


El rabdomiosarcoma es el sarcoma de tejidos blandos más frecuente en la edad pediátrica; en los adultos representa un reducido número de tumores malignos. Se localiza principalmente en cabeza y cuello, aparato genitourinario y extremidades. Existen diversos tipos histológicos de rabdomiosarcoma con distinto comportamiento biológico. El objetivo del presente artículo es informar el caso de un rabdomiosarcoma variedad seudovascular esclerosante de pared torácica posterior; el cual tiene pocos casos en el mundo; así como realizar una revisión integral de la literatura médica sobre el tema.
Se presenta paciente masculino de 56 años de edad, que acude a la Unidad con presencia de tumor intratorácico documentado por radiografía de tórax, quien es sometido a protocolo diagnóstico. Se detecta en tomografía axial computarizada (TAC) de tórax, tumor de gran tamaño dependiente de la pared torácica posterior izquierda. Se somete a resección tumoral a través de toracotomía postero-lateral izquierda, resecándose tumor en casi su totalidad, dejando R2. Cursa con evolución clínica satisfactoria en hospitalización, siendo egresado 12 días después a su domicilio. El reporte de patología con imunohistoquímica refiere rabdomiosarcoma seudovascular esclerosante, con infiltración a tejido pulmonar adyacente subpleural.
Se trata de una neoplasia maligna muy rara, con reporte de 15 casos a nivel mundial, la cual presenta una localización nunca descrita en la literatura, de ahí surge la importancia de tener en cuenta esta posibilidad diagnóstica, así como los conocimientos para su detección.
Rhabdomyosarcoma is the most common sarcoma of soft tissues in childhood; in adults represents a small number of malignant tumors. It is located mainly in the head and neck, genitourinary tract, and extremities. There are several histologic types of rhabdomyosarcoma with different biological behavior. The objective is to report the case of a variety pseudo-vascular sclerosing rhabdomyosarcoma posterior chest wall; of which there are few cases in the world; well as a comprehensive review of the literature.
A 56-year-old male attended the Unit in the presence of intrathoracic tumor documented by chest X-ray, who undergoes diagnostic protocol. Detected in thoracic tomography (CT) tumor size largely dependent left posterior thoracic wall. Subjected to tumor resection via left posterolateral thoracotomy, resecting tumor almost entirely, leaving R2. Presents with satisfactory clinical evolution and is a graduate hospital 12 days after his address. The pathology report with regards imunohistoquimica Pseudovascular sclerosing rhabdomyosarcoma with infiltration adjacent subpleural lung tissue.
This is a very rare malignancy, with 15 cases reported worldwide; and which present a location never described in the literature and hence, the importance of taking into account this possibility diagnosed and knowledge for detection.
Introducción
El rabdomiosarcoma es el sarcoma de tejidos blandos más frecuente en la edad pediátrica; mientras que en los adultos representa solamente un pequeño grupo de los tumores malignos. Se localiza principalmente en cabeza y cuello, aparato genitourinario y extremidades; puede presentarse en diversas partes del cuerpo, incluyendo aquellos sitios en los que no hay músculo estriado, como hueso y piel. Existen diversos tipos histológicos de rabdomiosarcoma importantes de distinguir, debido a que difieren en su comportamiento biológico1. Los tumores benignos de músculo esquelético son mucho menos frecuentes que sus contrapartidas malignas, y representan no más del 2% de todos los tumores malignos de músculo estriado. Básicamente, se emplean 2 denominaciones para definir los tumores de músculo esquelético: 1) rabdomioma para las forma benignas, con una subdivisión en forma fetal y adulta; y 2) rabdomiosarcoma para la forma maligna con sus variantes de embrionaria, alveolar y pleomórfica, además del reconocimiento más reciente de una forma esclerosante y de algunos subtipos especiales2.
El objetivo del presente artículo es describir el caso y el manejo de un paciente masculino con rabdomiosarcoma seudovascular esclerosante de pared torácica posterior izquierda; la cual constituye una neoplasia rara en nuestro medio y en el mundo, haciéndose meritorio realizar una revisión integral de la literatura médica sobre el tema.
Presentación del caso
Masculino de 56 años de edad, el cual tiene los siguientes antecedentes de importancia: niega historiales oncológicos familiares; es portador de diabetes mellitus tipo 2 con 8 años de evolución; quirúrgicos: vasectomía, orquiectomía izquierda, disección ganglionar mediastinal izquierda en 2009.
