



El adenocarcinoma de próstata es la segunda causa de mortalidad por cáncer en los hombres a nivel mundial. La terapia de deprivación hormonal ha sido por más de 50 años el estándar de tratamiento. Aunque la castración de manera inicial se considera una terapia efectiva, la mayoría de los pacientes progresará a pesar de los bajos niveles de testosterona (cáncer de próstata resistente a la castración CPRC). Los andrógenos intratumorales y extragonadales representan un medio para el crecimiento, mediado por el receptor androgénico (RA) en el CPRC, por lo que se han convertido en blancos terapéuticos.
El análogo de la pregnenolona, acetato de abiraterona (AA), es un medicamento de administración oral, potente inhibidor de la enzima CYP17, clave en la biosíntesis hormonal androgénica, incluyendo testosterona y dihidrotestosterona a nivel testicular, adrenal e intratumoral. En el estudio fase 3 del AA, se demostró que la administración de 1 000 mg del AA y prednisona, en pacientes con CPRC previamente tratados con docetaxel, incrementa la supervivencia global (SG) de 10.9 meses en grupo placebo, y a 14.8 meses en grupo control (IR=0.65; IC 95%=0.54-0.77; p<0001). Dada la extensión en la SG, la Administración de Alimentos y Fármacos de los Estados Unidos de América (FDA, por sus siglas en inglés), en Estados Unidos aprobó en abril del 2011 su uso para el tratamiento de pacientes con cáncer de próstata metastásico, previamente tratados con quimioterapia basada en docetaxel.
Adenocarcinoma of the prostate is the second leading cause of death for men worldwide. Hormone deprivation therapy has been the standard of care in these patients for more than 50 years. Although castration might initially be considered an effective therapy, many patients will have disease progression despite low levels of serum testosterone (Castration Resistant Prostate Cancer). Intratumoral and extragonadal androgens are a source of androgen receptor-mediated growth in CRPC, reason for which they might be good targets to look at.
Abiraterone acetate (AA), a pregnenolone analog, is an orally administered potent CYP17 inhibitor, key to androgen byosynthesis including the synthesis of dihidrotestosterone and testosterone at a testicular, adrenal and intratumoral level. A phase III study of AA 1000 mg daily dose, in patients with CRPC previously treated with docetaxel, showed an increase in overall survival from 10.9 in the placebo arm to 14.8 months in the control arm (IR=0.65; IC 95%=0.54-0.77; p<0001).
Because of prolongation in overall survival, the Food and Drug Administration approved Abiraterone acetate in April 2011 for the treatment of patients with metastatic prostate cancer who had been previously treated with docetaxel.
Pagina nueva 1
¿ INTRODUCCIÓN
La importancia del adenocarcinoma de próstata radica de manera primordial en su epidemiologia, ya que constituye la segunda causa de muerte en hombres a nivel mundial, representa un 22% y 28% de los nuevos casos de cáncer en Europa y Estados Unidos de América (EUA), respectivamente.1,2
En los EUA se diagnosticaron aproximadamente 192 000 casos en el año 2009, y 27 300 pacientes fallecieron de esta enfermedad.3 Los hombres en los EUA tienen un 16% de probabilidad de ser diagnosticados con cáncer de próstata a lo largo de su vida, y aproximadamente un 3% de probabilidad de fallecer por esa causa. El índice de incidencia/mortalidad es de 7.1, en contraste con otros cánceres como mama, cuyo índice es de 1.2. Por la alta incidencia y mortalidad de esta patología, no existe ninguna duda acerca de la importancia de su tratamiento.4
En nuestro país, el Registro Hospitalario de Cáncer publicado en 2004 por Rizo y colaboradores, el cáncer de próstata ocupó el noveno lugar a nivel global y el segundo en hombres con 527 casos registrados del año 2000 al 2004, en el Instituto Nacional de Cancerología, lo que representa el 2.7% de todos los cánceres, presentándose con mayor frecuencia en adultos de edad avanzada, el 96.8% de los casos se presentó en adultos mayores de 50 años.5 De acuerdo con los reportes de defunciones en hombres por tumores malignos en el 2008 publicados por el INEGI, el mayor porcentaje de lesiones malignas fue en la próstata (8.1%). En nuestro país, el 70% de los casos de cáncer de próstata se diagnostican en etapas avanzadas.6 Una vez que se desarrolla enfermedad metastásica y resistencia hormonal, existen pocas opciones terapéuticas que prolonguen la supervivencia. La quimioterapia basada en docetaxel provee únicamente una ventaja en supervivencia de dos a tres meses, en comparación con otros esquemas de quimioterapia.7,8
El cáncer de próstata es un cáncer de crecimiento lento, muchos hombres con la enfermedad nunca experimentarán problemas derivados de ella y morirán sin que el cáncer alcance un significado clínico. La incidencia se incrementa a partir de los 40 años, llegando a su pico máximo a los 80 años. El 80% de los casos se diagnostican en hombres de más de 65 años, siendo la edad media del diagnóstico 72 años.7,8
La supervivencia específica del cáncer de próstata confinado al órgano no tratado es del 93.8% a los cinco años. En numerosos estudios prospectivos, la supervivencia global (SG) media de pacientes con cáncer de próstata metastásico ha permanecido relativamente estable durante las últimas cinco décadas, siendo aproximadamente de 24 a 36 meses.9
Desde los estudios iniciales de Huggins y Hodges en 1941, en donde se demostró el efecto benéfico de la castración quirúrgica y la administración de terapia hormonal basada en estrógenos en los pacientes con cáncer de próstata metastásico, la terapia de deprivación hormonal (TDH) ha constituido la piedra angular en el tratamiento del cáncer de próstata avanzado. Los tratamientos habituales están constituidos por la orquiectomía bilateral o la castración farmacológica, mediante la utilización de agonistas de la hormona liberadora de gonadotropinas (GHRH), ya sea solos o en combinación con algún antiandrógeno (bloqueo androgénico completo), teniendo como resultado disminución en el antígeno prostático específico (APE), regresión de masa tumoral y adecuada paliación de los síntomas.10
A pesar de esto, la terapia hormonal de manera exclusiva rara vez cura la enfermedad, y la recurrencia tumoral ocurre de un 30% a 40% de los casos, incluso cuando el tumor se encuentre bien localizado. Estas observaciones sugieren que las células tumorales son resistentes a la TDH, siendo capaces de replicarse y sobrevivir.10,11
¿ CÁNCER DE PRÓSTATA HORMONO REFRACTARIO
El término adecuado para nombrar a los pacientes con cáncer de próstata hormono refractario continúa siendo tema de discusión, sin embargo, el término "cáncer de próstata resistente a la castración" (CPRC) es el más aceptado, ya que se sabe que estos pacientes continúan siendo sensibles a manipulaciones hormonales secundarias. El CPRC se define como aquel cáncer que ha progresado a pesar de niveles de testosterona adecuados para la castración (< 50 ng/dL). Antes de 1990, los pacientes con CPRC se presentaban con pérdida de peso y sintomatología ósea. Actualmente, cuando se utiliza el APE para seguimiento de pacientes con CPRC, la mayoría de los pacientes son asintomáticos, manifestando progresión de la enfermedad únicamente por la elevación del marcador.12
Es necesario un mejor entendimiento de la biología del CPRC para proporcionar nuevos abordajes terapéuticos que sean clínicamente útiles, tanto en pacientes que han progresado después de un régimen de quimioterapia basado en taxanos, como en aquellos que no son candidatos para quimioterapia.
