguda (FAB) |
|---|
| M0. Leucemia mieloide aguda sin diferenciación |
| M1. Leucemia mieloide aguda con diferenciación mínima |
| M2. Leucemia mieloide aguda con diferenciación |
| M3. Leucemia promielocítica aguda hipergranular o típica |
| M3v. Leucemia promielocítica aguda hipogranular |
| M4. Leucemia mielomonocítica aguda |
| M4v. Leucemia mielomonocítica aguda con eosinofilia en médula ósea |
| M5. Leucemia monocítica aguda |
| M6. Eritroleucemia |
| M7. Leucemia megacariocítica aguda |
| B. Clasificación de la leucemia mieloide aguda (OMS) |
|---|
| Leucemia mieloide aguda con anormalidades genéticas recurrentes |
| Leucemia mieloide aguda con t(8;21)(q22;q22); RUNX1-RUNX1T1 |
| Leucemia mieloide aguda con inv(16)(p13.1q22) o t(16;16)(p13.1;q22); CBFB-MYH11 |
| Leucemia promielocítica aguda con t(15;17)(q22;q12); PML-RARA |
| Leucemia mieloide aguda con t(9;11)(p22;q23); MLLT3-MLL |
| Leucemia mieloide aguda con t(6;9)(p23;q34); DEK-NUP214 |
| Leucemia mieloide aguda con inv(3)(q21q26.2) o t(3;3)(q21;q26.2); RPN1-EVI1 |
| Leucemia mieloide aguda (megacarioblástica) con (t(1;22)(p13;q13); RBM15-MKL1 |
| Leucemia mieloide aguda con mutaciones en NPM1 |
| Leucemia mieloide aguda con mutaciones en CEBPA |
| Leucemia mieloide aguda relacionada con cambios mielodisplásicos |
| Relacionada al tratamiento de neoplasias mieloides |
| Leucemia mieloide aguda, sin ningún otra especificación |
Actualmente, los resultados citogenéticos permiten estratificar a los pacientes con LMA en 3 clases —favorable, intermedio y desfavorable— de acuerdo con el pronóstico clínico que se reporta en la bibliografía. De esta manera, los pacientes con t(8;21)(q22;q22) [RUNX1/RUNX1T1], inv(16)(p13q22) [CBFB/MYH11] y t(15;17)(q24;q21) [PML/RARA] tienen un pronóstico favorable con buena respuesta al tratamiento y remisiones completas. Por otra parte, los pacientes con t(9;11)(p22;q23) [MLLT3/MLL] se consideran con pronóstico intermedio, y en los pacientes con t(6;9)(p23;q34) [DEK/NUP214], inv(3)(q21q26) [RPN1/EVI1] y t(1;22)(p13;q13) [RBM15/MKL1] el pronóstico clínico es adverso debido a la agresividad del padecimiento y la baja respuesta al tratamiento. Estas alteraciones citogenéticas producen genes de fusión que codifican proteínas aberrantes con propiedades funcionales alteradas20,21.
La caracterización genético-molecular de estas anormalidades citogenéticas recurrentes reveló que de manera directa o indirecta interferían con el desarrollo y la homeostasis de las células sanguíneas normales. Así mismo, al identificarse los puntos de corte en estos rearreglos se logró identificar los genes afectados, la creación de genes de fusión y los productos anormales generados (proteínas quiméricas)22. En la mayor parte de estas modificaciones, al menos uno de los genes involucrados codifica un factor de transcripción; así, la proteína quimérica posee cualidades reguladoras de la transcripción, pudiendo activar o silenciar genes interactuando con diversos promotores, enhancers o silencers, o bien regulando la interacción entre las histonas y el ADN21. La interacción de las histonas con el ADN dentro de los nucleosomas puede modificarse al ocurrir reacciones en las colas de estas proteínas; por ejemplo, si algunos residuos lisina son acetilados, el grupo acetilo reduce la carga positiva de las histonas, lo que ocasiona que se desestabilice la relación con el ADN, con lo que tanto los factores de transcripción como la maquinaria de transcripción tendrán acceso a las secuencias reguladoras de la expresión genética. Por su parte, la metilación de las histonas no modifica la carga de estas, aunque sí altera su basicidad e hidrofobicidad, incrementando así su afinidad por moléculas aniónicas como el ADN, y de esta manera produce la compactación de la cromatina y el silenciamiento genético23.