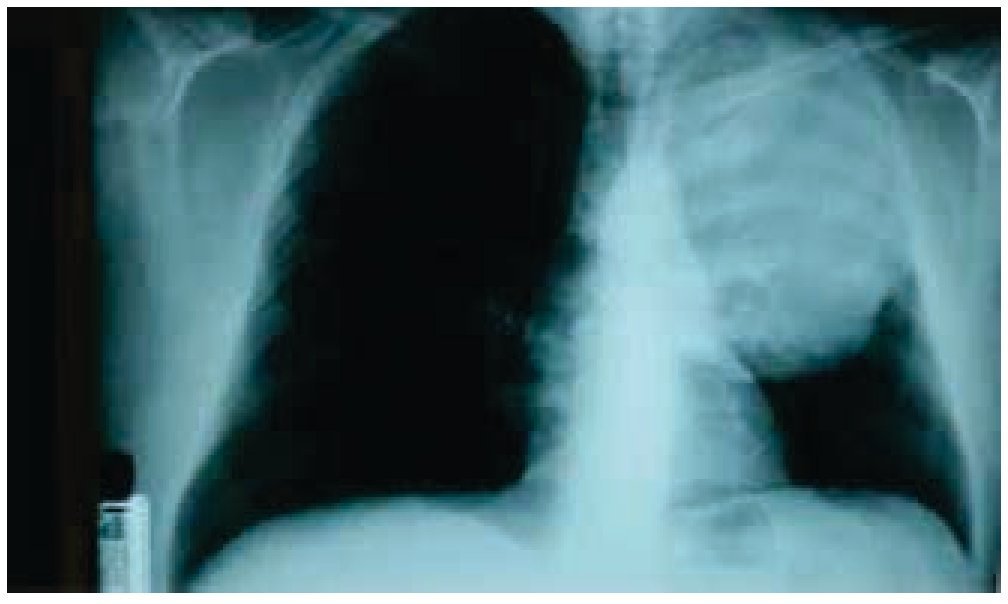
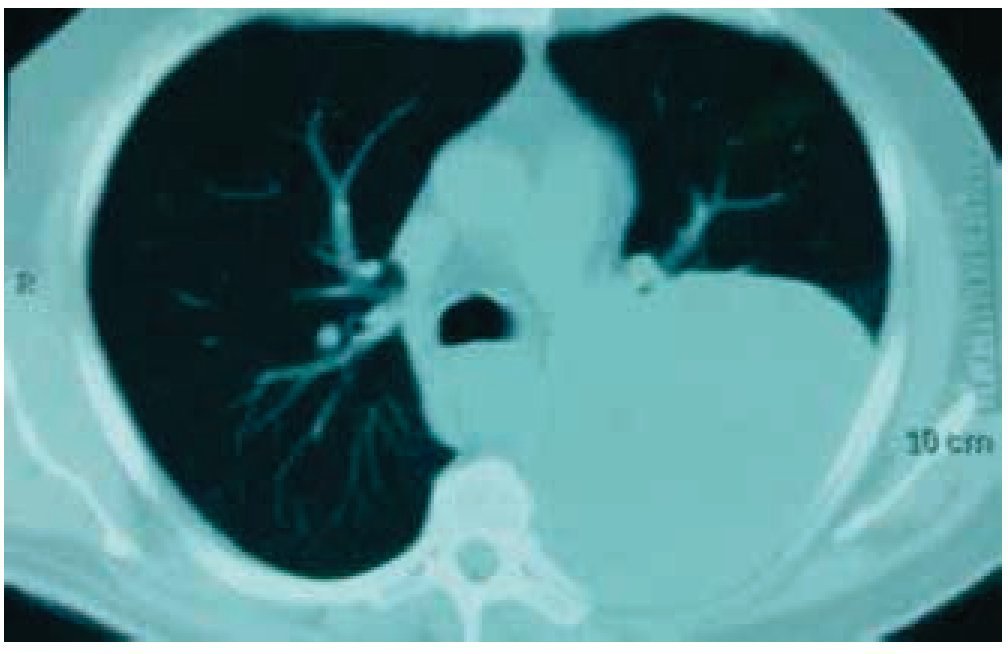
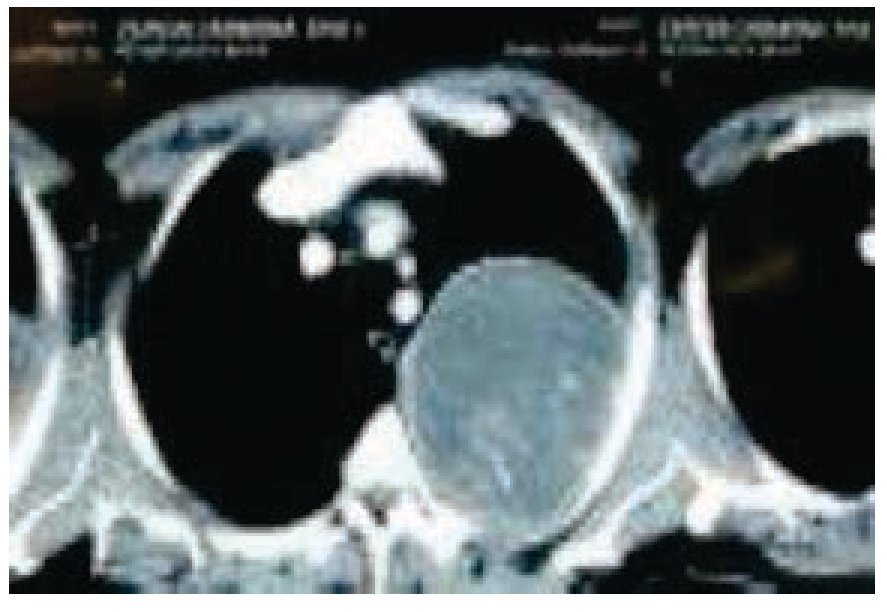
Es visto por primera vez el 25 febrero de 2014, en la Consulta Externa de Oncología con antecedentes de liposarcoma desdiferenciado (10%) y mixoide (90%) de región paratesticular izquierda, sometido a orquiectomía izquierda en 2009 sin tratamiento adyuvante, desconoce la estadificación ya que pierde control oncológico. Inicia en enero de 2014 con tos persistente, no cianosante, con escasa expectoración oscura, disnea a pequeños esfuerzos, dolor torácico izquierdo con irradiación a miembro torácico ipsilateral, de leve a moderada intensidad, sin ningún fenómeno acompañante, pérdida de peso 3 Kg en 15 días; acude a facultativo quien le indica una radiografía de tórax, donde se observa una lesión en hemitórax izquierdo que desplaza estructuras intratorácicas (fig. 1). Se solicitan estudios de extensión: tomografía axial computarizada (TAC) de tórax simple y contrastada, exámenes de laboratorio, marcadores tumorales (alfa-fetoproteína, fracción beta de gonadotropina coriónica humana y deshidrogenasa láctica se reportan como negativos). La TAC de tórax del 10 de junio de 2014 presenta tumor de 20 x 19 x 18 cm, dependiente de pared torácica posterior izquierda con infiltración a tejido pulmonar y pared torácica adyacente, sin presencia de adenopatías regionales, ni derrame pleural (figs. 2 y 3). Se envía a biopsia dirigida por TAC de dicha tumoración, la cual se realiza el 8 de abril de 2014 y se reporta por parte del Servicio de Patología como hemangioma. Ante este resultado, se envía a valoración por Cardiología y Neumología, quienes no contraindican procedimiento quirúrgico.