¿ MECANISMOS DE RESISTENCIA HORMONAL EN EL CÁNCER DE PRÓSTATA METASTÁSICO
El control el crecimiento tumoral en el cáncer de próstata es un elemento crucial. La testosterona es una hormona esencial para el crecimiento tumoral, por lo que es imperativo disminuir la síntesis de los andrógenos periféricos e intratumorales. Esta hormona se produce mediante el metabolismo de sus precursores androgénicos, dihidroepiandrosterona (DHE) y androstenediona, mediante la enzima CYP17, la cual convierte estos precursores esteroideos en andrógenos de 19 carbonos, posteriormente se convierte en el andrógeno 5 dihidrotestosterona (5 DHT), mediante la enzima 5α reductasa en la glándula prostática; tanto la testosterona como la 5 DHT, estimulan el crecimiento prostático.
La TDH estándar con orquiectomía o agonistas de hormona liberadora de la hormona luteinizante (LHRH), disminuye la producción testicular de andrógenos, sin embargo no tienen efecto en la producción adrenal. Los pacientes con cáncer de próstata que han sido sometidos a castración, ya sea quirúrgica o farmacológica, tienen de manera permanente un 10% de su basal de testosterona circulante, debido a la conversión de esteroides adrenales hacia testosterona.13 Se han detectado niveles elevados de RNA de la enzima CYP17 en pacientes con cáncer de próstata metastásico, cuya escala de Gleason es alta, esto sugiere que existen niveles residuales la enzima CYP17 involucrados en la síntesis autocrina de andrógenos, los cuales contribuyen a la detección de niveles elevados hormonales intratumorales.14 Se han postulado múltiples mecanismo de resistencia a la terapia androgénica, dentro de los cuales se incluyen la amplificación del receptor androgénico, hiperactivación del mismo sin ligando hormonal, mutación del receptor y activación por esteroides u otros ligandos,15 resultando en la producción continua adrenal e intratumoral de andrógenos, promoviendo la progresión de la enfermedad.16,17
¿ ABIRATERONA. BASES PARA EL DESARROLLO DE LA MOLÉCULA
La enzima clave que media la síntesis androgénica en los testículos y glándulas adrenales es la CYP17, cuyo gen se encuentra localizado en el cromosoma 19(q24.3), el cual codifica para la producción de la enzima, que se localiza en el retículo endoplásmico de las células de Leydig a nivel testicular, la región de la teca interna a nivel de los ovarios y en la zona fasciculada y reticular de las glándulas adrenales.18 Esta enzima microsomal cataliza dos reacciones esteroideas independientes, necesarias para la biosíntesis de andrógenos y estrógenos (17α hidrolixasa y 17,20 liasa) tanto a nivel testicular, adrenal e intratumoral.19 Debido al papel indispensable de esta enzima, la inhibición de la misma suprime la producción androgénica en todos los órganos endocrinos.
