


El quiste óseo aneurismático (QOA) es un tumor benigno de hueso, que consiste en cavidades llenas de sangre revestidas por septos de tejido conectivo. La conocida asociación del QOA con otras lesiones neoplásicas, así como la dificultad del diagnóstico diferencial, obligan al estudio exhaustivo con el examen histológico de todo el material obtenido por curetaje además de un seguimiento radiológico estrecho. Se reporta por su rareza y dificultad diagnóstica, el caso de un osteosarcoma telangiectásico (OT) localizado en fémur en una paciente con antecedente de QOA. No existe algún otro informe de este tipo en nuestro país.
Se describe el caso de una paciente que desarrolló un OTen el sitio de un QOA previamente tratado tras haberse realizado curetaje completo e injerto óseo del quiste. Se describen las manifestaciones clínicas, radiológicas y de microscopia de luz.
La evolución del presente caso ilustra la gran dificultad para diferenciar un QOA de un OT. En estos casos la evolución clínica puede orientar hacia la conducta más adecuada, sin llegar a sobre tratar una lesión benigna, ni dejar una neoplasia maligna sin tratamiento. Poco se sabe del tiempo que transcurre desde el origen de un osteosarcoma (OS) hasta el momento de su manifestación clínica, que en este caso fue precedida por un QOA, pero no podemos asegurar que la neoplasia no haya estado presente desde el inicio. No se puede excluir la posible alternativa de que el QOA se transformó en una lesión maligna.
Aneurysmal bone cyst (QOA) is a benign bone tumor consisting of blood-filled cavities lined by connective tissue septa. The known association with other neoplastic lesions QOA and the difficulty of differential diagnosis, requiring the comprehensive study on histological examination of the material obtained by curettage and a close radiological monitoring. We report for their rarity and diagnostic difficulty, a case of telangiectatic osteosarcoma (OT) located in the femur in a patient with a history of QOA. There is no report of this kind in our country.
We describe a patient who developed an OT at the site of a previously treated QOA after obtaining complete curettage and bone grafting of the cyst. We describe the clinical, radiographic and light microscopy.
The development of this case illustrates the great difficulty in differentiating a QOA of OT. In these cases the clinical course can be directed to more appropriate behavior, without being over treated a benign lesion or let a malignancy lesion without treatment. Little is known about the time from the origin of osteosarcoma at the time of clinical manifestation in this case was preceded by a QOA but we cannot ensure that the tumor was not present from the start. One cannot exclude the possible alternative of aneurysmal bone cyst was transformed into a malignant lesion.
¿ IntRoduccIón
El quiste óseo aneurismático (QOA) es un tumor benigno de hueso, que consiste en cavidades llenas de sangre revestidas por septos de tejido conectivo. Los QOA ocurren en pacientes menores de 20 años, con afección de huesos largos en un 50% de los casos. Los síntomas más comunes son dolor local y edema. La duración de los mismos es de tres meses con diagnóstico de hasta un año. Radiológicamente, se observan como lesiones centrales y fusiformes o excéntricas y expansibles con bordes bien demarcados. Histológicamente son lesiones vasculares con espacios llenos de sangre, septos compuestos de fibroblastos, células gigantes tipo osteoclastos, histiocitos y trabéculas de hueso osteoide y lamelar. Los hallazgos citogenéticos sugieren que al menos algunos de los QOA son verdaderas neoplasias.1 Recientemente, se hizo evidente el origen neoplásico de los QOA por la demostración de translocaciones cromosómicas clonales de la banda 17p13, que ponen al oncogén USP6 bajo la influencia reguladora de la gran actividad promotora de CDH11.2 Estas lesiones se han designado como primarias o secundarias, con el último término utilizado cuando la evaluación histológica indica una lesión coexistente, que pueden ser benigna o maligna.2
El osteosarcoma telangiectásico (OT) es considerado una variante histológica del osteosarcoma (OS). Comprende el 12% de todos los OS. El grupo de edad más afectado corresponde a la segunda década de vida, y es más frecuente en los varones. Radiológicamente, aparece como una lesión lítica, con mínima calcificación o formación ósea. Tiene un crecimiento expansivo masivo con márgenes no delimitados y puede simular un quiste óseo. Microscópicamente su apariencia es quística; a la histología muestra espacios dilatados que contienen sangre y las células tumorales están confinadas a las bandas de tejido que circunscriben estos espacios Las características clínicas y radiológicas pueden confundirse con la presencia de un QOA, por lo que a menudo es difícil distinguir entre estas dos lesiones, especialmente en su etapa temprana. Por tanto, es necesario un estudio minucioso que abarque las características epidemiológicas, clínicas, radiológicas e histológicas de estas dos lesiones.3
Al igual que en otros tumores benignos, se ha informado que el QOA ha sido objeto de transformación maligna, aunque la mayoría de las transformaciones fueron inducidas por radiación.3 En una revisión de la literatura médica, sólo se encontraron seis informes bien documentados de la transformación maligna de un QOA, sin antecedentes de radiación.4-9
El OS es el tumor óseo primario más frecuente en la población pediátrica. En la actualidad, gracias a un diagnóstico oportuno y al uso de tratamiento multimodal, la posibilidad de curación rebasa el 70%.10
El QOA es una lesión benigna intraósea, representa el 1% a 2% de todos los tumores óseos primarios. Es más frecuente en las primeras dos décadas de la vida, con una prevalencia similar para ambos géneros. Sólo un tercio de los casos de QOA se presentan de forma secundaria, y aproximadamente el 70% ocurre de forma primaria. La mayoría de los pacientes tienen menos de 20 años de edad al diagnóstico, con un promedio de 13 a 17.7 años.11 La localización más frecuente del QOA primario es en pelvis y huesos planos hasta en un 50%, 30% en el esqueleto axial, el resto en los huesos del esqueleto cráneo-facial o apendicular.12 Habitualmente se presentan como una lesión radiotransparente, bien circunscrita con marcada expansión ósea. Está constituido por espacios cavernosos rellenos de sangre, de tamaños variables. Histológicamente, la imagen muestra espacios cavernosos, sin revestimiento epitelial, rodeados por un estroma fibroso que contiene células gigantes multi-nucleadas y matriz osteoide.13
El objetivo del artículo es presentar el caso clínico de una paciente con antecedente de QOA que recibió tratamiento con procedimientos de curetaje, y después de tres años desarrolló un OT. No existe algún otro informe de este tipo en nuestro país.