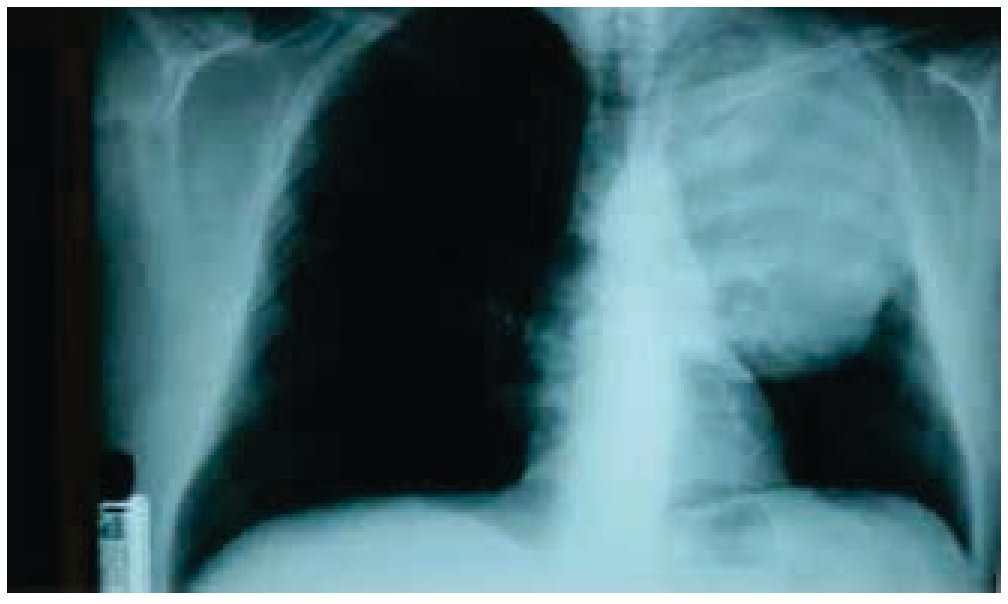
Figura 1 Radiografía de tórax, donde se observa una lesión en hemitórax izquierdo que desplaza estructuras intratorácicas.
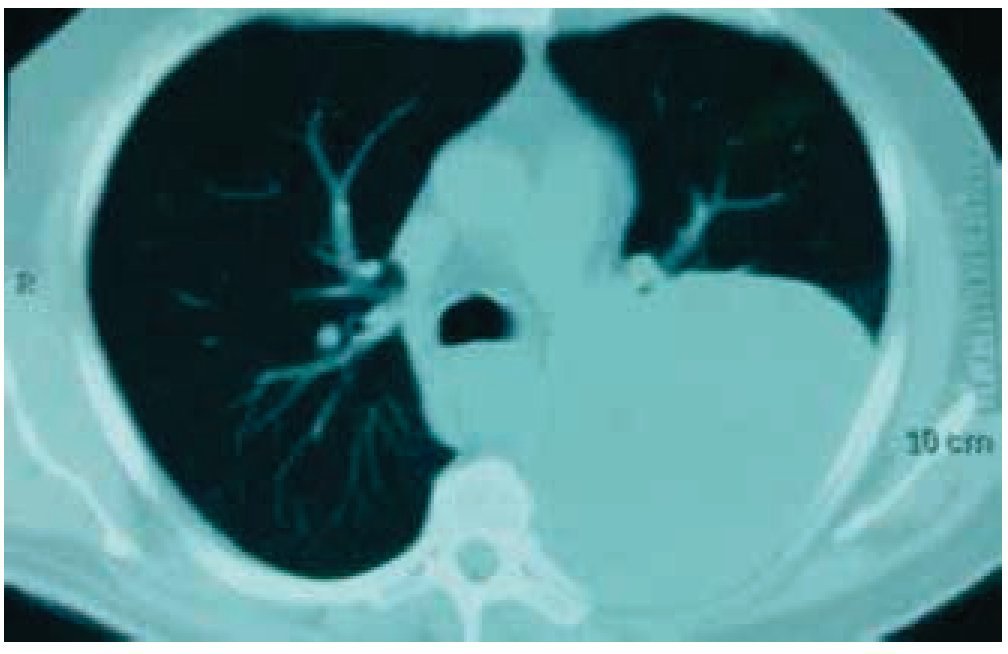
Figura 2 Tomografía de tórax con ventana pulmonar. Se observa tumor de 20 x 19 x 18 cm, dependiente de pared torácica posterior izquierda con infiltración a tejido pulmonar adyacente.
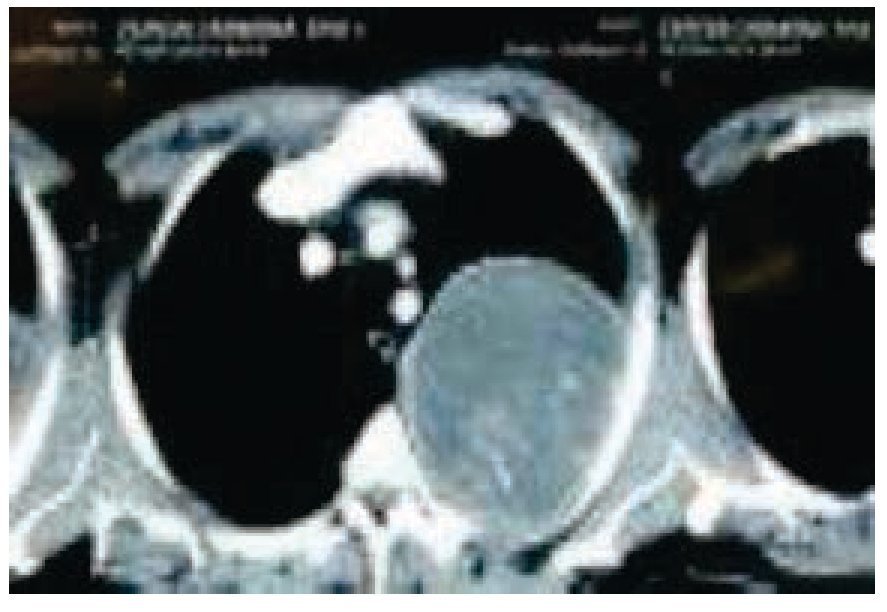
Figura 3 Tomografía de tórax contrastada. Se observa tumor dependiente de pared torácica posterior de hemitórax izquierdo, sin presencia de adenopatías regionales ni derrame pleural.