Los pacientes que cursan con deficiencia congénita de la enzima CYP17 no tienen insuficiencia adrenocortical, ya que la síntesis de cosrticosterona se encuentra intacta, sin embargo, la pérdida de la función de la CYP17 interrumpe la retroalimentación negativa con la hormona adrenocorticotrópica (ACTH), resultando en incremento en los niveles de la misma, así como de los niveles de precursores esteroideos.20
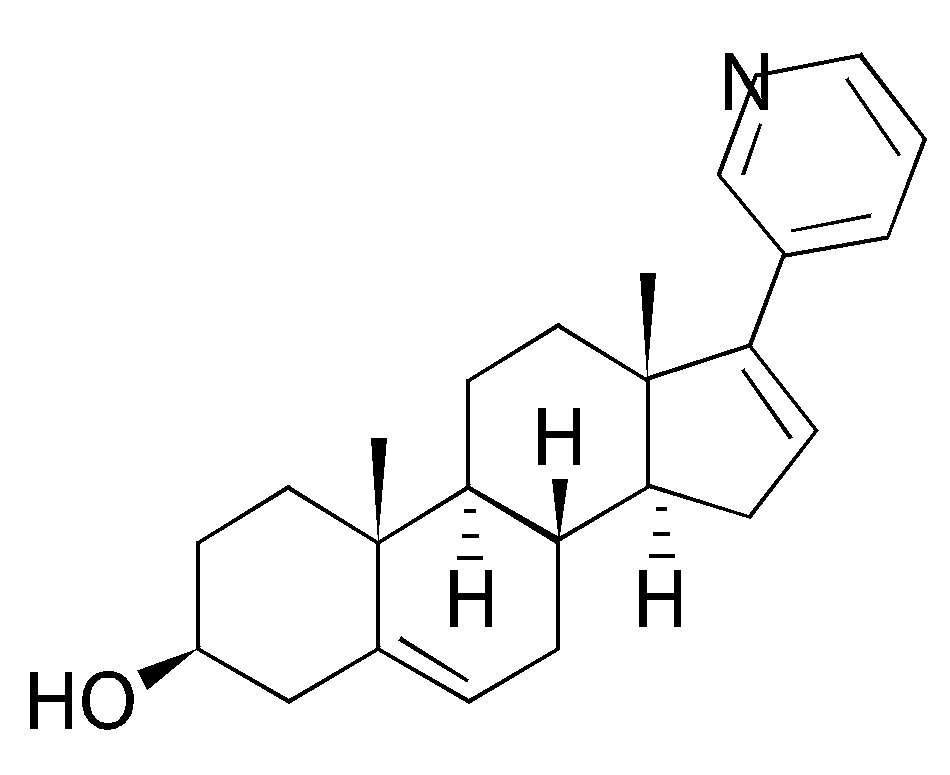
A pesar de que el ketoconazol es un inhibidor no selectivo de la enzima CYP17A1, este no ha demostrado un beneficio en la supervivencia en CPRC.21 Sin embargo, continúa siendo ampliamente utilizado en esta indicación. La eficacia del ketoconazol en el CPRC en pacientes que han progresado a la quimioterapia con docetaxel, únicamente ha sido evaluada en estudios retrospectivos,22 en los cuales la respuesta del APE (reducciones > 50%) se ha observado en aproximadamente 25% de los pacientes. Las toxicidades del ketoconazol son poco favorables, dentro de éstas se incluyen fatiga, náusea, vómito, efectos cutáneos, neuropatía periférica y elevación importante de las transaminasas.23 Las limitaciones del tratamiento con ketoconazol y el deseo de optimizar la TDP ha motivado el desarrollo de nuevos inhibidores más selectivos de la CYP17, siendo el compuesto más extensamente estudiado el AA24 (Figura 1).
¿ FARMACOLOGÍA
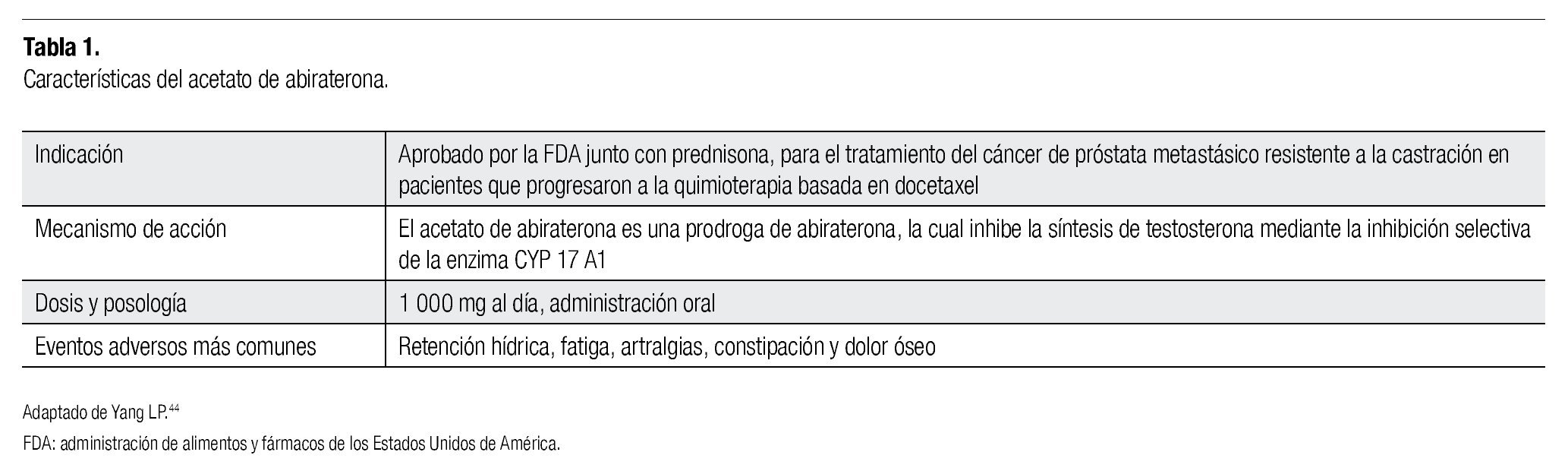
En la Tabla 1 se muestra un resumen de la actividad del AA, el cual es un compuesto esteroideo 3-piridil, derivado de la pregnenolona, desarrollada en el Instituto de Investigación de Cáncer del Reino Unido; es un potente inhibidor selectivo de la enzima CYP17 (Figura 1).25 Contiene dobles uniones en posición 16-17, la cual es necesaria para lograr la inhibición irreversible de la enzima CYP17, que junto con la sustitución 3- piridil resulta en una inhibición de 10 a 30 veces más selectiva de la