Mutaciones citogenéticasGracias al estudio de las diferentes mutaciones que acompañan a la LMA, la humanidad ha comprendido, aunque no completamente, el papel que desempeñan estos genes críticos. En el caso de la t(8;21)(q22;q22) e inv(16)(p13q22), cuyos productos de fusión son RUNX1/RUNX1T1 y CBFB/MYH11, respectivamente, se encuentran involucradas proteínas de la familia de factores de transcripción de unión nuclear (core binding factors [CBF]), los cuales son requeridos en la ontogenia hematopoyética y son reguladores claves en diversos pasos de la hematopoyesis. La familia CBF consiste de 3 subunidades CBFA de unión al ADN (RUNX1, RUNX2 y RUNX3) y una subunidad común, CBFB, que no tiene interacción con el ADN pero incrementa la afinidad de las otras subunidades por este. Aunque los mecanismos por los cuales estos genes de fusión contribuyen a la patogénesis de la leucemia no son completamente comprendidos, se ha encontrado una actividad inhibitoria dominante sobre los genes blanco del complejo CBF silvestre, a través del reclutamiento del complejo correpressor nuclear. Fenotípicamente, la t(8;21)(q22;q22) se encuentra asociada predominantemente a la LMA con maduración (subtipo M2 de la FAB), mientras que la inv(16)(p13q22) tiene relación con la LMA mielomonocítica con eosinofilia (subtipo M4v de la FAB). Afectaciones en el gen KIT se encuentran asociadas como mutaciones secundarias20,24.
Por su parte, la t(15;17)(q24;q21) es característica de la LMA promielocítica (LPA, subtipo M3 de la FAB) tanto de la variante hipergranular como de la microgranular (dependiendo del tamaño y de la cantidad de los gránulos azurófilos presentes en los promielocitos). Involucra la fusión de los genes PML, un factor de transcripción y supresor tumoral que regula la progresión del ciclo celular e induce la muerte celular, y RARA, receptor nuclear de ácido retinoico α que se une a los elementos de respuesta a ácido retinoico en los promotores de muchos genes, originando el gen de fusión PML/RARA. Normalmente, RARA se une con el receptor retinoicoX formando un heterodímero, el cual actúa como represor transcripcional reclutando al complejo correpresor nuclear histona desacetilasa, facilitando así el ensamblaje de los nucleosomas y el silenciamiento de varios promotores. La unión de su ligando (ácido retinoico) ocasiona un cambio conformacional que resulta en la activación transcripcional de genes requeridos para la diferenciación de los promielocitos. PML/RARA reprime a los promotores blanco de la cadena de señalización de la misma manera que RARA cuando no se encuentra unido a su ligando; sin embargo, a diferencia de la variante silvestre, requiere una mayor concentración del ligando para que esta represión sea eliminada, debido a que mantiene una interacción más estable con el complejo correpresor, así como con algunas metilasas como DNMT1 y DNMT3a. Además, PML/RARA tiene importantes efectos sobre la apoptosis debido a que interfiere de manera dominante negativa con la función del PML silvestre y su regulación de p53. De esta manera, los defectos en la apoptosis permiten la activación oncogénica y la persistencia de la inestabilidad genómica. La mayoría de los pacientes con LPA son sensibles al tratamiento con ácido transretinoico, que permite la transcripción del ADN y, por consiguiente, la maduración celular. Mutaciones secundarias involucran principalmente a FLT320,21,25,26.