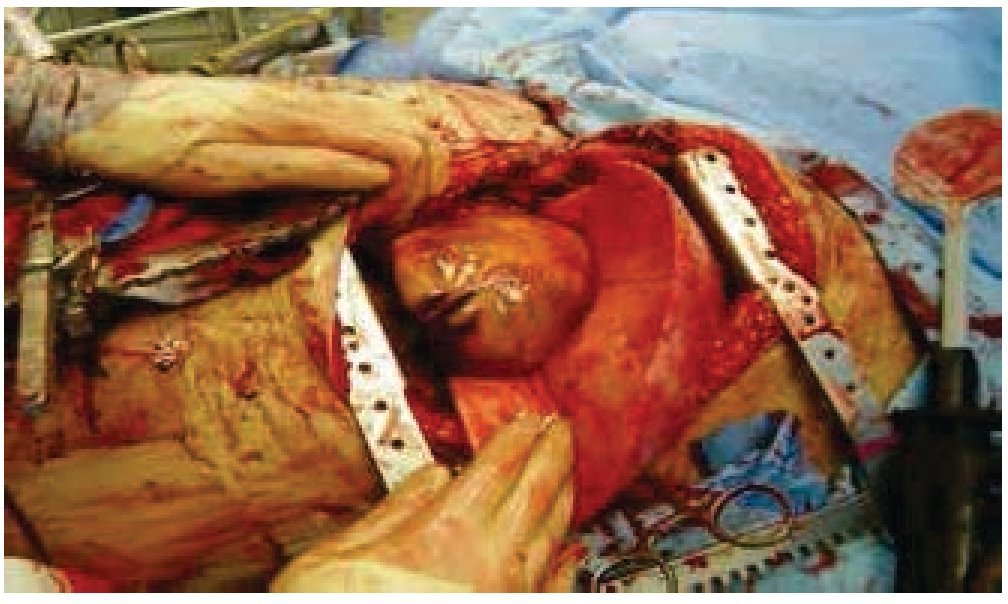
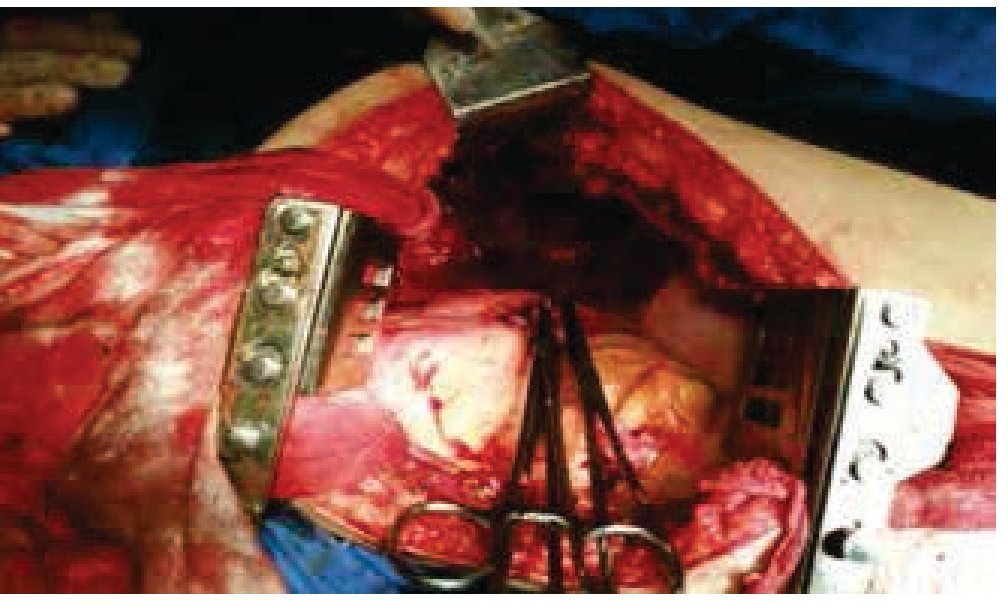
Se somete a toracotomía posterolateral izquierda y neumonectomía radical izquierda, resección tumoral de pared torácica posterior completa tipo R2, colocación de sonda endopleural ipsilateral el 19 de junio de 2014; con hallazgos de tumor dependiente de pared torácica posterior izquierda, de 25 cm, con infiltración de tejidos adyacentes: hueco axilar, parénquima pulmonar en zona del hilio pulmonar izquierdo con abundantes vasos de neoformación y consistencia pétrea (figs. 4 y 5). Es ingresado al Servicio de Terapia Intensiva el 19 de junio de 2014 con asistencia mecánica ventilatoria por choque hipovolémico grado IV, insuficiencia respiratoria, acidosis metabólica. Durante su estancia se mejoran condiciones clínicas con reversión del estado de choque, acidosis y mejoría de la ventilación mecánica. Se egresa el 01 de julio de 2014 de Terapia Intensiva, extubado y en buenas condiciones clínicas. Durante su estancia en hospitalización cursa con buen estado clínico, se mantiene vigilancia del gasto de sonda endopleural siendo de 500 mL en promedio, con disminución progresiva del gasto hasta el 07 de julio, en 50 mL. Se mantiene en vigilancia clínica y con signos vitales dentro de parámetros normales, buen estado clínico, herida de toracotomía cicatrizada.