enzima, en comparación con el ketoconazol. Para incrementar la disponibilidad oral de la abiraterona, se desarrolló la prodroga 3β-O-Acetato de Abiraterona, la cual es rápidamente metabolizada a su metabolito activo. El AA inhibe de manera primordial a la enzima CYP17 (IC50 72 nM), ya que se encontró que las enzimas CYP que están involucradas en las síntesis de glucocorticoides (CTP11B1), mineralocorticoides (CYP11B2) y metabolismo hepático de ciertas drogas (CYP3A4), no se inhiben con la misma intensidad que las CYP17. En modelos animales, el AA redujo la concentración de testosterona a ≤ 0.1nmol/L, y a pesar de incrementos de hasta cuatro veces en la hormona luteinizante (HL), no se encontró crecimiento de glándulas adrenales, ni aumento compensatorio en la ACTH.26
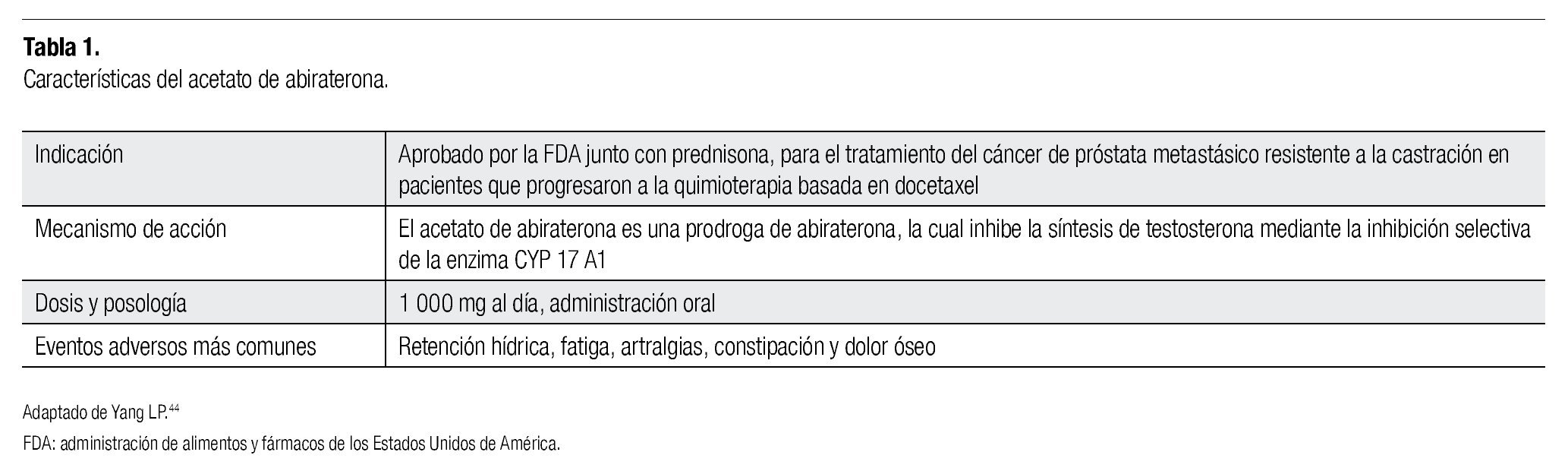
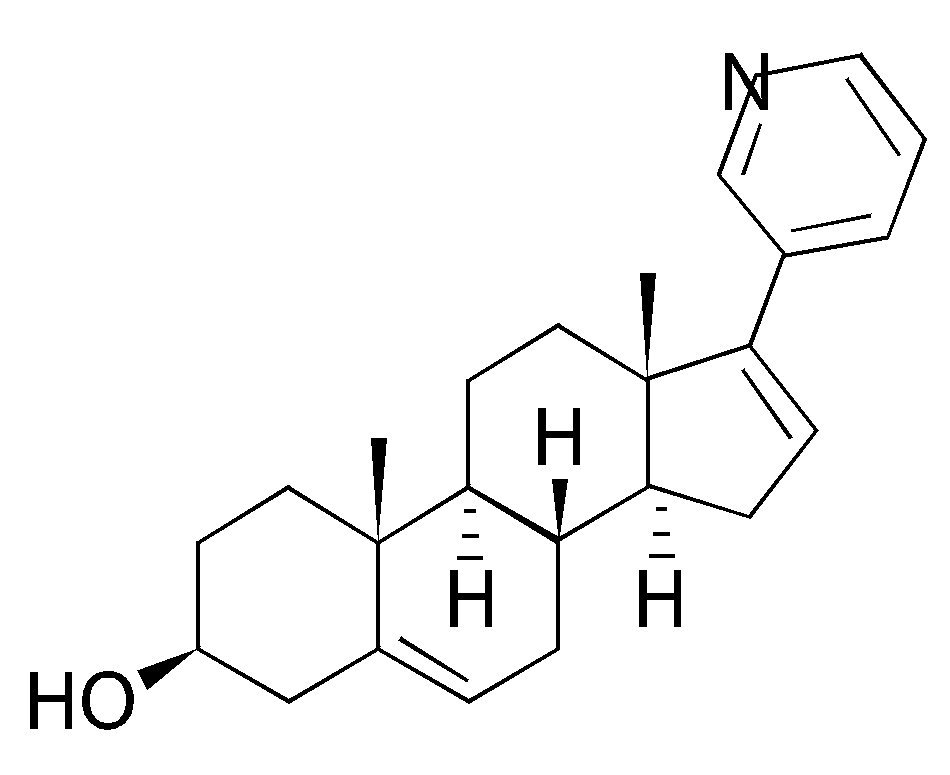
Figura 1. Molécula de acetato de abiraterona
¿ SEGURIDAD Y TOLERABILIDAD
El primer estudio fase I sobre el AA en humanos fue publicado por O`Donell en el 2004,27 reclutó 16 pacientes y tuvo como finalidad demostrar la habilidad del AA administrado como dosis única o múltiple de suprimir los niveles de testosterona en pacientes con cáncer de próstata metastásico, castrados como no castrados, que no hubieran recibido ningún tipo de quimioterapia previo. La dosis única del AA en aquellos pacientes sometidos a castración, ya sea quirúrgica o farmacológica, resultó en la supresión de los niveles de testosterona de dos a cinco días sin supresión de la 17α-OH progesterona o cortisol, aunque existió hipocorticolismo subclínico (pobre respuesta a la administración de ACTH). Se requirieron múltiples dosis > 800 mg al día en aquellos pacientes no sometidos a castración, para mantener los niveles de testosterona suprimidos, la mayoría de los pacientes alcanzaron la concentración plasmática máxima a las cuatro horas, con una vida media de aproximadamente 29 horas.
