


La investigación genómica y proteómica del cáncer aporta nuevos niveles funcionales de información y análisis sobre los cambios moleculares que se llevan a cabo en el tumor. La estructura de la secuenciación del DNA humano, que se realizó gracias al Proyecto Genoma Humano concluido en 2003, junto con la identificación de sus perfiles de expresión (RNAm) conforman las dos plataformas disponibles más importantes con base en las cuales se puede correlacionar el estado genómico con el estado celular funcional. Una tercera plataforma, que se encuentra en desarrollo, consiste en la identificación del estado dinámico de las proteínas, sus modificaciones postraducción, sus interrelaciones proteína-proteína, proteína-DNA, así como sus vías y redes funcionales. La integración de estas tres plataformas servirá para entender los principales eventos moleculares que participan en el desarrollo normal y en la fisiopatología de las enfermedades. En un futuro próximo, esta integración de información y análisis constituirá la base del diagnóstico clínico y del diseño terapéutico racional, dirigido a los blancos moleculares que ocasionan las enfermedades.
Los niveles funcionales de información, macroscópicos y microscópicos, en el estudio del paciente con cáncer han mejorado en forma progresiva y significativa en las últimas décadas, gracias al esfuerzo de investigadores clínicos y biomédicos. Inevitablemente, este avance ha tenido repercusiones en el tratamiento de los pacientes, y ha permitido que disfruten de una mejor calidad de vida y sobrevida. Si no se avanza en el entendimiento molecular de la fisiopatología de los diferentes tipos de cánceres, se postergará la posibilidad de lograr aún mejores resultados.