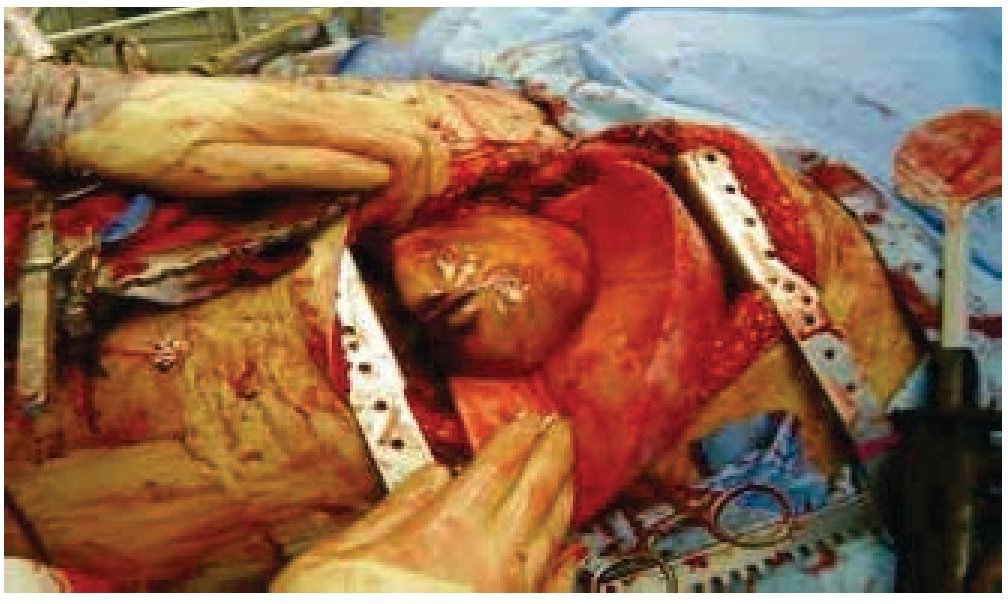
Figura 4 Tumor dependiente de pared torácica posterior izquierda, con diámetro de 25 cm, así como infiltración a parénquima pulmonar e hilio pulmonar izquierdo.
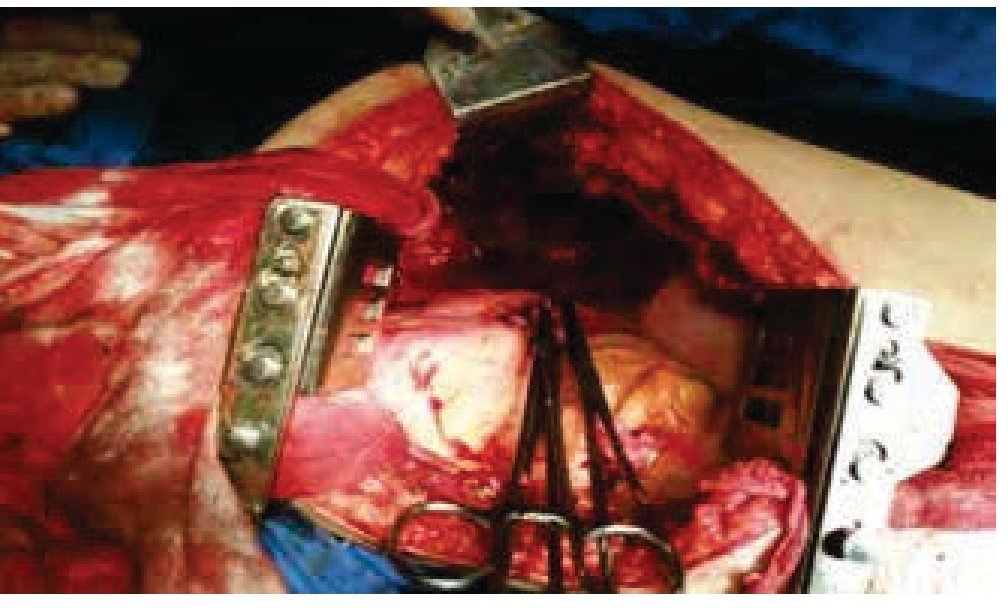
Figura 5 Cavidad torácica izquierda posterior a realización de neumonectomía.