¿ PResentAcIón del cAso
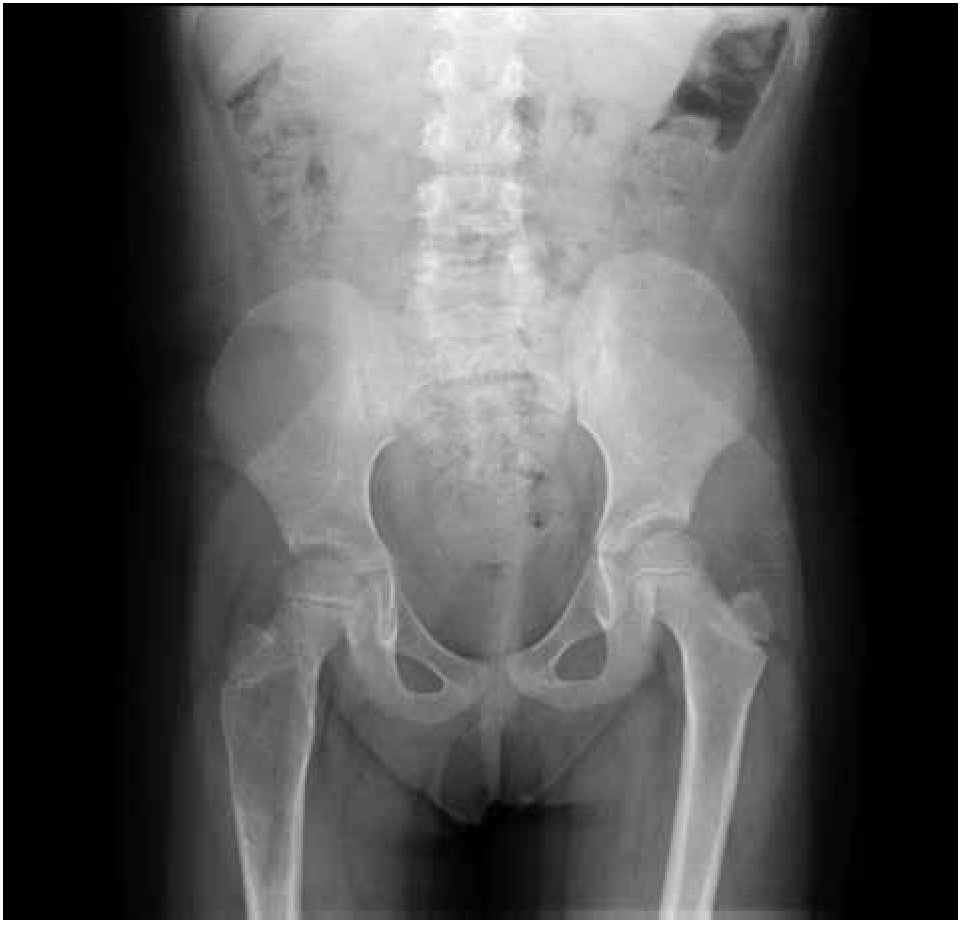
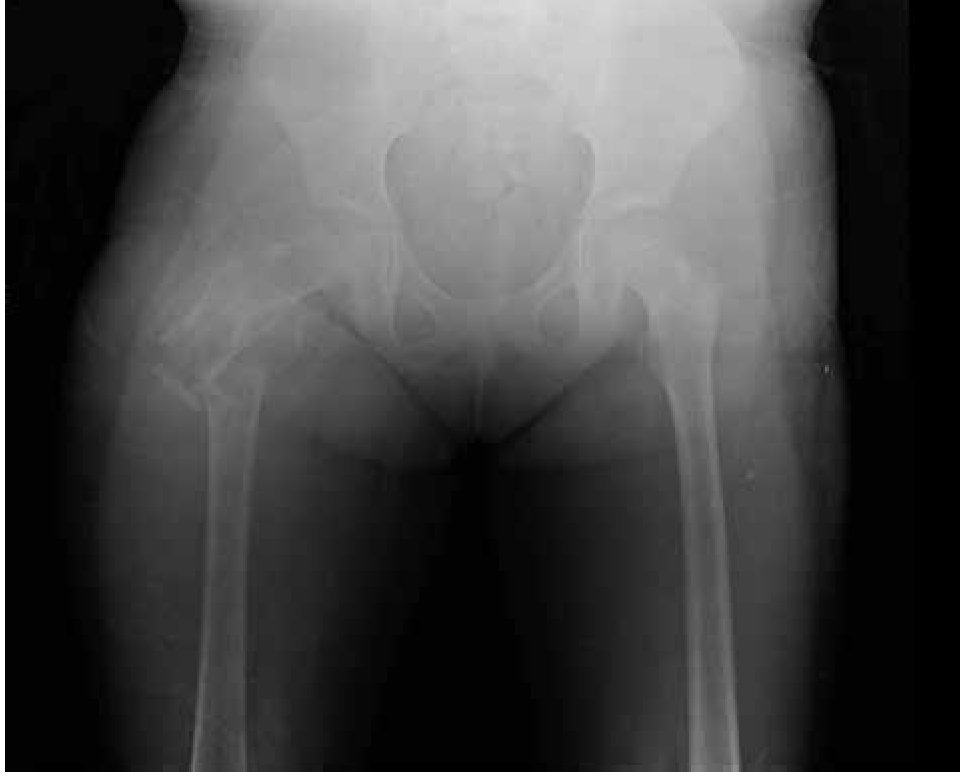
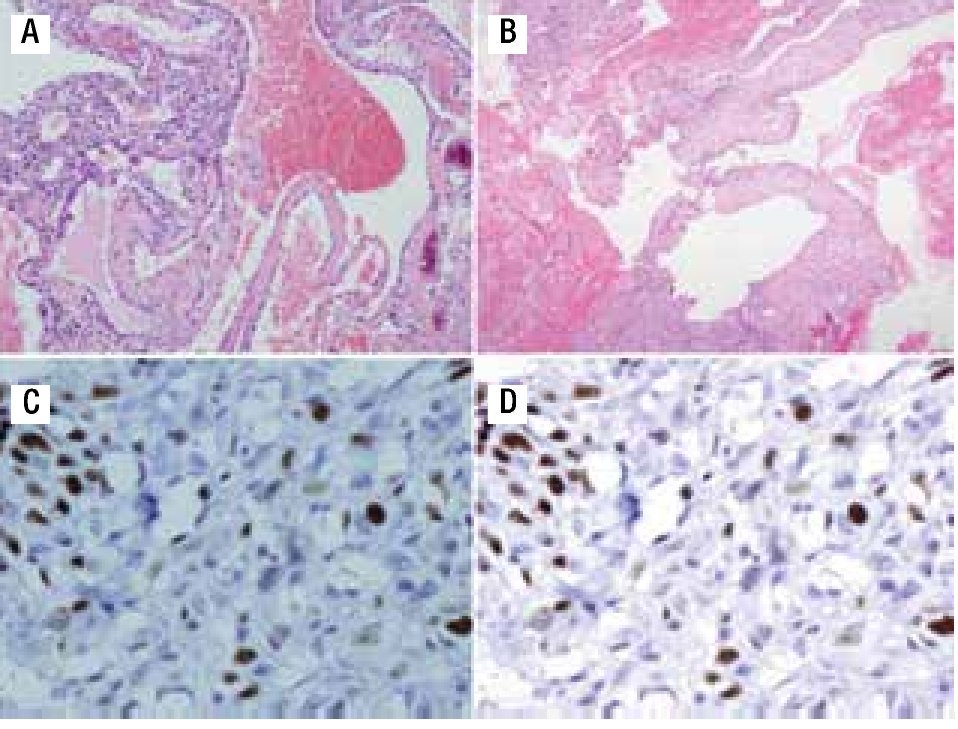
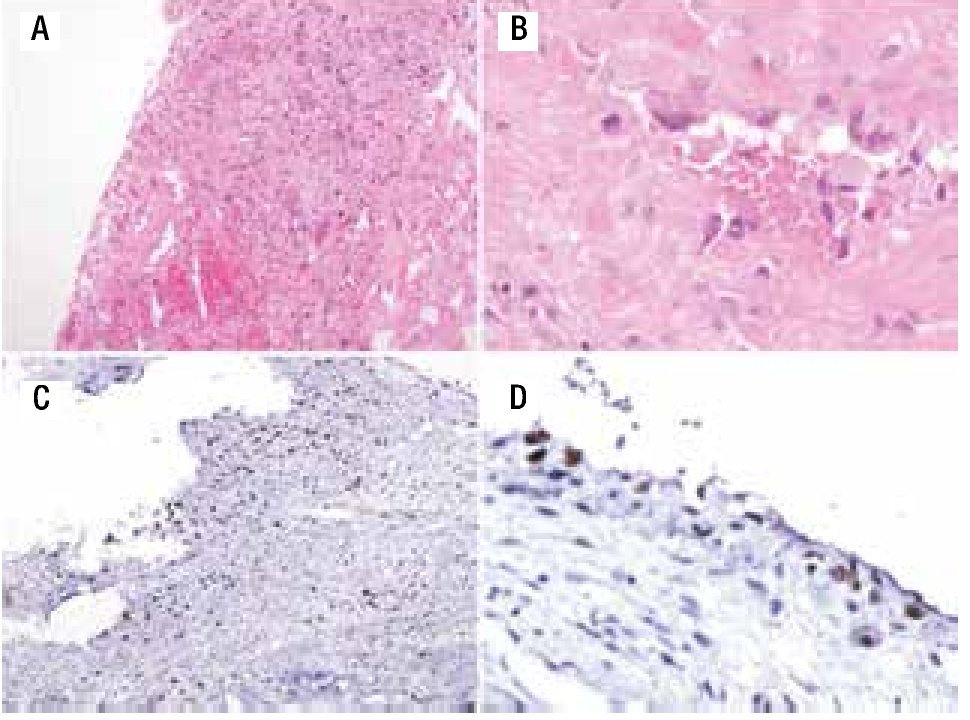
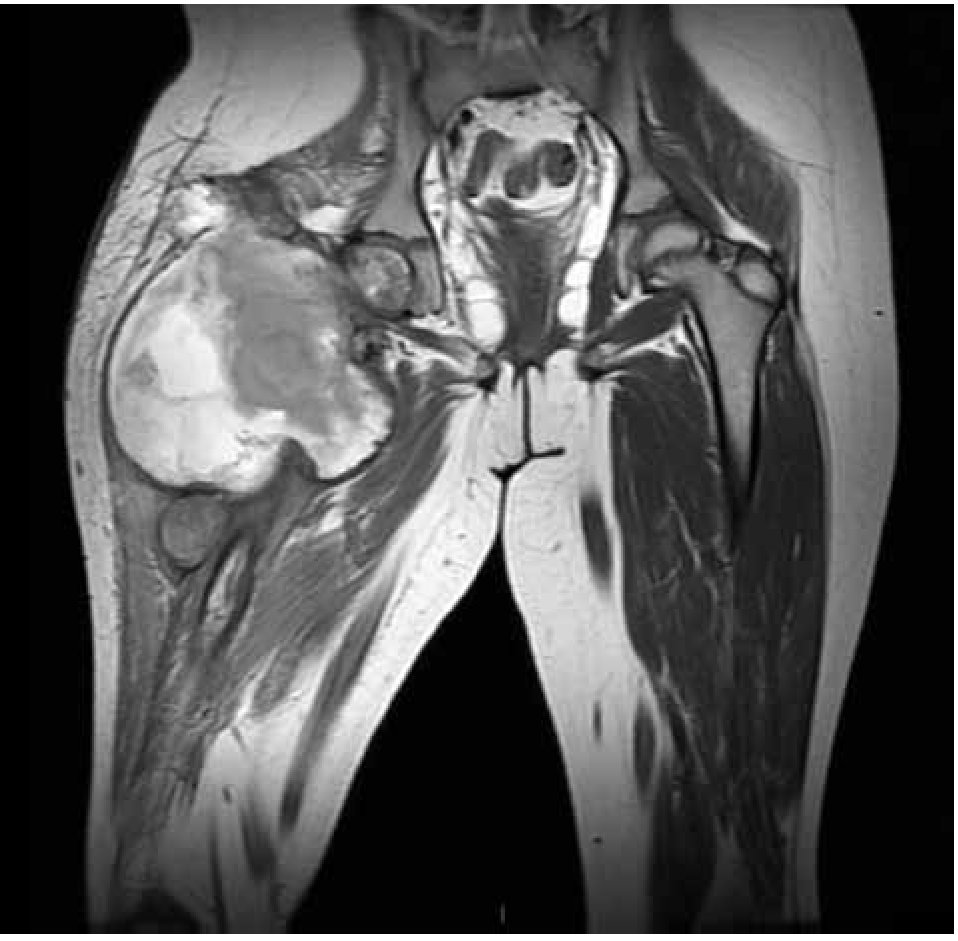
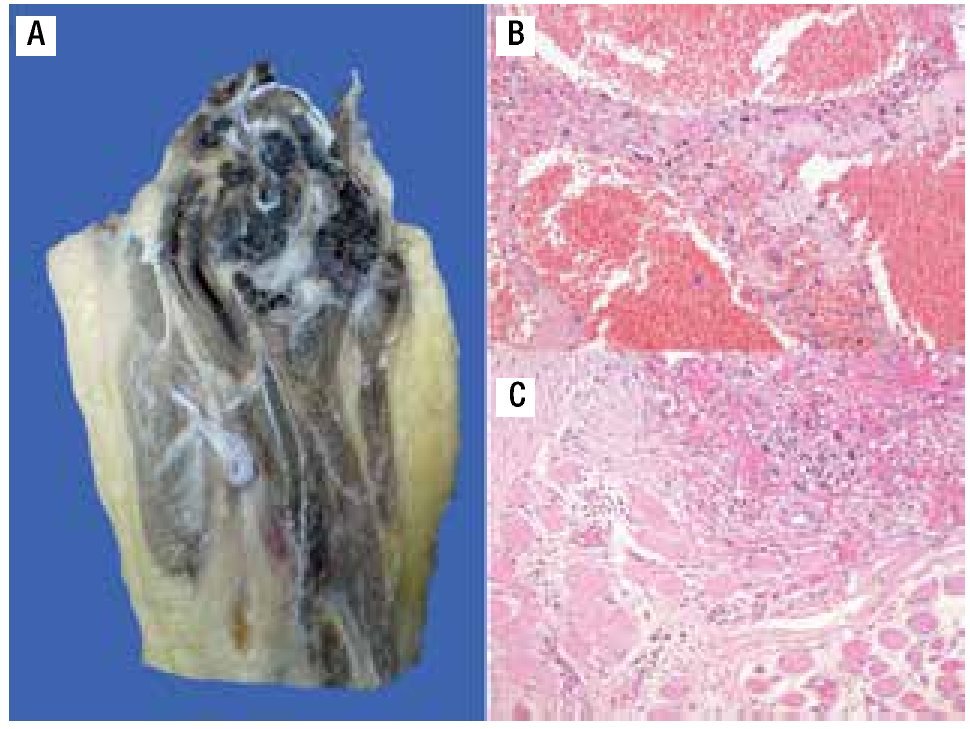
Paciente femenina, quien a los seis años ocho meses de edad, presentó aumento progresivo de volumen, dolor y limitación funcional del tercio proximal del muslo derecho en diciembre de 2007. La radiografía simple de fémur derecho al ingreso mostró una lesión osteolítica, metafisiaria, radiolúcida y con bordes mal definidos, sin afección de la cortical localizada en el tercio proximal del fémur derecho (Figura 1). Se realizó tratamiento con legrado óseo y colocación de injerto óseo, con mejoría de sus condiciones clínicas. En la biopsia se realizó diagnóstico de QOA de tercio proximal de fémur derecho. Dos años después la paciente acude nuevamente por dolor localizado en extremidad pélvica derecha y en la radiografía se observó una fractura de tercio proximal de fémur, resorción del injerto y destrucción de la cabeza femoral y del tercio proximal de la diáfisis (Figura 2), colocándose clavo intramedular. Siete meses después presentó aumento de volumen de muslo derecho