La t(9;11)(p22;q23) involucra a los genes MLLT3 y MLL, y usualmente se asocia a leucemias con características monocíticas como LMA monocítica (subtipo M4 de la FAB) y LMA mielomonocítica (subtipo M5 de la FAB). El gen de fusión MLLT3-MLL es el rearreglo más común en LMA del gen MLL (aunque existen otros rearreglos menos comunes), el cual codifica una proteína histona metiltransferasa que, al asociarse con complejos proteicos, regula la transcripción a través del remodelamiento de la cromatina. MLL es un regulador positivo de la expresión de los genes HOX, factores de transcripción que participan en el desarrollo de múltiples tejidos, incluyendo el sistema hematopoyético. Los rearreglos de MLL tienen varios mecanismos para activar la expresión leucemogénica; en el caso de MLLT3-MLL, donde existe la pérdida del dominio de metilación de histonas H3K4 por parte de MLL, la región correspondiente a MLLT3 contiene dominios para el control transcripcional, interactúa con DOT1L, otra histona metiltransferasa que metila el residuo lisina 79 en la histona 3 (H3K79), y con MENIN1, factor transcripcional que se une a diversos promotores, provocando así el incremento de la transcripción de los genes HOX, lo que conlleva la proliferación celular y permite reactivar, en al menos algunos aspectos, la capacidad celular de autorrenovación20,27,28.
La translocación balanceada t(6;9)(p23;q34), con el gen de fusión DEK/NUP214 como marcador, se presenta en LMA con o sin características monocíticas y a menudo asociada con basofilia y displasia multilinaje, principalmente se relaciona con LMA con maduración (subtipo M2 de la FAB) y LMA mielomonocítica (subtipo M4 de la FAB), aunque se puede presentar en algunos otros fenotipos. DEK/NUP214 codifica una proteína nucleoporina que actúa como factor de transcripción aberrante, y además altera el transporte nuclear por unirse a factores de transporte solubles. Adicionalmente se pueden presentar mutaciones en FLT3 como alteraciones complementarias20.
La inv(3)(q21q26) involucra al gen EVI1, un factor de transcripción que presenta un patrón de expresión específico en CGH, el cual resulta esencial regulando el proceso de autorrenovación. Notablemente, EVI1 regula a factores de transcripción como GATA2, PBX1 y PLM, puede realizar modificaciones epigenéticas para silenciar ciertos genes por interactuar con histona desacetilasas y enzimas modificadoras de la cromatina, así como activar otros genes asociados a acetiltransferasas. RPN1 actúa como enhancer de la expresión de EVI1, con lo que el gen de fusión RPN1/EVI1 produce un incremento de la proliferación y bloquea la diferenciación celular, induciendo la transformación leucémica. Puede presentar cualquier patrón morfológico, a excepción de LPA, pero comúnmente se presenta como LMA sin maduración (subtipo M1 de la FAB), LMA mielomonocítica (M4) y LMA megacarioblástica (subtipo M7 de la FAB)20,29.
La última anormalidad citogenética recurrente en LMA es la t(1;22)(p13;q13), la cual presenta como marcador al gen de fusión RBM15/MKL1, donde se unen el motivo de unión al ARN de RBM15 con el motivo de unión al ADN involucrado en la remodelación de la cromatina de MKL1. Este gen de fusión, por lo tanto, modula la remodelación de la cromatina, la diferenciación asociada a HOX, así como interfiere en algunas vías de señalización extracelular. Su fenotipo sugiere principalmente una LMA megacariocítica (subtipo M7 de la FAB)20.
Mutaciones genéticasUn grupo importante de pacientes (aproximadamente el 45%) con diagnóstico de LMA presentan un cariotipo normal. Estos pacientes se clasifican con un pronóstico clínico intermedio debido a que clínicamente no se tiene un marcador de referencia y su origen biológico aún es desconocido. Recientemente, con el desarrollo de metodologías de secuenciación masiva se han identificado nuevas mutaciones genéticas asociadas con la LMA. Algunos de los genes identificados incluyen a: KIT, FLT3, NPM1, CEBPA, RAS, WT1, BAALC, ERG, MN1, DNMT, TET2, IDH, ASXL1, PTPN11 y CBL. De todos ellos destacan los que afectan a los genes FLT3, NPM1 y CEBPA, porque se han asociado con la respuesta al tratamiento y el progreso de esta enfermedad7,8,10,20,30.