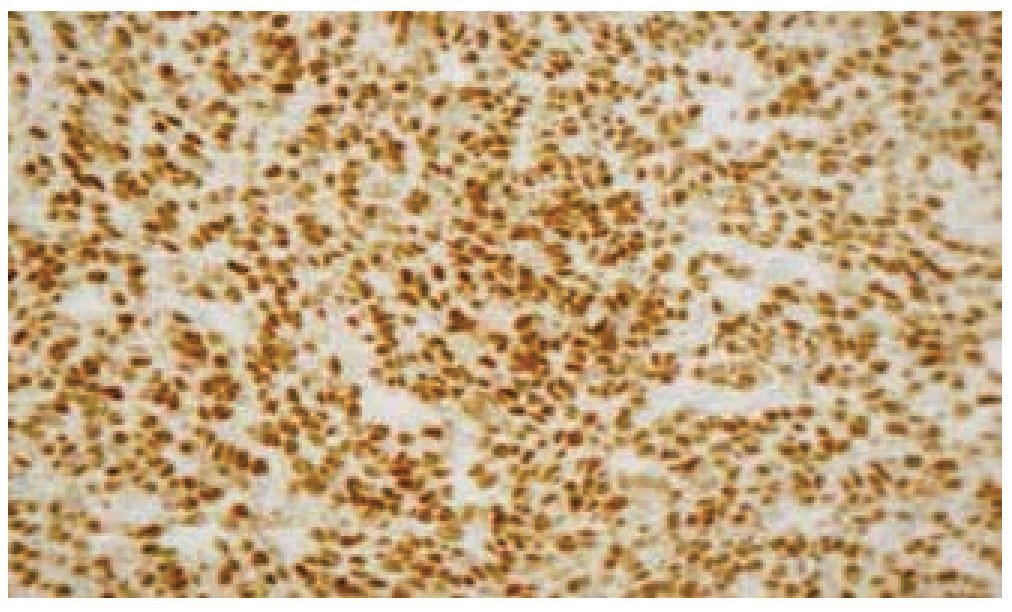
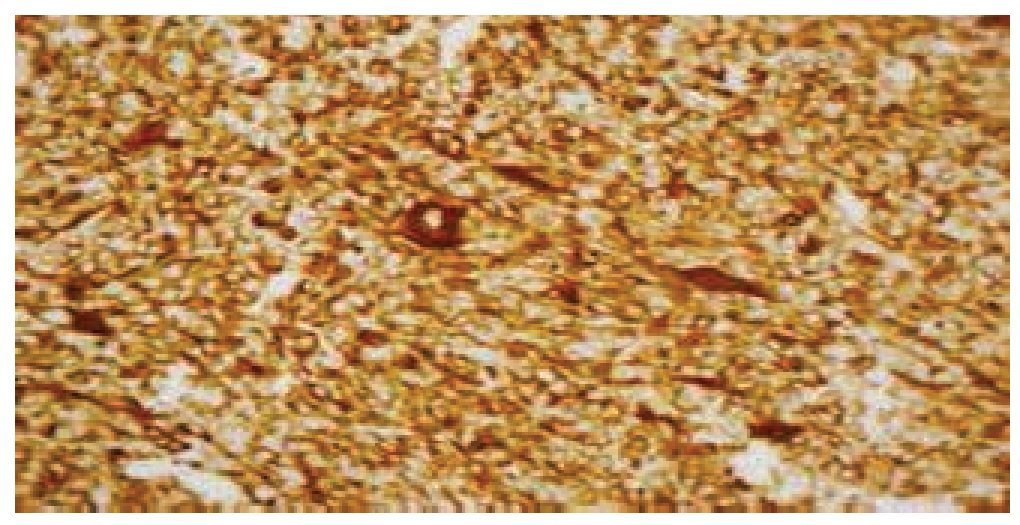
El reporte de patología con imunohistoquímica del 07 de julio de 2014 concluye: rabdomiosarcoma seudovascular esclerosante con infiltración al tejido pulmonar adyacente subpleural. Ganglios linfáticos hiliares con hiperplasia sinusoidal y antracosis. Inmunohistoquímica: desmina (+), Myo D1 (+), miogenina (+) débilmente focal, S-100 (-), CD34 (-), PAN CK (-), Ag AML (-) (figs. 6 y 7).
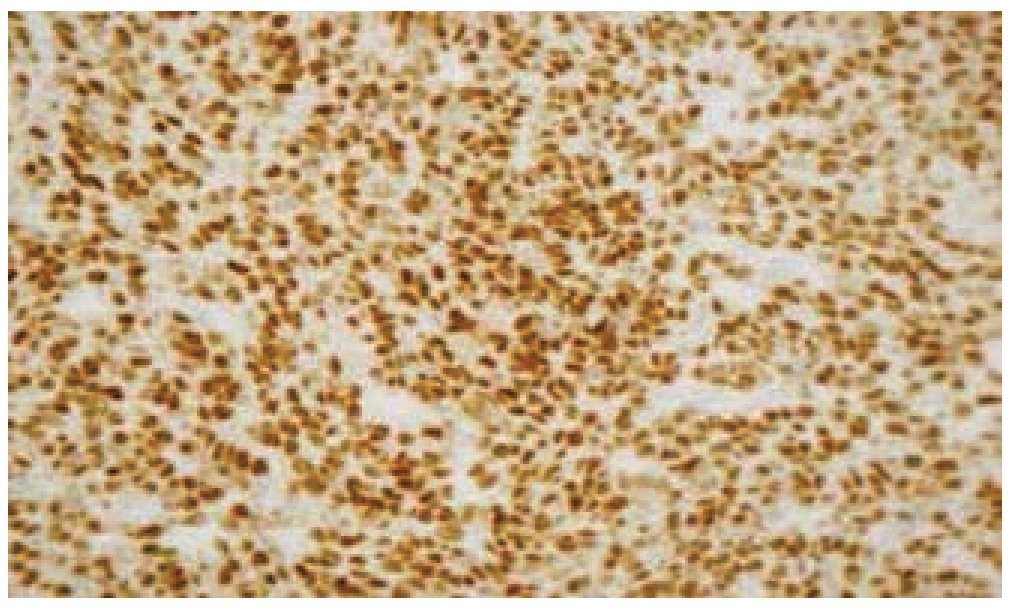
Figura 6 Microfotografía de inmunohistoquímica, donde se observa la reacción intensa a Myo D1.
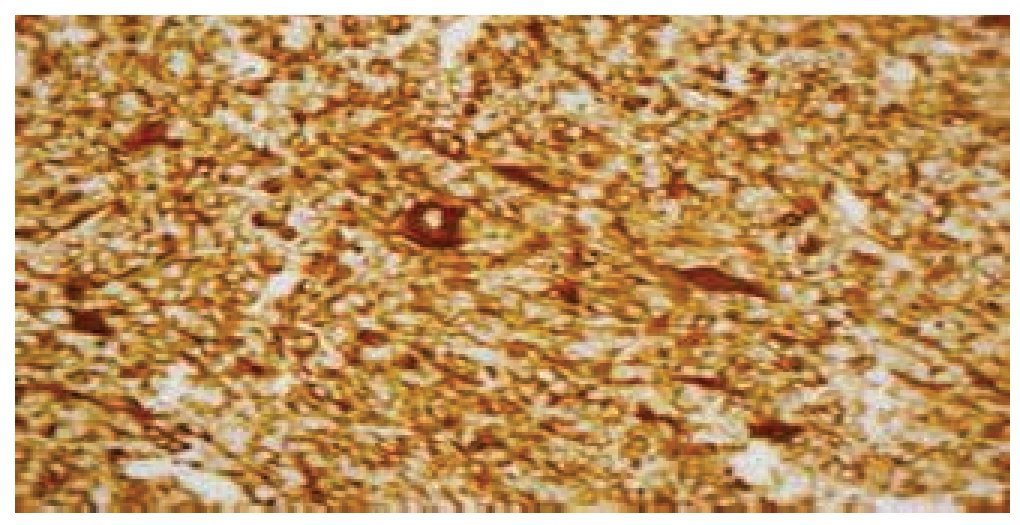
Figura 7 Tinción con inmunohistoquímica, donde se expone reacción intensa a desmina.