y dolor. Se realizó exploración quirúrgica con toma de biopsia de la cabeza femoral y de la diáfisis, así como drenaje de hematoma (1 500 mL). En esta segunda biopsia se aprecia en la mayoría de los fragmentos, septos fibrosos rodeados por hemorragia, células gigantes multinucleadas y hueso de neoformación concluyentes de QOA con áreas focales hipercelulares con atipias con un Ki67 del 50% y un p53 de 20%, por lo que se sugirió la posibilidad de malignidad (Figura 3). De forma retrospectiva y conociendo la existencia de una lesión radiológica e histológica probablemente maligna, se revisó de forma intencionada el material de la biopsia original del diagnóstico de QOA, encontrándose aisladas células atípicas con un índice de proliferación de 30% y positividad focal a p53. (Figura 4). En la imagen por resonancia magnética (RM) se observó lesión de tercio proximal de fémur, dependiente de la cortical, con reacción perióstica e involucro de tejidos blandos, que abarcaba hasta la unión del tercio medio con el tercio proximal (Figura 5). Se realizó una tercera biopsia guiada por tomografía axial computarizada (TAC), confirmando la presencia de sarcoma osteogénico. En la evaluación de la búsqueda de metástasis, se observó presencia de nódulo pulmonar basal izquierdo de 7 mm que ha permanecido sin cambios hasta el momento actual. Inició tratamiento con quimioterapia neoadyuvante con cisplatino (120 mg/m2) y doxorrubicina (75 mg/m2) por dos cursos, presentando posterior a los mismos, progresión local de la enfermedad por lo que se intensificó tratamiento sistémico con ciclofosfamida a dosis escalas (iniciando con 300 mg/m2/dosis por seis, aumentando 50 mg/m2/dosis en cada curso) y etopósido (200 mg/m2/día por tres días) durante cuatro cursos, y se realizó amputación de extremidad. El producto de la amputación mostró un tumor con cavidades llenas de sangre limitadas por delgadas láminas de hueso (Figura 6A). Histológicamente, se aprecian septos limitados por células pleomórficas de aspecto neoplásico. Hay áreas de formación de osteoide. El diagnóstico histopatológico fue de OT con 10% de necrosis tumoral posquimioterapia con invasión a tejidos blandos (Figura 6B y 6C). Posteriormente se administró quimioterapia con altas dosis de metotrexate (12 g/m2/dosis semanal por tres) más ifosfamida (3 g/m2/día por cinco) y doxorrubicina (75 mg/m2/dosis) por tres cursos. Se envió a vigilancia en noviembre de 2011 y actualmente se encuentra en remisión completa de la enfermedad.