No se conocen por completo los eventos de señalización intracelular inherentes al fenotipo de las células tumorales. El paradigma de la oncología molecular consiste en la identificación de los genes, las proteínas y otras moléculas que participan en forma colectiva y dan origen a un comportamiento biológico específico, en cada paciente con cáncer.1
El Proyecto Proteoma Humano inició en 2001, sin embargo, debido a su magnitud y complejidad, sólo se han alcanzado metas parciales. En 2008 principió un proyecto de genómica funcional denominado, "Proyecto de los 1000 genomas", el cual ayudará a predecir el riesgo que tiene una persona de padecer ciertas enfermedades, su sensibilidad a los factores ambientales y su respuesta a determinados fármacos.
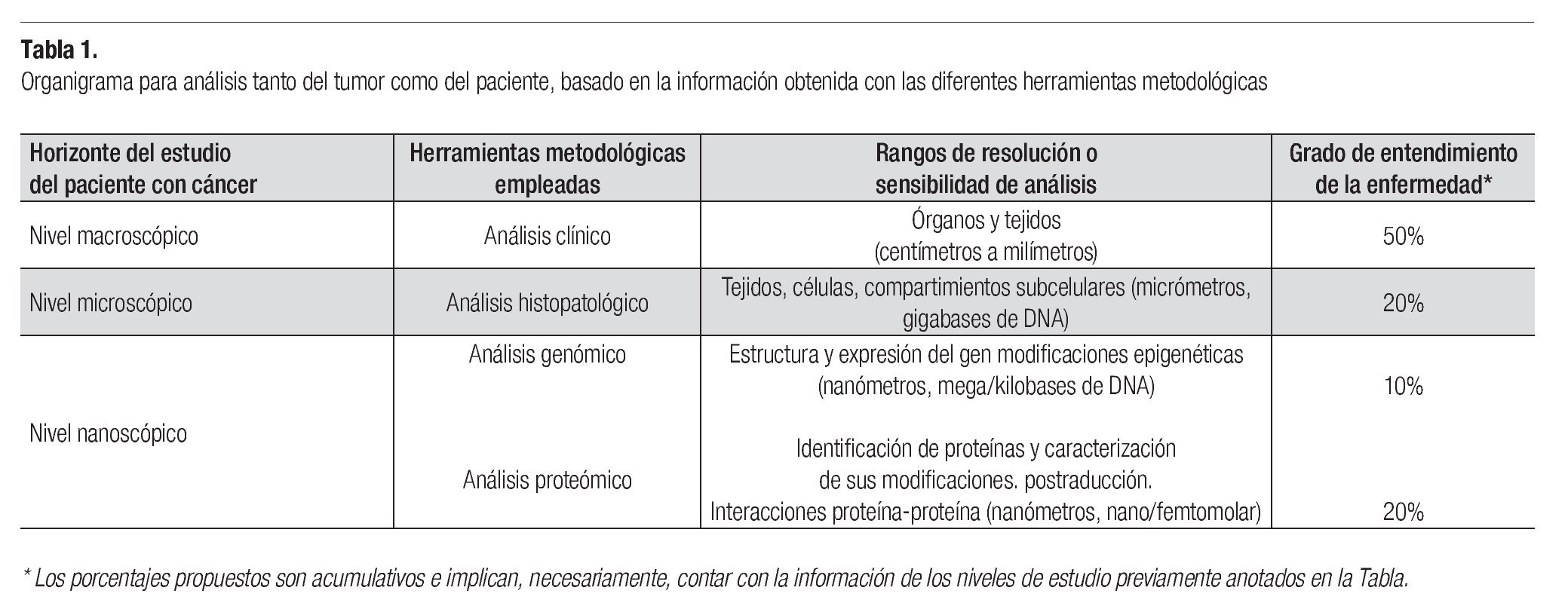
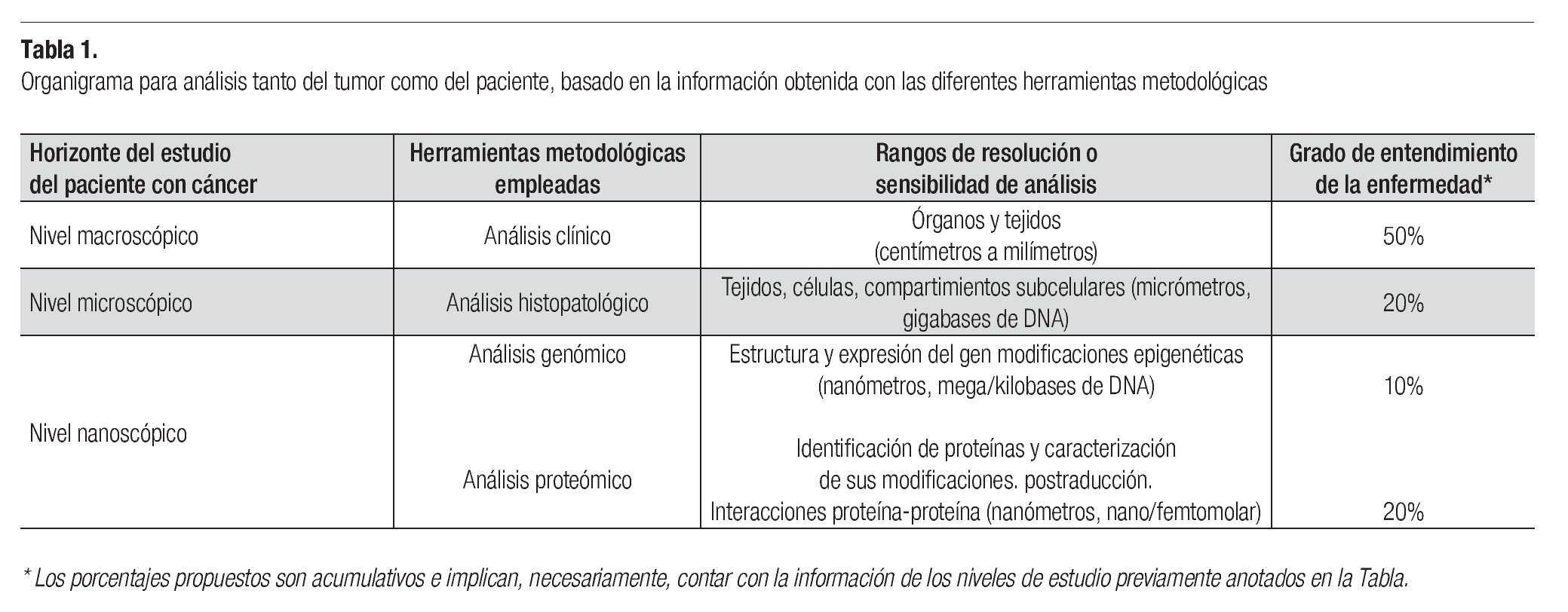
El presente artículo hace una revisión somera sobre los principales aspectos conceptuales de los niveles de información macroscópica, microscópica y, de forma más amplia, sobre los aspectos a nivel nanoscópico en los cuales con frecuencia trabajan los profesionales clínicos y biomédicos que se dedican al estudio y tratamiento de pacientes con cáncer. De manera complementaria, se hace énfasis en la información sobre los principales aspectos de oncología proteómica (Tabla 1).