Discusión
El rabdomiosarcoma es el sarcoma de tejidos blandos más frecuente en la niñez. Es poco frecuente por arriba de los 50 años. Es el sarcoma de tejidos blandos que con mayor frecuencia se localiza en la región de cabeza y cuello en los niños2. Representa el 75% de todos los sarcomas en niños de 0-14 años y constituye menos del 3% de los tumores malignos de tejidos blandos del adulto. Es un tumor maligno que se origina de células mesenquimatosas primitivas y tienen diferenciación hacia músculo esquelético. En los niños se localiza predominantemente en cabeza y cuello (40%), aparato genitourinario y tejidos blandos de las extremidades (40%), y con menor frecuencia en tronco, órbita, intratorácico y retroperitoneo. Existe controversia sobre el sitio predominante en adultos, aunque varios estudios han demostrado preponderancia en cabeza y cuello (42.85%), seguida de las extremidades (23.8%) y el aparato genitourinario (14.8%), similar a los casos pediátricos1.
Los sarcomas de tejidos blandos primarios de la pared torácica son una enfermedad poco frecuente. Abarcan menos del 10% de todos los casos de sarcomas reportados. La mayoría de las series en la literatura médica son pequeñas, se extienden por décadas y presentan un rango amplio de abordajes terapéuticos3. La mayor parte de los sarcomas de tejidos blandos de tórax son superficiales, no obstante la malignidad tiene un pronóstico favorable. Para obtener el control local es indispensable un margen quirúrgico adecuado; por lo tanto, la toracotomía asociada a cistotomía y transferencia de colgajos puede llegar a ser necesaria. Sólo el 17.9% de los sarcomas se originan en el tronco3.
La clasificación histológica abarca muchas histologías, como angiosarcoma, leiomiosarcoma, mesenquimoma, histiocitoma, rabdomiosarcoma, desmoide, sarcoma sinovial y liposarcoma. La clasificación de Stojadinovic, donde casi la mitad (47%) son desmoides, ha llevado a proponer que se consideren como grupo independiente. El 15% corresponde a dermatofibrosarcoma protuberans, y el 40% son histologías comunes a extremidades y comparten porcentajes relativamente similares. El histiocitoma fibroso maligno parece ser una entidad particular, no asociado a las características del resto de sarcomas3. Desde el punto de vista histológico, dentro de los rabdomiosarcomas se identifican varios tipos: el embrionario, el botrioides (variante del embrionario), el alveolar (con su variante sólida), el pleomórfico y los subtipos recientemente descritos como el fusocelular y el esclerosante seudovascular; este último, por sus características inmunohistoquímicas y citogenéticas, parece ser una variante del rabdomiosarcoma embrionario4,5. La distinción de subtipos de rabdomiosarcoma es importante debido a que difieren en su comportamiento biológico6.
En todos los subtipos de rabdomiosarcoma, la célula característica es el rabdomioblasto caracterizado por la presencia de un citoplasma amplio, eosinófilo y granular con núcleo excéntrico7,8. Las células pueden ser redondeadas, poligonales o alargadas, estas últimas tienen la forma de raqueta o renacuajo. La diferenciación muscular se puede demostrar con microscopía electrónica o con técnicas inmunohistoquímicas. Ultraestructuralmente, el rabdomiosarcoma se caracteriza por la presencia de bandas Z sarcoméricas e inmunohistoquímicamente, por la reactividad para la desmina, actina, mioglobina y Myo D1. La proteína miogénica reguladora Myo D1 y la mioglobina, son los marcadores más específicos de diferenciación muscular estriada9,10.
El rabdomiosarcoma esclerosante es una nueva variante de rabdomiosarcoma caracterizada por una matriz esclerosante hialina y un patrón seudovascular que afecta preferentemente a adultos11,12. Fue descrito por primera vez en el 2000 por Mentzel et al. Este es un tumor con diferenciación mioide, áreas de esclerosis y patrón seudovascular. Forma matriz osteoide o condroide (nunca hueso ni cartílago). En 2004, Vadgama et al. describe el primer caso en niños12. Al inicio fue denominado rabdomiosarcoma esclerosante seudovascular, posteriormente como rabdomiosarcoma esclerosante en adultos13-15. Esta entidad ha sido descrita en base a la publicación de 15 casos a nivel mundial y se caracteriza por afectar preferentemente a adultos. Los estudios moleculares realizados no han detectado los genes de fusión descritos en la forma alveolar. La casuística es pequeña para intentar establecer un comportamiento biológico de esta variante de rabdomiosarcoma. En un trabajo más reciente se ha revisado una serie de 13 casos de esta variante de rabdomiosarcoma descritos en niños12. En la serie de 7 casos publicados, 3 casos ocurrieron en la región de cabeza y cuello (órbita y base del cráneo, nasofaringe y mandíbula superior izquierda)15. Los restantes 4 tumores se desarrollaron en tejidos blandos de extremidades y uno dentro de la primera vértebra sacra14.