Existen dos estudios fase 1 que fueron determinantes para la realización de estudios posteriores con AA, el primer estudio incluyó pacientes no sometidos de manera previa a ketoconazol, mientras que el segundo incluyó pacientes con exposición previa.19,28 El primer estudio publicado por Attard y colaboradores en el 2008, tuvo como objetivo determinar la seguridad y tolerabilidad del AA, incluyó 21 pacientes con CPRC vírgenes a quimioterapia, se encontró que los valores de eliminación plasmática variaron entre 4 943.3 a 1 347.2 L/h. El área bajo la curva (AUC) y la concentración máxima (Cmax) se incrementaron al aumentar la dosis, aunque no de manera proporcional (R2=0.186 y R=0.049, respectivamente). La vida media fue de 10.3 horas de manera constante, cuando el AA se administró con alimentos de abundante contenido de grasa se incrementó de manera importante la exposición a la droga (4.4 veces) en comparación con la administración de la droga en ayuno (p=0.049). No existió incremento en la Cmax, sin embargo la absorción se incrementó de manera importante después de la administración con alimentos. Este fue el primer estudio que demostró que la inhibición selectiva de la enzima CYP17 es segura, y se asocia con una supresión sérica de los niveles de andrógenos duradera. El incremento de la dosis hasta 200 mg fue exitoso y no se encontraron toxicidades grado tres o cuatro, las toxicidades encontradas en este estudio fueron de manera predominante, los asociados a un exceso de mineralocorticoides y se trataron de manera exitosa con el antagonista del receptor de mineralocorticoides eplerrenona. Ningún paciente desarrolló insuficiencia suprarrenal y los autores concluyeron que, el AA debe administrarse con una dosis diaria de prednisona de 5 mg.
En el segundo estudio fase 1 realizado en la Universidad de California, un estudio de incremento de dosis, se reportó el impacto del tratamiento previo con ketoconazol.36 De los 33 pacientes, 18 (55%) experimentaron una disminución del APE ≥ 50%, de los 14 pacientes que no recibieron ketoconazol de manera previa, ocho (61%) respondieron al AA. De los 19 pacientes tratados con ketoconazol de manera previa, 10 (53%) experimentaron ≥ 50% de disminución en el APE, mientras se encontraban en tratamiento con AA y la media del tiempo de progresión fue de 21 semanas. Tres de cuatro pacientes que suspendieron el ketoconazol por toxicidad, experimentaron una disminución del APE. Siete pacientes de 15 que presentaron progresión de la enfermedad con ketoconazol tuvieron una disminución del APE con la administración del AA. Estos datos sugieren que los pacientes tratados de manera previa con ketoconazol no tienen un riesgo incrementado de toxicidad con el AA, y gran parte de estos pacientes responderán a esta terapia.