¿ INFORMACIÓN Y ANÁLISIS DEL PACIENTE CON CÁNCER A NIVEL MACROSCÓPICO (DE CENTÍMETROS A MILÍMETROS)
Los estudios epidemiológicos, ya sean descriptivos (observacionales) o analíticos (experimentales), sobre las diferentes variables clínicas e histopatológicas que se encuentran en los pacientes con distintos tipos de cánceres han constituido las bases para la identificación de causas de la enfermedad, asociaciones causales, factores de riesgo (hábitos, costumbres, estilos de vida, exposiciones), entre otros, a partir de las cuales se identifican grupalmente las condiciones del paciente que predisponen al desarrollo de un tipo particular de enfermedad. De manera análoga, en los próximos años la epidemiología molecular se integrará a las herramientas para el diagnóstico clínico de las diferentes enfermedades, debido a que las interacciones entre el medio ambiente y los genes desempeñan una función significativa en el origen y desarrollo del cáncer.2 De esta manera, será posible determinar los perfiles genómicos y proteómicos del tumor y del paciente, que sirvan de apoyo para formular el diagnóstico clínico y para tomar las mejores decisiones terapétuicas.3
En los últimos 50 años, la tasa de incidencia y la mortalidad, causadas por las enfermedades neoplásicas en países que cuentan con sistema de salud de cobertura amplia, se han reducido en 12% y 25%, respectivamente.4 El mejoramiento del diagnóstico clínico y el tratamiento del paciente con cáncer se ha debido, en parte, al empleo de una tecnología diagnóstica más precisa (de segunda o tercera generaciones) con mayor sensibilidad y especificidad (con resoluciones milimétricas) y, en especial, al proceso de sistematización (estandarización) del diagnóstico y tratamiento, gracias a la aplicación de los sistemas de estadiaje-TNM (propuestos por organizaciones internacionales reconocidas como la UICC y la AJC), que valoran la enfermedad local (tamaño tumoral), regional (número de ganglios afectados) y la diseminación distante. Dichos sistemas, además, funcionan como guías referenciales que hacen posible valorar la progresión macroscópica de la enfermedad.
Otro valioso elemento que ha beneficiado a los pacientes con cáncer, es la investigación científica y tecnológica, que en forma constante añade al armamentarium terapéutico nuevos tratamientos cuya utilidad ha sido demostrada en protocolos clínicos de fase III, los cuales han sido aprobados por organizaciones internacionales reguladoras como la FDA y la EMEA. La aplicación de otros sistemas de evaluación clínica complementarios al sistema de estadiaje clínico del tumor-TNM en los pacientes con cáncer, como el uso de nomogramas, también han favorecido dicho progreso. Los nomogramas clínicos son propuestas de evaluación, que consideran las principales variables clínicas, histopatológicas de subgrupos particulares en pacientes con cáncer (una etapa clínica específica) y ayudan a predecir una condición particular (sobrevida a cinco años). Dichos nomogramas además de considerar los parámetros de los sistemas de estadiaje-TNM e histopatológicos, toman en cuenta los niveles séricos de biomarcadores, condiciones particulares del estado global del paciente y tipo de tratamiento aplicado. Toda esta información se traduce en coeficientes numéricos, que en la sumatoria final reflejan el riesgo pronóstico o predictivo de cada paciente. La aplicación de nomogramas ha demostrado gran utilidad para predecir tasas de sobrevida postratamiento en pacientes con cánceres de estómago y próstata, por medio de pruebas de validación internas y externas.5,6
¿ INFORMACIÓN Y ANÁLISIS DEL PACIENTE CON CÁNCER A NIVEL MICROSCÓPICO (DE MILÍMETROS A MICRAS)
Todos los tumores, benignos y malignos, comprenden dos componentes básicos en su estructura: el parénquima, constituido por las células neoplásicas proliferantes, y el estroma de sostén, conformado por tejido conectivo y vasos sanguíneos. La nomenclatura histopatológica tumoral se basa en el componente parenquimatoso. Una célula animal típica mide entre 10 a 20 μm de diámetro, el microscopio de luz permite observarlas con un límite de resolución del orden de 0.2 a 0.7 μm, que son las dimensiones correspondientes al tamaño de una bacteria o una mitocondria. Las células tumorales tienen una morfología alterada que se determina, principalmente, por su diferenciación o grado histológico (grado en el que las células cancerosas se asemejan a las células normales de las que proceden, tanto morfológica como funcionalmente) y de su anaplasia o ausencia de diferenciación que conlleva una falta de especialización o de la función celular. El diagnóstico clínico de cáncer se basa en el estudio histopatológico de la biopsia del tumor; el microscópico, en patrones morfológicos reconocidos por anatomopatológos bien entrenados (patrón de tinción, grado de diferenciación del tumor, forma y contorno nuclear, configuración celular y pleomorfismo, entre otros). El estadiaje anatomopatológico o quirúrgico consiste en el análisis histológico de todos los tejidos extirpados durante la cirugía, y constituye uno de los indicadores más importantes en la predicción de la respuesta al tratamiento en los niveles macro y microscópico.