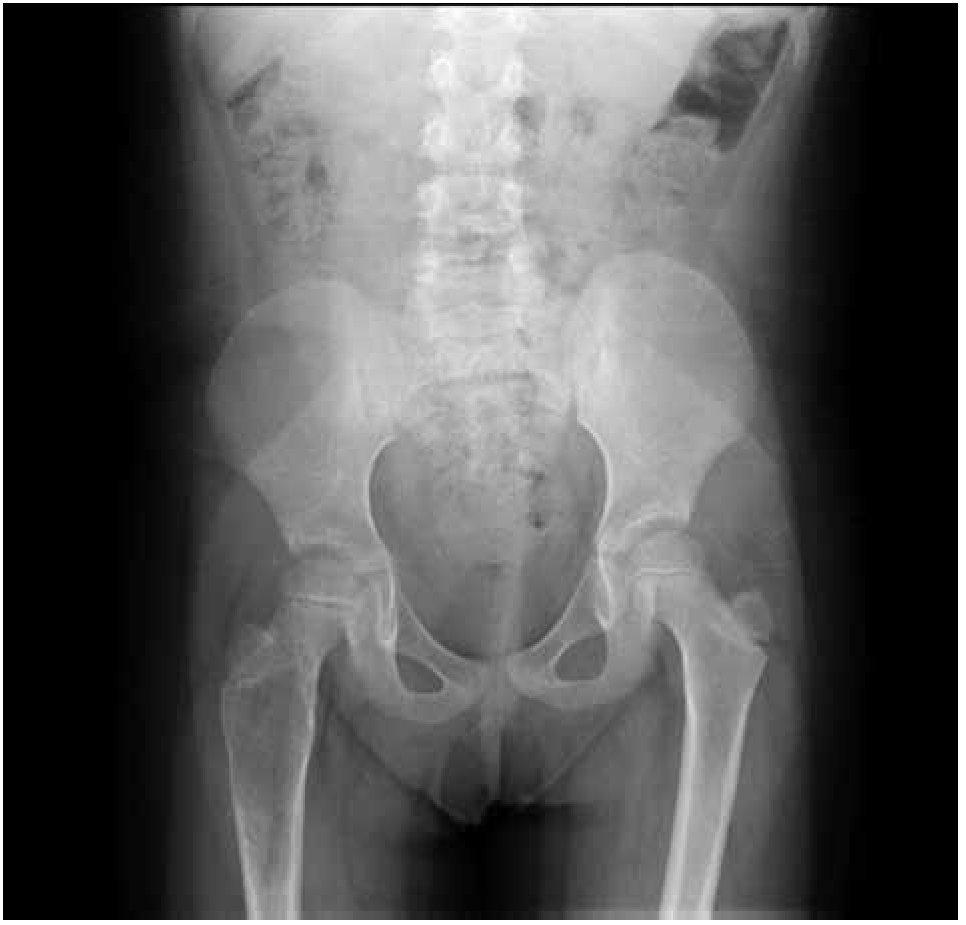
Figura 1. Radiografía antero-posterior de fémur derecho con quiste óseo aneurismático.
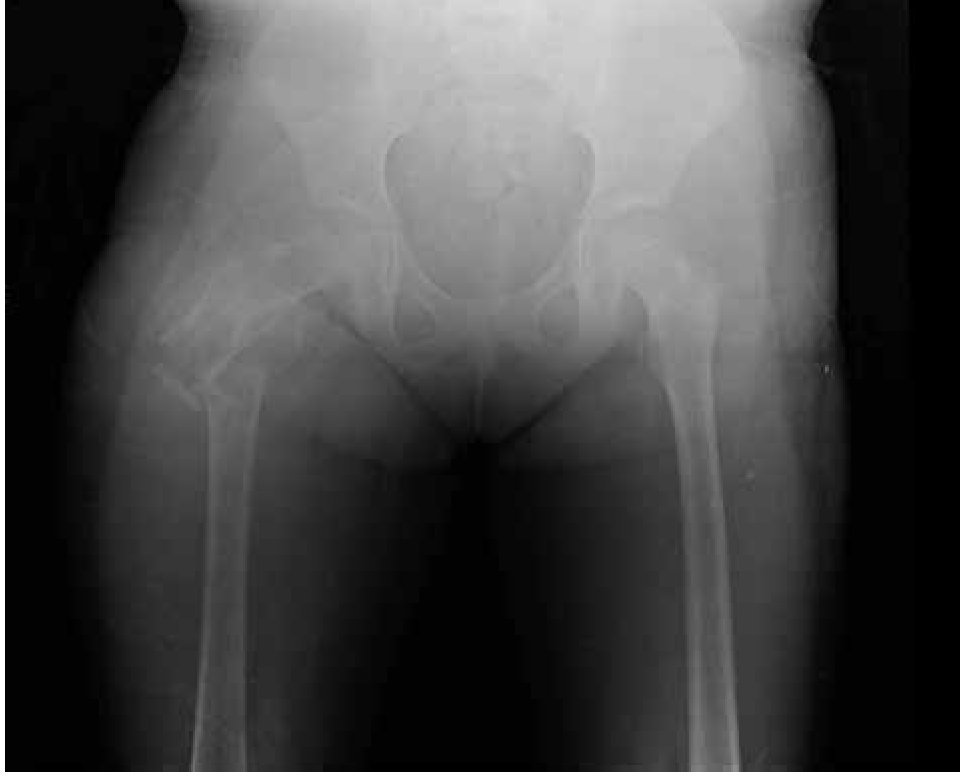
Figura 2. Radiografía antero-posterior de fémur derecho que presenta fractura de cuello de fémur, resorción del injerto óseo y destrucción de la cabeza femoral y tercio proximal de la diáfisis.