¿ EFICACIOA
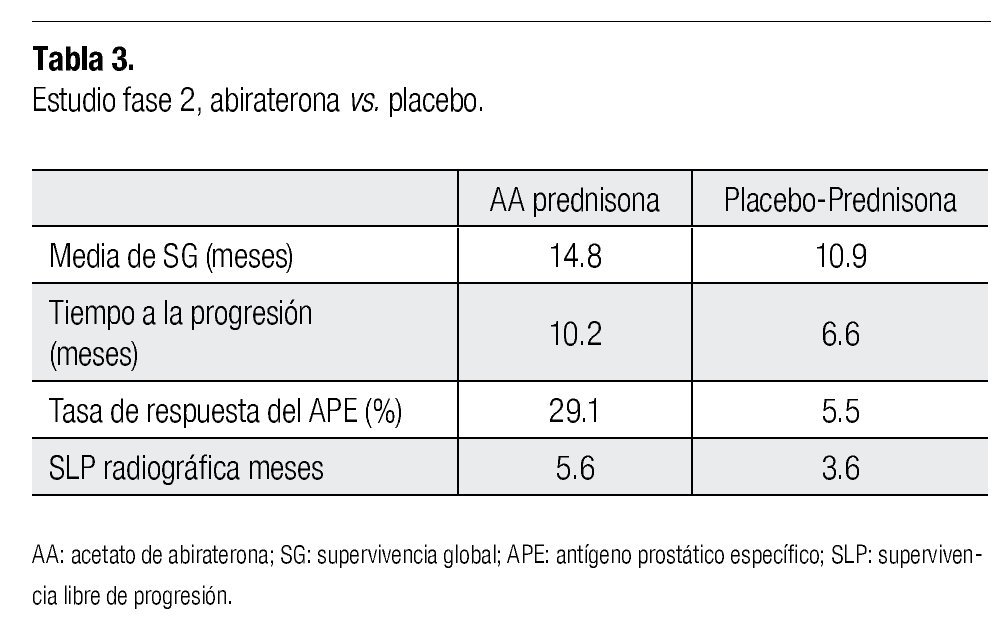
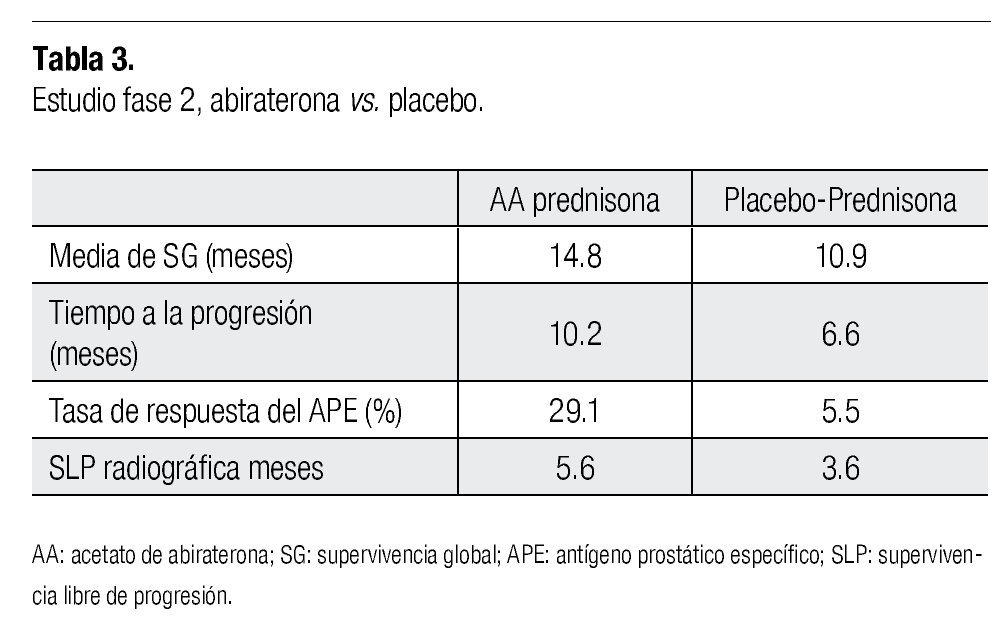
En un estudio fase 2,29 se reportó la eficacia del AA en dosis de 1 000 mg al día en pacientes con CPRC progresivo, antes y después del uso de docetaxel. En éste, ocho de los 47 pacientes reclutados habían recibido ketoconazol previamente y 18 pacientes se encontraban recibiendo dosis estables de esteroides, para mantener la clase funcional. El 45% de los pacientes tratados con AA alcanzaron una disminución del APE ≥ 50%. El tiempo promedio a la progresión fue de 169 días (IC 95%=82-200) y 12 pacientes (25.5%) permanecieron en el estudio al término de 48 semanas. Hipocalemia, hipertensión y retención hídrica ocurrieron en 26 (55%), ocho (17%) y siete (15%) de los pacientes, respectivamente, los cuales fueron tratados con eplerrenona y dosis bajas de esteroides. Un segundo estudio fase 2 que incluyó 58 pacientes con CPRC con progresión a docetaxel, demostró que el tiempo a la progresión (TTP) en aquellos pacientes que no habían recibido ketoconazol de manera previa, fue mayor que en aquellos con exposición anterior (198 vs. 99 días, respectivamente).30
La actividad del AA fue establecida en un estudio fase 3 de asignación aleatoria (estudio COU-AA-301), multicéntrico, en el cual se incluyeron 1 195 hombres previamente tratados con quimioterapia basada en docetaxel. Fueron asignados de manera aleatoria en un radio 2:1, para recibir AA (1 000 mg/día) más prednisona (5 mg dos veces al día) vs. placebo más prednisona, con el mismo horario de administración31 (Tabla 1).
El tratamiento se continuó hasta la progresión de la enfermedad (basado en el incremento del APE en suero, hallazgos por imagen o clínicos) o muerte. Se determinó la progresión de la enfermedad cuando existió incremento del dolor y deterioro en el estado general, así como cuando existió progresión objetiva, ya sea por imagen. Un incremento en el APE (o la falta de disminución del mismo), no se consideraron criterios para terminación del tratamiento. El ensayo clínico fue terminado basado en un análisis interino cuando los resultados excedieron los criterios previamente establecidos. Después de un seguimiento a 13 meses, el tratamiento con abiraterona mejoró significativamente la SG, considerado el objetivo primario del estudio, comparado contra placebo (media de 14.8 vs. 10.9 meses; tasa de riesgo (HR) 0.65; IC 95%=0.54-0.77). Los beneficios fueron similares para todos los análisis pre-establecidos. Se encontraron también mejorías estadísticamente significativas en el tiempo de progresión del APE, supervivencia libre de progresión (SLP) y respuesta en la disminución del APE (10.2 vs. 6.6 meses, 5.6 vs. 3.6 meses y 29% vs. 6%, respectivamente).