El advenimiento de la técnica inmunohistoquímica, así como el uso de anticuerpos monoclonales conjugados a un marcador, ha permitido hacer subclasificaciones histológicas de los tumores, que tienen como sustento la identificación de la expresión diferencial de proteínas específicas en una imagen celular morfológicamente similar, observada con el microscopio de luz. La técnica inmunohistoquímica permite conocer la localización histomorfológica y subcelular de las proteínas. Sin embargo, no es cuantitativa, amén de que es poco sensible en condiciones de baja concentración del analito. Los estudios citogenéticos, de nivel microscópico, habitualmente se realizan en laboratorios especializados e investigan el cariotipo, el análisis de bandeo de los cromosomas (cromosomas en metafase) y la hibridación in situ con sondas fluorescentes de DNA. Estos estudios proporcionan información sobre mapas citogenéticos y sobre algunos cambios moleculares (mutaciones, fusión de genes, cambios cromosómicos). Distinguen aproximadamente 400 bandas cromosomales, cuando se realiza el bandeo de cromosomas en metafase, y cuando se efectúa en profase y prometafase, este número se incrementa al doble (los cromosomas se encuentran más extendidos); la resolución analítica en estos procedimientos es de giga a cientos de megabases de DNA.
¿ INFORMACIÓN Y ANÁLISIS DEL PACIENTE CON CÁNCER A NIVEL NANOSCÓPICO (DE DÉCIMAS DE MICRAS A NANÓMETROS/PICÓMETROS)
El requisito esencial para obtener información molecular de un grupo celular en particular consiste en obtener las células que se desea investigar. El estudio molecular de poblaciones celulares puras o específicas de los tejidos requiere el empleo de técnicas de microdisección, ya que las poblaciones puras por lo regular sólo constituyen una pequeña fracción del volumen tisular total. El problema de la heterogeneidad celular (contaminación celular, con células del estroma o de otros tipos) constituye una limitante importante para el análisis genómico o proteómico de grupos celulares homogéneos (células neoplásicas, preneoplásicas o normales). La técnica de microdisección mediante captura con láser (LCM) permite la diseccionar poblaciones celulares semejantes en secciones de tejido teñidas; su extracción se realiza por la acción de un pulso de láser. El acoplamiento de la LCM, para obtener poblaciones celulares puras, hace posible una mayor sensibilidad y especificidad de las técnicas de proteómicas y genómicas multiplex (PRC en sus distintas variedades, microarreglos de expresión de DNA, microarreglos de proteínas, secuenciación de proteínas por espectrometría de masas, etc.), con lo que se integra la biología molecular a la morfogénesis y la patología.1,3
Indicadores genómicos y proteómicos del tumor y del paciente. El cáncer es una enfermedad poligénica causada por alteraciones genéticas, epigenéticas o posgenéticas, que ocasiona diferentes alteraciones en los mapas de redes moleculares funcionales (predominantemente proteínas). El desarrollo del fenotipo celular neoplásico en cánceres de adultos se encuentra influido por múltiples condiciones como el microambiente tisular local, condiciones genéticas del hospedero (como polimorfismos del DNA), cambios metabólicos por de exposición a interacciones entre el ambiente y los genes, entre otras condiciones pro-cancerígenas (las cuales provocan alteraciones epigenéticas). Las redes moleculares que participan en el fenotipo de los diferentes procesos celulares están constituidas mayormente por proteínas. La proteómica corresponde a la genómica funcional de las proteínas, y comprende la identificación de las proteínas nativas celulares, así como de sus modificaciones estructurales o funcionales. De esta manera, el entendimiento completo del cáncer depende del análisis multidisciplinario, que combina aspectos de genética, patología, estructura y función de proteínas, biología celular, bioinformática y medicina clínica.7
Los datos que aporta la secuenciación del genoma humano constituyen una herramienta para entender las principales alteraciones genéticas que ocurren en el cáncer. Se ha calculado que una célula expresa aproximadamente 20,000 genes durante su vida, y que solo 4,000 se usan de manera cotidiana; de ellos sólo unos cuantos son susceptibles de ser modificados en la carcinogénesis (los que regulan proliferación, diferenciación o apoptosis). Durante el proceso de progresión del cáncer, las células tumorales pueden acumular una gran cantidad de translocaciones u otros tipos de alteraciones cromosómicas, así como mutaciones, hetero y homodeleciones o amplificaciones genéticas.