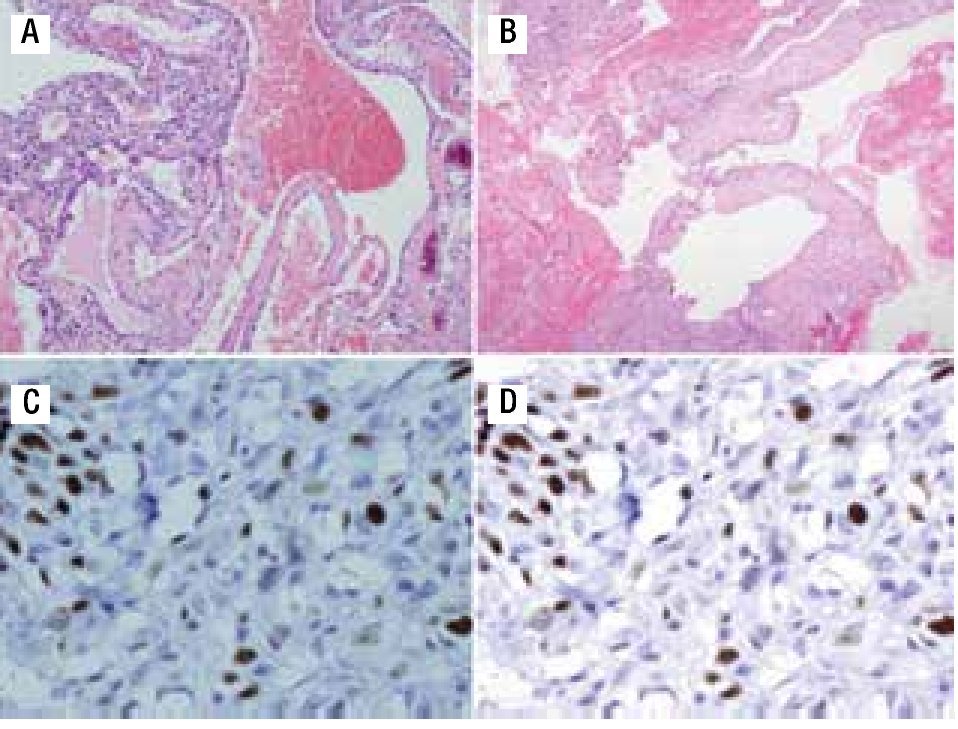
Figura 3. A) Material de biopsia que muestra células gigantes multinucleadas, pleomorfismo, presencia de osteoide maligno y zonas de necrosis con patrón telangiectásico. B) Patrón angiomatoide celular. C) Ki67 de 50%. D) p53 de 20%.
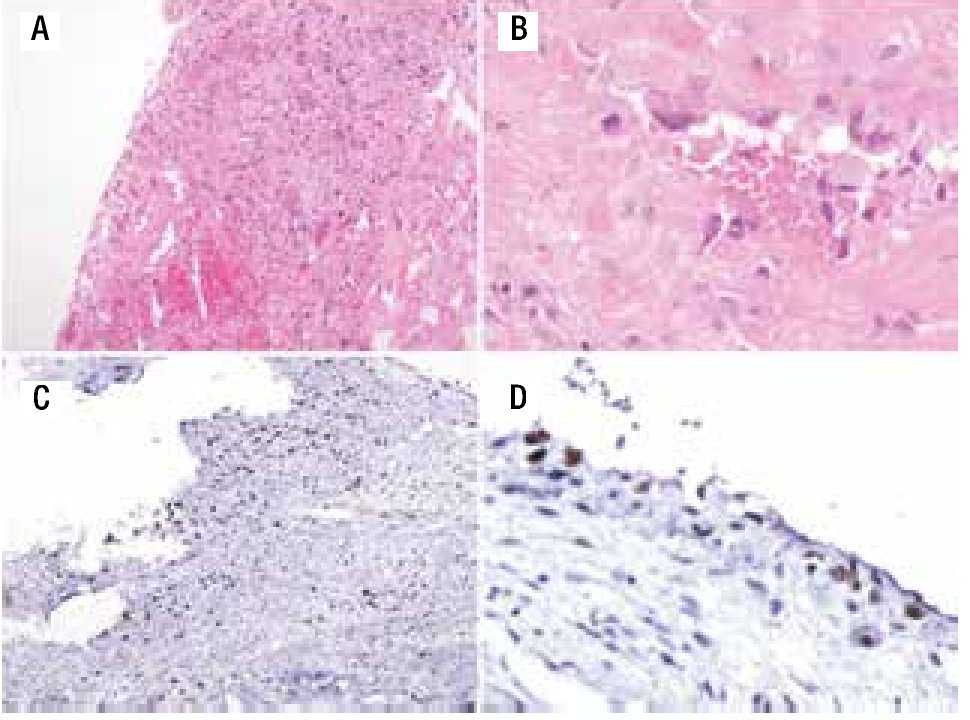
Figura 4. A) Material de curetaje de la biopsia inicial que muestra septos fibrosos y espacios sinusoidales vasculares. B) Células atípicas aisladas. C) Ki67 de 30%. D) p53 positivo focal.