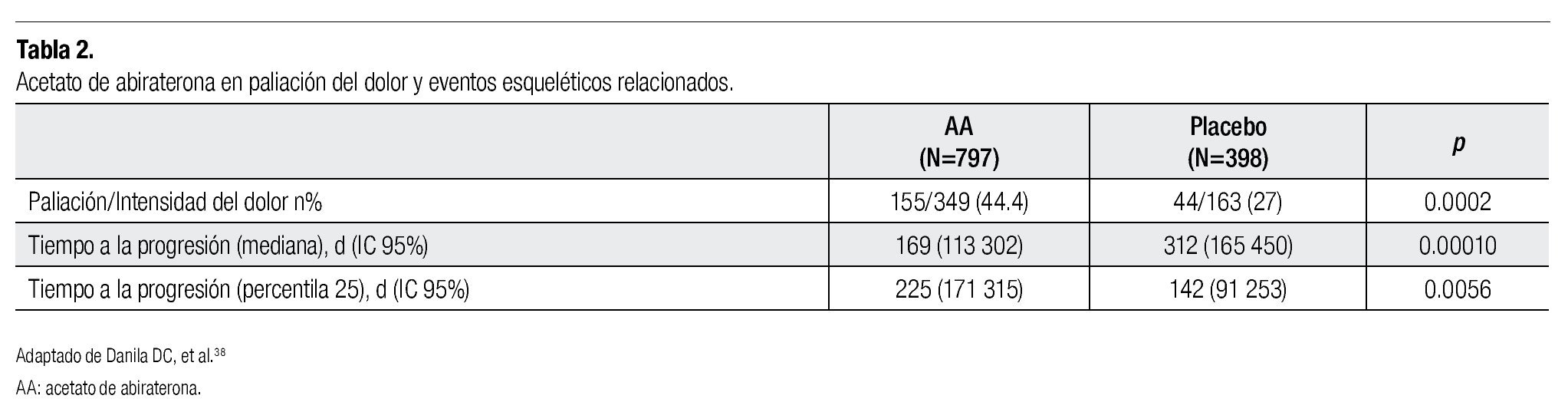
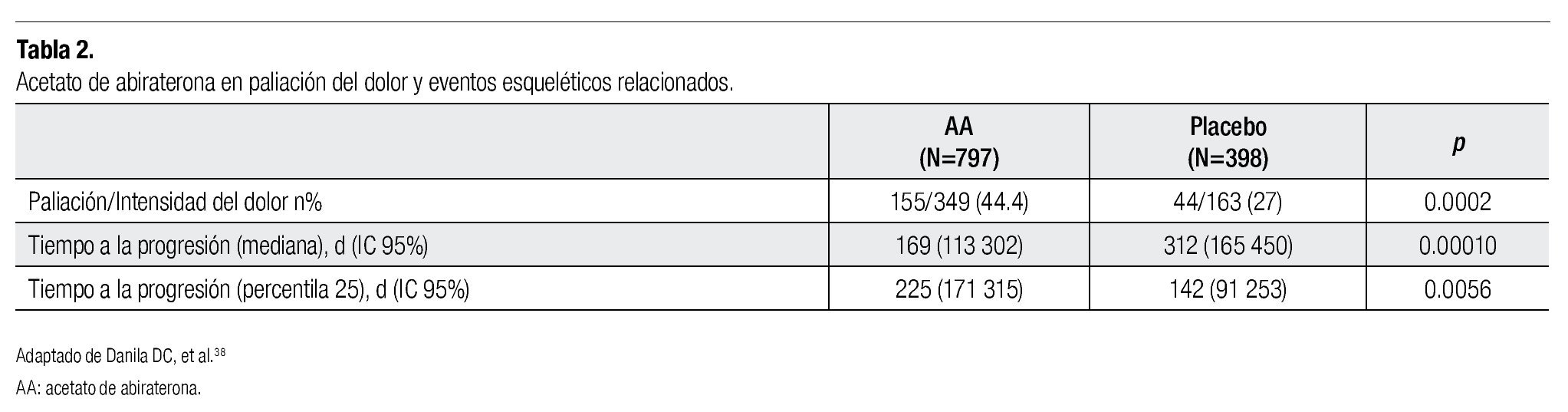
Los análisis secundarios de este estudio fase 3 se han enfocado en el impacto de abiraterona en la mejoría de los síntomas por metástasis óseas. De los 797 pacientes incluidos en el estudio, el 90% de los pacientes tenían metástasis clínicamente significativas, el 44% de los pacientes cursaron con dolor clínicamente significativo secundario a estas metástasis. En el brazo de abiraterona más placebo existió una mejoría significativa en la paliación del dolor en comparación con el brazo de placebo y prednisona, esto incluyó mejoría del dolor intenso en aquellos pacientes con al menos dos ciclos de tratamiento de paliación para el dolor (44 vs. 27%), tanto la mediana de duración de la mejoría del dolor como la mediana del tiempo de progresión de la intensidad del dolor fueron mejores en el brazo de abiraterona (10.3 vs. 5.6 meses y 7.4 vs. 4.7 meses, respectivamente). Existió un incremento significativo en el tiempo a la aparición de algún evento óseo (se definió como la presencia de alguna fractura patológica, síndrome de compresión medular o necesidad de radioterapia paliativa o cirugía). Se concluye en los análisis posteriores que en cuanto a paliación del dolor, el AA más prednisona ofrece beneficios clínicos significativos, incluyendo alivio del dolor y retardo en la recurrencia del dolor, previniendo también la aparición de nuevos eventos óseos relacionados31 (Tabla 2).
¿ TOLERABILIDAD
El perfil de toxicidad de abiraterona es dependiente de su mecanismo de acción, la inhibición selectiva de la enzima CYP17 regula la conversión de pregnenolona y esteroides relacionados en andrógenos.20
Los datos clínicos obtenidos hasta el momento han demostrado que la inhibición continua de la enzima CYP17 resulta en un incremento de la enzima liberadora de corticotropina, resultando a su vez en un aumento de hormonas esteroideas, incluyendo corticoterona y deoxicorticosterona; el incremento de la ACTH puede causar a su vez, producción adrenal de mineralocaorticodes que conducen a hipertensión e hipocalemia.33 Cuando el AA se administra sin glucocorticoides concomitantes, los pacientes no desarrollan insuficiencia adrenal, ya que la producción de cortisol se encuentra preservada. Los efectos producidos por el exceso de mineralocorticoides pueden ser atenuados mediante la coadministración de prednisona, la cual reduce la estimulación mediada por ACTH.34 En el estudio fase 3,31 la incidencia de eventos adversos graves (tres o cuatro) que llevó a la interrupción del tratamiento fue similar en el grupo de abiraterona y placebo. Los eventos adversos más comunes en el grupo de abiraterona fueron retención hídrica (31% vs. 22%) e hipocalemia (17% vs. 8%), hipertensión (9.7 vs. 7.9%), se observaron alteraciones en las pruebas de funcionamiento hepático (10.4 vs. 8.1%) y alteraciones cardiacas (13.3 vs. 10.4%), siendo más frecuentes la taquicardia y la fibrilación auricular, la tasa de eventos cardiacos grado 5 fue idéntica en los brazos del estudio: 1.3% (n=5) en el brazo de AA y 1.3 % (n=5) en el brazo placebo (Tabla 3).