La exploración de la estructura (DNA) y expresión de genes (RNA) en seres humanos se ha desarrollado gracias a la construcción de sondas nucleotídicas de alta especificidad y afinidad. A partir de esta infraestructura, los investigadores biomédicos han empleado tecnologías recientes de cobertura numérica amplia, para explorar conjuntos de genes, desde cientos hasta miles, denominados microarreglos o chips. Éstos estructuralmente comprenden espacios micrométricos donde se adhieren las sondas nucleotídicas específicas de genes conocidos, organizados en una retícula cuadricular (columnas y renglones) de silicón, vidrio o nitrocelulosa, y a los cuales se añade el DNA o el RNA mensajero del tumor, que previamente se marca con una sonda fluorescente. De esta manera, se realiza la hibridación (conforme al principio de bases complementarias) bajo condiciones específicas (parecidas al de la PCR). La intensidad de fluorescencia que emite el híbrido en cada sitio corresponde a la cantidad del cDNA complementario a la sonda y, por ende, refleja la cantidad o el nivel de expresión de ese gen.
Para llevar a cabo el análisis de su identificación estructural o de su expresión genética, se emplean métodos estadísticos bioinformáticos multivariables, como el de agrupamiento jerarquizado. Con una metodología parecida, es posible identificar los micro RNA (miRNA), que corresponden a RNAs pequeños de entre 19 a 24 nucleótidos no codificantes, los cuales regulan negativamente la expresión de los genes, y participan en diferentes procesos celulares como apoptosis, diferenciación y desarrollo. Los miRNAs también participan en el inicio y progresión del cáncer, por lo cual algunos perfiles de su expresión han permitido mejorar las clasificaciones de ciertos tumores y la predicción de la sobrevida de los pacientes.8 Los resultados obtenidos en los ensayos de los microarreglos-DNA deben ser validados o verificados por medio de ensayos individuales en PCR de tiempo real.
De la genómica funcional a la proteómica. Como antes se expuso, el DNA es un archivo de información y las proteínas son las moléculas encargadas del trabajo celular, a través de su participación en las vías de señalización intracelular y extracelular de los principales procesos. El hecho de que exista una secuencia génica del DNA no garantiza la síntesis de su correspondiente proteína, ni sus modificaciones postraducción. La fosforilación de las proteínas indica su estado activo; la sobreactivación de varias de ellas se ha asociado a la patogénesis de algunas enfermedades y puede representar un blanco terapéutico en protocolos clínicos de tratamiento. El término proteoma deriva de las proteínas expresadas por el genoma; éste refleja una lista de productos genéticos que pueden estar presentes, mientras que el proteoma refleja una imagen dinámica las proteínas presentes y de las funcionales. Las células expresan miles de proteínas diferentes, y cada una de ellas puede experimentar numerosas modificaciones en respuesta a microambientes diferentes. Se ha proclamado que la Proteómica es el siguiente paso después de la Genómica, la meta de los investigadores consiste en integrar una biblioteca completa de todas las proteínas celulares, ya que actualmente sólo se ha catalogado una pequeña cantidad de ellas. No hay una metodología de amplio uso y de amplia reproducibilidad para estudiar las proteínas, como la PCR en el estudio del DNA, debido a la variabilidad de los 20 aminoácidos que comprende su estructura, y a que la secuenciación de cada proteína es relativamente lenta y requiere de un gran esfuerzo metodológico.1,7
El primer paso para estudiar la función de las proteínas es su purificación, y la principal técnica para separar las proteínas aún es la electroforesis en dos dimensiones (G2D), que separa las proteínas por su peso, carga, solubilidad y afinidad. Así, las proteínas individuales son separadas de acuerdo a su peso y carga en capas de gel; a continuación se separan del gel, para luego analizarlas por medio de otro tipo de ensayos. La visualización de manchas de proteínas en el gel requiere elevadas cantidades de proteína (del orden de miligramos) equivalentes al del contenido de miles de células específicas que interesa estudiar, por lo que una manera óptima de obtener proteínas representativas de poblaciones celulares puras es emplear la LCM.
Aplicaciones de ensayos de expresión del DNA en la práctica clínica. Aunque muchos investigadores se han mostrado entusiasmados al trasladar el uso de los ensayos de expresión génica a la práctica clínica, otros advierten que esto sólo podrá hacerse cuando se instituyan controles de alta calidad. Ntzani e Ioannidis 9 y Dupuy y Simon10 analizaron un numeroso grupo de estudios sobre los perfiles de la expresión génica en pacientes con cáncer, y encontraron que algunos patrones específicos de dicha expresión se asociaron a respuestas particulares de tratamiento. No obstante, observaron en su análisis que varios de estos estudios presentaban limitaciones metodológicas en su realización, y por ello concluyeron que se necesitan estudios adicionales que incluyan series numerosas, con diseños clínicos adecuados, aplicación de métodos de validación y con análisis estadísticos adecuados, antes de que puedan ser trasladados a la práctica clínica.