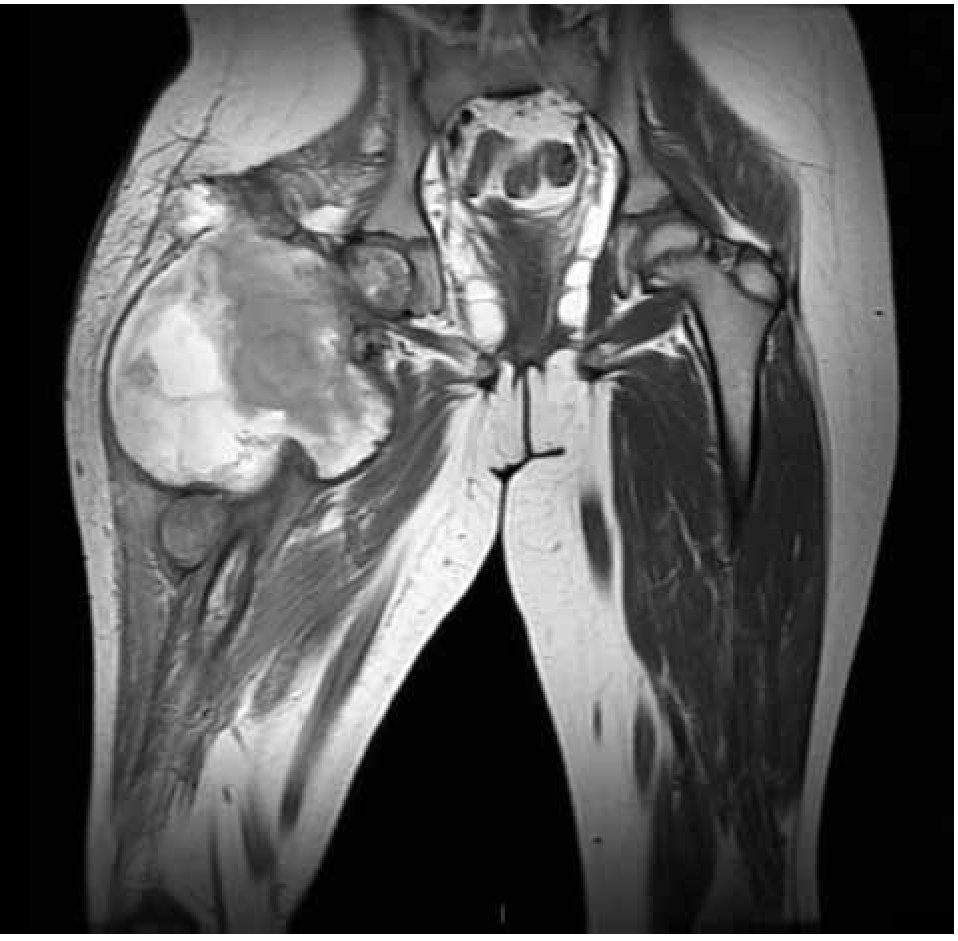
Figura 5. Imagen por RMN corte coronal, observando la lesión en tercio proximal de fémur con reacción perióstica e involucro de tejidos blandos.
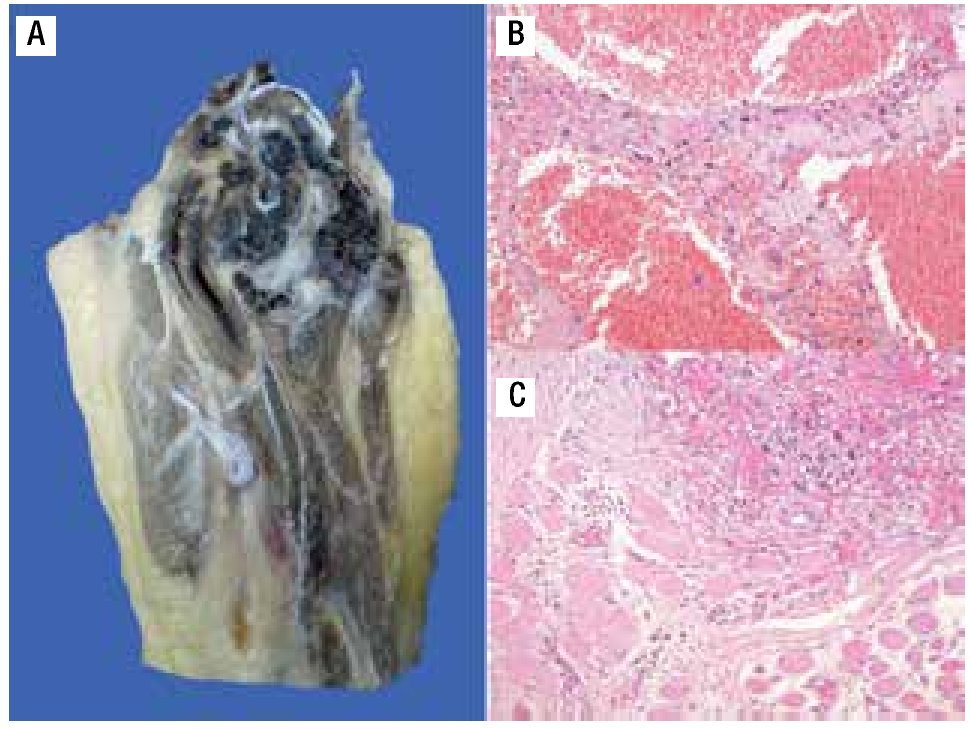
Figura 6. A) Imagen macroscópica del producto de amputación con un tumor con cavidades llenas de sangre, limitadas por delgadas láminas de hueso. B) Áreas de anaplasia. C) Invasión a tejidos blandos (músculo).