¿ ACETATO DE ABIRATERONA EN PACIENTES CON CPRC METASTÁSICO VÍRGENES A GUIMIOTERAPIA
En un estudio fase 1/2 que incluyó 54 hombres con CPRC vírgenes a tratamiento (dos con tratamiento previo con ketoconazol), 42 de los 54 hombres en la fase extendida recibieron 1 000 mg de AA al día.33 Las diminuciones del APE ≥ 50% y del 90% fueron observadas en 28 (67%) y ocho (19%) de los 42 pacientes, respectivamente. Además, nueve (37.5%) de los 24 pacientes con enfermedad medible tuvieron una respuesta parcial (RP) de acuerdo a los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST). De los 17 pacientes con enfermedad medible por tomografía, 10 pacientes (59%) tuvieron disminución de ≥ 5-7.5 mL a < 5-7.5 mL y 12 pacientes (70%) tuvieron disminución ≤ 30%. La mediana de progresión del APE (TTP) fue de 225 días. La administración de dexametasona en dosis de 0.5 mg a día, al momento de la progresión disminuyó la resistencia en 33% de 39 pacientes.19,33 El AA demostró excelente tolerabilidad como monoterapia, aunque los síntomas de exceso de mineralocorticoides fueron evidentes, hipocalemia (88%), hipertensión (40%) y retención hídrica ocurrieron de manera frecuente, sin embargo, fueron manejados de manera exitosa con 50-200 mg de eplerrenona al día. Los bochornos se presentaron en cuatro de 42 pacientes y dos pacientes (5%) desarrollaron transaminasemia asintomática grado 3, la cual se resolvió con la suspensión del tratamiento.
Ryan y colaboradores publicaron un estudio fase 3, en el que se aleatorizaron 1 088 pacientes con CPRC, para recibir 1 000 mg de abiraterona más prednisona 5 mg al día contra placebo más prednisona, se demostró un beneficio en la supervivencia libre de progresión radiográfica de 16.5 contra 8.3 meses. Aunque a los 22 meses de seguimiento se encontró un beneficio en supervivencia global a favor de grupo control, no se alcanzó la significancia estadísitca. La administracion de abiraterona y prednisona demostró superioridad en el tiempo de inicio de quimioterapia, uso de opioides para tratamiento del dolor asociado a cancer, y progresion del APE comparado contra placebo. 35
¿ PERSPECTIVAS FUTURAS
En la Tabla 4 se enumeran los estudios clínicos en desarrollo para AA. Un segundo estudio fase 3 internacional en pacientes con CPRC metastásico compara el AA (1 000 mg/día) más prednisona (5 mg dos veces al día) vs. placebo más prednisona, en pacientes asintomáticos o mínimamente sintomáticos vírgenes a tratamiento con quimioterpia y ketoconazol, únicamente con metástasis a ganglios linfáticos (NTC00887198) (Tabla 4). Los pacientes se aleatorizaron en un radio 1:1, con 1 000 pacientes planeados para participar en el estudio, con un poder estadístico para detectar el 20% de mejoría en la supervivencia de 22 a 27.5 meses. El reclutamiento fue terminado en el 2009. El objetivo primario es la SG. Estudios del AA combinado con terapia de deprivación androgénica (TDA) convencional y radioterapia en enfermedad localmente avanzada se están llevando a cabo (Tabla 4). Debido a la excelente tolerancia se esperan estudios en combinación con agentes biológicos, quimioterapia y otros agentes de inhibición de la vía androgénica como la molécula MV3100. De manera notable, la combinación de docetaxel con ketoconazol cada tres semanas ha sido factible, sin embargo se requiere una disminución considerable de la dosis.36 A diferencia de ketoconazol, el AA no modula de manera significativa la enzima CYP3A4, permitiendo su combinación en dosis completas.36

¿ CONCLUSIONES
El CPRC es una enfermedad altamente letal, la cual depende en gran medida de la producción de andrógenos y las vías de señalización que determinan el crecimiento tumoral. Siendo una enfermedad heterogénea, se requieren tratamientos individualizados basados en un mejor entendimiento de la biología tumoral. El AA es un inhibidor selectivo de la biosíntesis de la enzima CYP17, el cual ha demostrado inhibir la síntesis androgénica de manera persistente tanto adrenal como intratumoral, suprimiendo el crecimiento tumoral en el CPRC, lo que constituye el primer agente terapéutico de administración oral aprobado por la FDA, que ha demostrado incrementar la SG y mejorar la calidad de vida, en pacientes con cáncer de próstata metastásico previamente tratados con quimioterapia basada en taxanos.
CONFLICTO DE INTERESES
La Dra. Lucía Martínez Hernández, es médico internista certificado por el Consejo de Medicina Interna. Al momento de redactar el documento, se desempeñaba como Enlace Médico Científico para Jannsen México en el área de oncología.
La Dra. Marcela Martínez Prieto es médico internista y oncóloga certificada por el consejo de ambas especialidades. Desde hace 4 años es gerente para el área de oncología en Janssen México.
En la preparación y redacción de este manuscrito no existió ningún tipo de apoyo financiero, ni en material de investigación, por parte de Janssen México. No existe por parte de los autores ningún conflicto de intereses para la publicación de este artículo.
Correspondencia:
Dra. Lucía Martínez Hernández.
Janssen de México S. de R.L de C.V.
Av. Miguel Ángel de Quevedo N° 247, Colonia Romero de Terreros,
C.P. 04310, Delegación Coyoacán, México D.F., México.
Celular: (04455) 16862019.
Correo electrónico: lmarti41@its.jnj.com