En el 2008, la OMS publicó la actualización de la clasificación de neoplasias mieloides, siendo uno de los principales cambios en esta revisión la incorporación de las mutaciones en NPM1 y CEBPA como entidades dentro del grupo de LMA con anormalidades genéticas recurrentes. La mutación en FLT3 no fue incluida como una entidad independiente debido a que esta se asocia con varias entidades; sin embargo, su significancia no debe ser subestimada, ya que su identificación en pacientes con cariotipo normal o con alguna anormalidad cromosómica puede establecer el pronóstico de la leucemia20.
El gen FLT3 codifica un receptor tipo tirosina cinasa (RTK) que juega un rol crítico en la hematopoyesis y el crecimiento celular, debido a que regula diversos procesos celulares como proliferación, diferenciación y apoptosis celular. Normalmente reside en la membrana celular de manera monomérica, con una configuración que impide su activación31,32. La mutación más común en FLT3 involucra una duplicación en tándem interna (ITD) entre los exones 14 y 15 en el dominio yuxtamembrana, la cual varía en longitud y posición de paciente a paciente32. Se ha sugerido que el cambio conformacional ocasionado por el segmento de la duplicación de FLT3-ITD es responsable de eliminar el impedimento estérico que normalmente bloquea la dimerización sin estimulación del ligando, exponiendo diversos sitios dentro de los dominios tirosina cinasa que inducen su autofosforilación10,31,32. El principal impacto de FLT3-ITD es su asociación con altas cuentas blásticas, incremento del riesgo de recaída y disminución de la supervivencia. FLT3-ITD es especialmente frecuente en pacientes con cariotipo normal, t(15;17)(q22;q12) [PML-RARA] y t(6;9)(p23;q34) [DEK-NUP214]. Otras mutaciones asociadas ocurren en NPM1 y DNMT3a31–34. En contraste con la proteína FLT3 silvestre, FLT3-ITD activa la vía STAT5 significativamente. La proteína STAT5 induce la expresión de genes como la ciclina D1, c-MYC y p21, los cuales son importantes para la proliferación celular. Por otra parte, las proteínas Pu.1 y CEBPA, involucradas en la regulación de la diferenciación en células hematopoyéticas, son significativamente reprimidas, lo que sugiere su contribución en el bloqueo de la diferenciación10,31–33,35. El segundo tipo común de mutación en FLT3 son mutaciones puntuales missense en el exón 20 del loop de activación en el dominio tirosina cinasa (TKD). Casi todas estas mutaciones involucran la sustitución de un aspartato por una tirosina en el codón 835 (D835Y) por una mutación puntual (GAT→TAT). El aspartato en la posición 835 pertenece al dominio aspartato-fenilalanina-glicina (DFG) del loop de activación, que juega un rol crítico en la prevención de la unión eficiente del ATP, pudiendo adoptar una forma cerrada (inactiva) o abierta (activa). Estas mutaciones producen un cambio conformacional en la proteína, perturbando el balance energético requerido para estabilizar la forma cerrada, eliminando su función autoinhibitoria que provoca su activación constitutiva. También se han identificado otras sustituciones, deleciones e inserciones dentro de este codón y otros aledaños10,31,36.