¿ DIscusIón
El QOA ha sido considerado una entidad no neoplásica, que se produce ya sea como un fenómeno reactivo vascular o secundario a una lesión o trauma. La etiología permanece incierta en un gran número de casos, aunque cada vez hay más evidencia de alteraciones citogenética en los QOA primarios, lo que sugiere que sí representan una neoplasia primaria de hueso.14
Recientemente, se ha demostrado una base genética de la formación de los QOA, lo que contradice la teoría vascular reactiva. Se describieron reordenamientos de genes relacionados con los oncogenes USP6 y CDH11, que han sido identificados en la presentación primaria pero no así en la presentación secundaria.15 Se han encontrado aberraciones clonales en los cromosomas 16 y 17 de los tejidos blandos y la superficie de los QOA.16
Cuando la evolución clínica de este caso cursó con un crecimiento agresivo después del tratamiento del QOA, buscamos la presencia de otro tumor subyacente, en particular un OT, aunque el patrón histológico característico fue insuficiente en la primera biopsia. Desafortunadamente, incluso la presencia de tejido osteoide dentro de un QOA no es suficiente para el diagnóstico de OS. Sólo un aspecto característico de las estructuras celulares con polimorfismo y un incremento de figuras mitóticas pueden ser aceptados como criterios para el diagnóstico de OT. Sin embargo, estas características no fueron definitivas de forma inicial en el presente caso.
Se han descrito algunos casos con un curso clínico agresivo del QOA. Sin embargo, estos casos fueron reportados como QOA debido a la falta de formación de osteoide, independientemente de su comportamiento clínico agresivo. Kyriacos y colaboradores informaron un caso de OS pleomórfico en la literatura que surgió de un QOA, fue tratado con curetaje en varias ocasiones, y posteriormente presentó OS tres años después del tratamiento inicial.4
Saito y colaboradores reportaron la presencia de un OT del fémur derecho en un paciente de 20 años, quien inicialmente fue diagnosticado como QOA. En este caso se describió la recurrencia de la lesión en dos ocasiones, y al realizar la resección completa de la lesión se diagnosticó un OT. En la revisión retrospectiva de la lesión primaria se observaron características compatibles con QOA, y la presencia de células atípicas sin evidencia de mitosis.3
Existen dificultades en obtener el diagnóstico histopatológico. La interpretación de pequeñas muestras de biopsia puede contener poco material sólido. Es difícil localizar tumor viable, que puede estar presente sólo en la periferia de la lesión que rodea los espacios quísticos en una cáscara gruesa o nodular.17 Incluso si estas áreas son biopsiadas con éxito, pueden presentar sólo atipia escasa. La situación se complica aún más por la rara posibilidad de la transformación maligna del QOA primario, incluso aquellos que no se han tratado con radioterapia.4 La pregunta a menudo en estos casos es si un pequeño foco de OS se perdió en el momento del diagnóstico inicial. Si éste fuera el caso, la historia natural del OS cursa con rápido crecimiento del tumor primario y presencia de metástasis.10
El QOA se percibe generalmente como un tumor benigno. Sin embargo, se han informado casos excepcionales convincentes que se comportan de una manera más agresiva, desde la presencia de implantes en tejidos blandos regionales18 hasta la transformación maligna.4 Se ha descrito un caso único con presencia de metástasis, que conservaron las características histopatológicas benignas del QOA primario, cuya evolución clínica condujo a la muerte del paciente.19 El escenario más común para la transformación maligna de los QOA es el desarrollo de un sarcoma en un sitio previamente radiado.20 Sólo un pequeño número de casos han sido reportados, en los cuales hay una transformación maligna de un QOA primario sin historia de radiación.4-9 La falta de evidencia de un componente maligno en la lesión primaria y la evolución clínica larga y prolongada en estos reportes, orienta a favor de una verdadera transformación maligna a través de un diagnóstico de sarcoma primario. En el caso reportado por Van de Luijtgaarden y colaboradores,19 se documentó la presencia de un locus de reordenamiento de USP6, ilustrando una evolución inusual de metástasis de un QOA de aspecto benigno.
En este caso planteamos la posibilidad de la presencia de un OT, que se desarrolló en un QOA primario o secundario en un OT. En apoyo de la primera posibilidad está el tiempo de evolución de casi tres años entre el diagnóstico de QOA y el desarrollo de la lesión destructiva de características malignas. Desafortunadamente no tenemos documentación radiográfica de la evolución del quiste hasta el momento de la fractura patológica. La segunda posibilidad se basa en la reconocida coexistencia de un QOA con alguna otra lesión ósea, y en la dificultad del diagnóstico diferencial entre QOA y OT. Dado que la biopsia original fue muy pequeña puede no haber sido representativa, además del hecho de que en el examen retrospectivo ya conociendo la existencia de una lesión maligna y en la búsqueda intencionada, se encontraron aisladas células atípicas con un índice de proliferación alto y positividad a p53. Poco se sabe del tiempo que transcurre desde el origen de un OS hasta el momento de su manifestación clínica, que en este caso se expresó por el quiste asociado, pero no podemos asegurar que la neoplasia no haya estado presente desde el inicio.
¿ ConclusIones
Los tumores óseos son entidades de alta complejidad diagnóstica. Se recomienda realizar un adecuado muestreo en las biopsias y especímenes quirúrgicos, que permita poner en evidencia todos los componentes histológicos de los tumores óseos y realizar la búsqueda exhaustiva de características malignas. Hacemos hincapié en las similitudes de la apariencia microscópica del OT y el QOA. Un análisis cuidadoso de las características radiográficas e histológicas contribuye al diagnóstico correcto.
Correspondencia:
Dra. Araceli Castellanos Toledo.
Insurgentes Sur 3700-C, Colonia Insurgentes Cuicuilco, Delegación Coyoacán,
C.P. 04530. México D.F., México.
Teléfono: (55) 1084 0900, ext. 1312.
Correo electrónico: lourdesromrdz@gmail.com