Entre algunos estudios de expresión de genes que cumplieron estos criterios, se encuentran los de van't Veer, van de Vijver y colaboradores,11,12 sobre la identificación de 70 genes asociados al riesgo de desarrollar metástasis distantes en pacientes con cáncer de mama y ganglios negativos (a partir de microarreglos que contenían 25,000 oligonucleótidos); su utilidad clínica fue validada interna y externamente (con una cohorte de 307 pacientes a quienes se hizo seguimiento durante 13.6 años). Para facilitar el uso de este microarreglo en la práctica clínica, los 70 genes identificados como marcadores pronósticos, se trasladaron a un microarreglo simplificado denominado MammaPrint, el cual emplea sólo 1900 sondas. La prueba del MammaPrint mide el nivel de expresión de cada uno de los 70 genes en la muestra del cáncer de mama, y emplea un algoritmo específico para predecir la sobrevida del paciente a 10 años.13 En 2007, esta prueba fue validada por la Food and Drug Administration para su aplicación clínica, con lo cual se convirtió en el primer microarreglo de expresión de genes en cáncer que se emplea como herramienta diagnóstica.
Las pruebas de microarreglos de expresión de genes empiezan a formar parte de la rutina clínica. Sin embargo, cabe la posibilidad de que la información del transcrito (RNAm) no corresponda al perfil de la activación de las proteínas en las vías de señalamiento molecular intracelular. Esto se debe, en parte, a que el RNA transcrito no correlaciona con los eventos funcionales pos traducción como la fosforilación, glicosilación o las interacciones proteína-proteína (la fosforilación constituye el evento de activación que con más frecuencia se identifica en las proteínas). Por ello, el transcriptoma proporciona una imagen incompleta de la red molecular.
El Proyecto de los 1000 Genomas, iniciado en 2008, se propone secuenciar los genomas de al menos mil personas de distintas etnias de todo el mundo. Su objetivo es generar una base de datos más detallada y que sea de mayor utilidad que la disponible hasta la fecha, sobre la variación genética humana llamada "variantes estructurales". Mejorará y ampliará la información obtenida por el Proyecto HapMap, el cual identificó 100 regiones asociadas al riesgo de enfermedades como diabetes, cánceres de mama, próstata, y otras. Asimismo, el Proyecto de los 1000 Genomas analizará el genoma humano a un nivel de detalle no realizado anteriormente, para lo cual empleará secuenciadores masivos de alto rendimiento y generará tan sólo en tres años, 60 veces más información sobre los mapas del DNA humano acumulados en los últimos 25 años, la cual estará accesible al público de manera gratuita (www.1000genomes.org).
¿ APLICACIONES DE LOS ENSAYOS PROTEÓMICOS EN LA PRÁCTICA CLÍNICA
En teoría, el camino más eficiente para identificar a los pacientes que responderán a una terapia dirigida molecularmente consiste en determinar (antes de iniciar el tratamiento) cuál vía de señalamiento intra/extracelular se halla sobreactivada en cada tumor.7 Esto idealmente se podría obtener con el análisis del material tisular tomado por biopsia. En general, las tecnologías proteómicas tienen limitaciones cuando la muestra es pequeña (menor al equivalente de miles de células) y/o contiene células no representativas (población heterogénea).
La manera más fácil de realizar un estudio protéomico consiste en comparar los proteomas en dos condiciones, tejido normal y tejido tumoral, observar sus diferencias, para lo cual se debe emplear como apoyo la información contenida en bases de datos como Protein Data Bank. Para realizar el análisis proteómico, inicialmente se separa la proteína de interés del resto, mediante electroforesis bidimensional en geles de poliacrilamida, luego se purifica/ concentra por medio de diversos tipos de cromatografía (de filtración en gel, de intercambio iónico, de afinidad) o por cromatografía liquida de alta presión (HPLC), para finalmente secuenciarla (por fraccionamiento bioquímico en péptidos y luego por degradación de Edman) o seleccionarla por la técnica de espectrometría de masas (separando los núcleos por su relación masa : carga, empleando en particular la espectrometría de desorciónionización en matriz inducida por láser-MALDI, y de ionización por electrospray ESI) o por medio de los inmunoensayos con empleo de anticuerpos específicos. En virtud de que no hay tecnología alguna que amplifique las proteínas, con frecuencia se requiere obtener un lisado celular de muchos miles de células. Una segunda limitación que tiene la mayoría de las tecnologías proteómicas es su incapacidad para analizar proteínas nativas, ya que dichas tecnologías desnaturalizan y alteran su configuración tridimensional.1,7,14
Los métodos inmunológicos para el estudio de proteínas aprovechan la especificidad de los anticuerpos por sus proteínas diana, un anticuerpo reconoce un dominio o grupo de aminoácidos en la molécula diana. Su uso se encuentra limitado, porque el número de anticuerpos es menor en relación al mayor número de proteínas a explorar.7 No obstante, cuando se logre bioproducir una amplia cobertura de anticuerpos, los inmunoensayos con anticuerpos específicos seguramente identificarán los blancos, así como los defectos en las vías de señalización oncogénicas de las células tumorales.