NPM1 es una proteína que originalmente fue identificada como una fosfoproteína expresada en altos niveles en la región granular del nucléolo. NPM1 reside principalmente en el nucléolo, aunque se transporta rápidamente entre el núcleo y el citoplasma, lo que le lleva a tener parte en diversos procesos celulares, que incluyen el transporte de partículas prerribosomales y biogénesis de los ribosomas, la respuesta contra estímulos estresantes como radiación UV e hipoxia, el mantenimiento de la estabilidad genómica a través del control de la ploidía celular y la participación en procesos de reparación del ADN, la regulación de la transcripción a través del moldeamiento de los eventos de condensación y descondensación de la cromatina; previene la agregación proteica en el nucléolo y participa en la regulación de la actividad y la estabilidad de supresores tumorales cruciales como p53 y ARF. En realidad NPM1 funciona como histona chaperona, capaz de realizar el ensamblaje de histonas y del nucleosoma, así como promover un incremento de la acetilación dependiente de la transcripción37,38. Las mutaciones en el gen NPM1 son consistentemente heterocigotas, presentándose principalmente en el exón 12, con algunas pocas excepciones reportadas en el exón 11 y el exón 9. Aproximadamente 50 variantes genéticas han sido descritas, sin embargo en un 95% de los casos ocurren en la posición del nucleótido 960, siendo la mutación más común la duplicación de los nucleótidos TCTG en las posiciones 956 a 959, que es conocida como varianteA. Independientemente de la variante de la mutación, todas ellas generan modificaciones en el extremo C terminal de la proteína, generando un dominio de exportación nuclear adicional rico en leucina, y segundo, la pérdida de los residuos aromáticos 288 y 290, que son cruciales para la localización nucleolar. Por esta razón, una de las características distintivas de las mutaciones en NPM1 es su sobreexpresión en el citoplasma de células leucémicas con LMA (NPM1c+)39–41. Las mutaciones en NPM1 son muy estables, y la pérdida de la mutación generalmente se asocia con el cambio de cariotipo, de normal a anormal, y con una buena respuesta a la terapia y supervivencia a 5años. La presencia de NPM1 se correlaciona significativamente con la presencia de FLT3-ITD; en contraste, las mutaciones en tándem dentro del gen MLL son usualmente excluyentes con NPM1. Fenotípicamente se asocia con LMA mielomonocítica (subtipo M4 de la FAB) y LMA monocítica (subtipo M5 de la FAB)39,41,42.
CEBPA es un factor de transcripción que juega un rol fundamental en estados tempranos de la diferenciación mieloide y es particularmente expresado en células mielomonocíticas y específicamente es sobrerregulada durante la diferenciación granulocítica. CEBPA da lugar a 2 diferentes transcritos, usando 2 diferentes secuencias de inicio AUG dentro del mismo marco de lectura; la primera secuencia de inicio codifica una isoforma de 42KDa (p42), mientras que la segunda secuencia de inicio codifica otra isoforma de 30KDa (p30). Las células regulan la relación de p42/p30 a través la señalización celular desencadenada por rapamicina y la proteína cinasa R de la siguiente manera: bajo condiciones de crecimiento favorables, los factores de iniciación de la transcripción elF2α y elF4E incrementan su actividad, posiblemente a través del incremento de la actividad de c-MYC; a su vez, aquellas actúan promoviendo la transcripción de p30, que inicia el proceso de proliferación celular. De la misma manera, cuando existen bajos niveles de elF2α y elF4E se promueve la transcripción de p42, la cual induce diferenciación celular43,44. Las mutaciones en CEBPα son mutaciones puntuales que pueden afectar la transcripción de la variante p42, permitiendo la sobreexpresión de la isoforma p30, o bien la región de zipper de leucina (bZIP) y el dominio de unión al ADN, de manera que se afecta su interacción con el ADN en el surco mayor, su dimerización e interacción con otras proteínas. La mayoría de los pacientes poseen más de una mutación en C/EBPα, y el escenario más frecuente es la combinación de 2 mutaciones en alelos diferentes (una mutación que bloquea la transcripción de p42 y otra en el bZIP), las cuales se asocian con un pronóstico favorable, así como a la LMA sin maduración (subtipo M0 de la FAB) y a la LMA con maduración (subtipo M2 de la FAB)43,45,46.