Los microarreglos, micromatrices de proteínas o biochips constituyen una nueva tecnología de la proteómica, que permite identificar las interacciones proteína-proteína, modificaciones postraducción, la variación que sufren las proteínas en distintas condiciones ambientales, los factores de transcripción activados o los blancos biológicamente activos de pequeñas moléculas. Además, los microarreglos de proteínas emplean comúnmente anticuerpos monoclonales para identificar proteínas específicas, detectan su concentración en rango de femtomolas, por ello su sensibilidad y especificidad son muy elevadas.1,14 Otras moléculas empleadas en la captura de las proteínas en los biochips son ácidos nucléicos, receptores, enzimas, aptámeros u otras proteínas. En los chips de proteínas, los métodos de detección más frecuentes son similares a los de los chips de DNA, como fluorescencia, quimioluminiscencia o colorimetría (con cromógenos). El bioanálisis de los datos se representa gráficamente en forma de mapa "caliente" (heat map), y se organiza a partir de un análisis bayesiano en el que se infiere la estimación. Hay dos principales clases de microrreglos proteómicos: (1) los ensayos de fase hacia delante (FPA) donde las moléculas cebadoras se depositan en la superficie del chip (generalmente anticuerpos) y atraen a la proteína de interés, que es reconocida por un segundo anticuerpo marcado; (2) ensayos de fase inversa, (RPA), donde el lisado celular (que puede contener muchas proteínas) se adhiere a la laminilla y funciona como cebador, luego se agregan diferentes anticuerpos específicos marcados que identifican las proteínas de interés.7,14 Los microarreglos FPA generalmente emplean dos anticuerpos para reconocer un proteína, uno cebador y otro marcador (contra diferente epítope); esta característica los torna menos prácticos.
En contraste, los RPA sólo utilizan un anticuerpo; la muestra a examinar (lisado en diferentes diluciones) se inmoviliza en la rejilla del microarreglo y de esta manera el ensayo sirve para múltiples muestras, para lo cual emplea uno o diferentes anticuerpos, en forma simultánea. Con los RPA es posible identificar diferentes proteínas, así como sus modificaciones postraducción (proteínas fosforiladas con anticuerpos específicos anti-fosfoproteína), y se puede lograr una sensibilidad de detección en picomoles (contenido equivalente al de 10-100 células). El análisis con los RPA de los lisados celular o tisular, permite explorar cuantitativamente las vías de señalamiento intracelular y extracelular con alta sensibilidad y especificidad.15 Estos microarreglos también se utilizan en el análisis de expresión diferencial de proteínas en células cancerosas o en tejidos tumorales con el fin de identificar biomarcadores, monitorear la respuesta celular a diferentes drogas o para explorar y mapear las vías de señalamientos moleculares. La gran ventaja de los RPA es su capacidad de detección simultánea de una gran cantidad de proteínas (en concentraciones de nano-picomolas) y/o muestras -lo que no se puede lograr con los ensayos convencionales de Western blot o ELISA (que además requieren concentraciones de micro-nanomolas- y la alta reproducibilidad de sus resultados (en comparación con Western blot y FPA). La implementación exitosa de esta tecnología requiere la disponibilidad de anticuerpos específicos de alta calidad, en particular que sean capaces identificar las modificaciones postraducción. Antes de emplear los anticuerpos específicos contra proteínas específicas en los microarreglos proteómicos, los anticuerpos deben validarse mediante ensayos de Western blot, bajo idénticas condiciones a las del ensayo de RPA. Para llevar a cabo este último, se diluye el lisado en diferentes proporciones, luego se coloca y fija en las placas de microarreglo, posteriormente se agrega el anticuerpo primario seguido del anticuerpo secundario, y se realiza la detección con un sistema de amplificación de la señal.
El reto actual que enfrentan los consorcios internacionales, como el Human Proteome Organization y el Human Protein Atlas,16,17 consiste en generar de bibliotecas integrales de anticuerpos, ligandos y sondas específicas que identifiquen todas las proteínas celulares. La lista de anticuerpos que ha mostrado adecuada especificidad a diversas proteínas a través de ensayos de Western blot, se encuentra en las bases de datos http://discover.nci.nih. gov/abminer y http://mtt.uscancer.org.