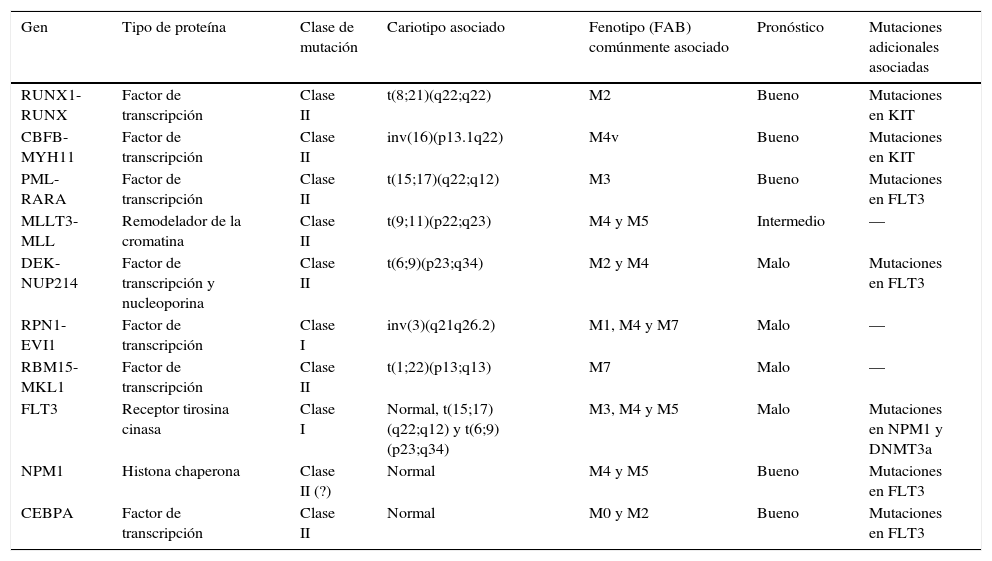
En la tabla 2 se simplifican las características de las diferentes mutaciones citogenéticas y genéticas presentadas.
Principales características de las anormalidades genéticas recurrentes en la Leucemia mieloide aguda
| Gen | Tipo de proteína | Clase de mutación | Cariotipo asociado | Fenotipo (FAB) comúnmente asociado | Pronóstico | Mutaciones adicionales asociadas |
|---|---|---|---|---|---|---|
| RUNX1-RUNX | Factor de transcripción | Clase II | t(8;21)(q22;q22) | M2 | Bueno | Mutaciones en KIT |
| CBFB-MYH11 | Factor de transcripción | Clase II | inv(16)(p13.1q22) | M4v | Bueno | Mutaciones en KIT |
| PML-RARA | Factor de transcripción | Clase II | t(15;17)(q22;q12) | M3 | Bueno | Mutaciones en FLT3 |
| MLLT3-MLL | Remodelador de la cromatina | Clase II | t(9;11)(p22;q23) | M4 y M5 | Intermedio | — |
| DEK-NUP214 | Factor de transcripción y nucleoporina | Clase II | t(6;9)(p23;q34) | M2 y M4 | Malo | Mutaciones en FLT3 |
| RPN1-EVI1 | Factor de transcripción | Clase I | inv(3)(q21q26.2) | M1, M4 y M7 | Malo | — |
| RBM15-MKL1 | Factor de transcripción | Clase II | t(1;22)(p13;q13) | M7 | Malo | — |
| FLT3 | Receptor tirosina cinasa | Clase I | Normal, t(15;17)(q22;q12) y t(6;9)(p23;q34) | M3, M4 y M5 | Malo | Mutaciones en NPM1 y DNMT3a |
| NPM1 | Histona chaperona | Clase II (?) | Normal | M4 y M5 | Bueno | Mutaciones en FLT3 |
| CEBPA | Factor de transcripción | Clase II | Normal | M0 y M2 | Bueno | Mutaciones en FLT3 |
A pesar de los grandes avances en la caracterización genético-molecular de la leucemia mieloide aguda, existen todavía muchas preguntas que esperan respuesta. Los estudios de secuenciación masiva han abierto la puerta para poder analizar gran cantidad de genes y sus mutaciones, sin embargo aún es necesario vislumbrar entre aquellas que solo forman parte del contexto de la enfermedad y aquellas que controlan los procesos celulares claves de la patología. El entendimiento del rol que juegan estás mutaciones en la leucemogénesis debe proveer las bases para el desarrollo de mejores y más específicas formas de prevención y tratamiento, que además se puedan extrapolar a otras tipos de cáncer.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.