La tecnología RPA ha hecho posible la identificación de las proteínas sobreactivadas en las vías de señalamiento oncogénicas en 90 líneas celulares de cáncer,18 establecer la relación de algunos perfiles proteómicos con la sobrevida de pacientes con cáncer de próstata, linfomas y rabdomiosarcoma,19-21 así como la determinación de las diferencias en las vías de señalamiento intracelular entre el tumor primario y sus lesiones metastásicas (como en cáncer de colon y sus metástasis hepáticas o en el cáncer de ovario y sus metástasis en el epiplón).7,22 Los perfiles moleculares entre el tumor primario y sus metástasis son diferentes, y estos cambios pueden funcionar como biomarcas críticas para diseñar terapias moleculares. En la actualidad, esta tecnología se aplica en diferentes protocolos clínicos de tratamiento para diferentes tipos de cáncer; la respuesta terapéutica puede monitorearse mediante el registro de los cambios en el nivel proteómico de las moléculas blanco en las vías de señalamiento oncogénicas. Probablemente los principales requisitos para emplear los RPA en el laboratorio clínico son la estandarización de las actuales o futuras plataformas de chips de proteínas, así como la unificación, simplificación y estandarización de sus sistemas de análisis bioinformático para la simplificar la interpretación clínica.
Una de las aplicaciones prometedoras de las metodologías proteómicas es la detección de cánceres en sus etapas incipientes mediante la identificación de biomarcas (proteínas o péptidos fosforilados) en fluidos del cuerpo como suero, orina o saliva. Para la determinación en suero de la diferentes variedades de proteínas, sus fragmentos, isoformas y péptidos, se ha denominado a los que son menores de 50,000 dalton, peptidoma sérico (perfil peptídico), dado que la información que éste refleja permite reconocer miles de fragmentos peptídicos, derivados de los eventos enzimáticos celulares y extracelulares. En la circulación, diferentes especies de proteínas pueden adoptar diferentes isoformas, debido a las modificaciones postraducción, que varían en tamaño o en su unión a proteínas transportadoras (como albúmina). Pongamos por caso, cuando existe desequilibrio entre proteasas e inhibidores de proteasas extraceulares, se detecta un peptidoma sérico alterado. Algunos de los primeros estudios han podido encontrar cambios en el peptidoma sérico de pacientes con ciertos tumores malignos. Lowenthal y colaboradores.23 demostraron cambios en los fragmentos proteína BRCA2, que tenían diferentes tamaños, en mujeres con riesgo elevado para desarrollar cáncer de ovario, en pacientes con cáncer de ovario en etapas incipientes y en quienes lo padecían en etapas avanzadas. Kim y colaboradores.24 examinaros 35 biomarcas séricas de 98 pacientes con cáncer de mama, y encontraron que las concentraciones del factor de crecimiento epidermoide, del ligando soluble de CD40 y de la proapoliproteína A1 fueron mayores en comparación con los del grupo control.
Dadas las limitaciones actuales de las metodologías proteómicas para la identificación de biomarcadores en cáncer, se requiere el descubrimiento de nuevas generaciones de tecnologías probablemente basadas en inmunoensayos (como la inmuno-espectrometría de masas y la espectrometría de masas marcada con isótopos),25-27 que tengan la capacidad de determinar con mayor sensibilidad y especificidad (altos niveles de resolución y reproducibilidad) la identidad y el tamaño de estos biomarcadores.
Los estudios proteómicos presentes y futuros tendrán gran aplicación traslacional en los pacientes con cáncer; sus análisis basados en perfiles proteómicos podrán ser aplicados a corto plazo por el equipo clínico que atiende al paciente, como complemento de los estudios histopatológicos y de imagen, ya que será posible identificar las principales moléculas en las vías oncogénicas de los tumores de cada individuo. El análisis proteómico y genómico de lesiones tumorales recurrentes puede constituir la base para darle una nueva dirección racional a la terapéutica. Este cambio tendrá repercusiones directas en la práctica clínica, ya que impacta en todas las fases cruciales del cuidado y manejo del paciente con cáncer.
¿ AGRADECIMIENTOS
Hago patente mi reconocimiento a la Sra. Adriana Medina por su valiosa colaboración en la búsqueda documental.
Asimismo a la Sra. Margarita Castillo por su valiosa colaboración en la revisión de la redacción de este documento.
Correspondencia:
Dr. Víctor M. Valdespino.
Andrés Molina Enríquez 361 Col. Ampliación Sinatel México D. F. 09479 México.
Teléfono: 5674 3439.
Correo electrónico:valdespinov@yahoo.com