Respecto a la inmunohistoquímica, las células tumorales muestran una fuerte y difusa reactividad para Myo D1, miogenina y marcadores específicos de tumores músculo-esqueléticos14,15. Los anticuerpos antidesmina, actina y mioglobina han sido utilizados como marcadores musculares. La mioglobina es un marcador más sensible, pues se presenta sólo en rabdomioblastos diferenciados. La actina sarcomérica sólo se presenta en rabdomioblastos bien diferenciados y ha sido descrita ocasionalmente en leiomiosarcomas4,16,17. La desmina es positiva en una pequeña proporción de células y puede haber positividad a CD5614,15.
La genética molecular puede ser de ayuda para el diagnóstico de estos tumores. El rabdomiosarcoma alveolar puede presentar 2 translocaciones; t(2;13) en más del 50% de los casos y t(1;13) en 22%, mismas que no están presentes en otros tipos de rabdomiosarcoma. Estas translocaciones resultan en la fusión de los genes PAX (3 y 7) con el gen FKHR, fusión que produce proteínas quiméricas de función alterada y más potente que las normales, afectando el control del crecimiento celular, la apoptosis, la diferenciación y la motilidad. En los pacientes con enfermedad en estadio avanzado, la presencia de la fusión PAX3-FKHR es un factor de pronóstico adverso16-18.
Los sarcomas del tronco frecuentemente se presentan como una masa indolora y de lento crecimiento, en donde la sintomatología suele ser secundaria a la compresión ejercida por el tumor. Estos sarcomas troncales están asociados con diversos agentes carcinógenos y síndromes hereditarios. Entre los agentes carcinógenos se ha citado a los clorafenoles, al arsénico y a la radiación. Los síndromes genéticos relacionados son: síndrome de Li-Fraumeni, retinoblastoma, enfermedad de von Recklinghausen y síndrome de Gardner3.
Diagnóstico
La biopsia es el primer recurso utilizado. Aunque la biopsia con aguja fina es frecuentemente la modalidad de elección para otras masas tumorales, su uso está limitado en los sarcomas, debido a que no proporciona una muestra adecuada para el diagnóstico. Si la biopsia con aguja gruesa es insuficiente o imposible de practicar, el siguiente paso es realizar una biopsia incisional. Para evitar la diseminación durante la biopsia, la disección debe ser por fuera de la cápsula y es imprescindible evitar la formación de hematomas3.
La evaluación radiográfica se utiliza para determinar la extensión de las masas tumorales, y para dar seguimiento a la progresión de la enfermedad. La tomografía y la resonancia magnética son imprescindibles. La tomografía es también útil para determinar la existencia de enfermedad metastásica. La angiografía sólo es requerida cuando existe sospecha de invasión vascular3.
Tratamiento
El tratamiento en este tipo de pacientes es multimodal. Es primordial la resección completa de la lesión con márgenes negativos a tumor de aproximadamente 10 cm3,21,22. La resección en bloque con márgenes amplios, es el tratamiento de elección para todos los sarcomas, incluyendo los troncales. De cualquier modo, en nuestros días, el manejo multidisciplinario es quizá lo más importante. En cirugía de sarcomas troncales se debe considerar la localización, su tamaño, la invasión de estructuras importantes, la necesidad de reconstrucción y la profundidad. La cirugía puede ser radical, compartamental, resección amplia y resección multiestructural3,23-15. La linfadenectomía se realiza cuando existe evidencia clínica o radiográfica de involucramiento ganglionar. Diversos subtipos histológicos se asocian con un incremento en el riesgo de involucramiento ganglionar, sobre todo cuando se trata de sarcoma epitelioide, rabdomiosarcoma, sarcoma de células claras y angiosarcoma3.
Quimioterapia adyuvante: La quimioterapia adyuvante no es considerada estándar. Los pacientes sintomáticos pueden iniciar terapia sistémica, que de acuerdo a la severidad de los síntomas, puede ser en combinación o monoterapia26. La doxorrubicina ha sido el fármaco más utilizado en dicho escenario. La inclusión de ifosfamida a los regímenes adyuvantes ha mantenido los resultados en la tasa de periodo libre de enfermedad (PLE); sin embargo, no hay un beneficio contundente en supervivencia general (SG)26,27. Todo lo anterior se documenta en el estudio clásico publicado en 1997 (SMAC, por sus siglas en inglés, Sarcoma Meta-Analysis Collaboration), donde se reporta una disminución absoluta del riesgo de recaída a 10 años del 10%; sin embargo, el beneficio absoluto en SG fue de sólo de 4% a 10 años, lo cual no fue estadísticamente significativo26,27. El anterior meta-análisis fue actualizado en el 2008 por O'Connor et al., quien incluyó en el trabajo 4 estudios aleatorizados recientes donde se agrega ifosfamida al esquema basado en antraciclinas y se confirma el beneficio en PLE (HR=0.71; p=0.0001), siendo nuevamente imposible demostrar algún beneficio en SG (HR=0.87; p=0.12)28-30.
Quimioterapia neoadyuvante: Al igual que en el terreno adyuvante, los resultados son controvertidos. Sin embargo, la terapia neoadyuvante en sarcomas de tejidos blandos puede disminuir el volumen tumoral facilitando el éxito de la cirugía conservadora, y podría también erradicar la enfermedad microscópica, además de ser un excelente escenario para valorar la respuesta in vivo. Está indicada en pacientes con tumores voluminosos de etapas II y III, con alto riesgo de recurrencia, para facilitar el procedimiento quirúrgico conservador, sobre todo en histologías quimiosensibles. Esta neoadyuvancia ha demostrado un aumento en la tasa de PLE de aproximadamente 7%. La modalidad a utilizar es preferentemente quimio-radioterapia, con esquemas basados en antraciclinas. Se han realizado ensayos con quimioterapia de inducción seguida de perfusión aislada, aunque habrá que esperar la maduración de sus resultados. Como por ejemplo, el estudio EORTC 62961 (fase III), con 341 pacientes (149 con sarcomas de tejidos blandos de extremidades), donde se valoró un esquema de quimio-radioterapia con etopósido, ifosfamida y adriamicina, administrados con o sin hipertermia local, observándose una tasa de PLE de 70% vs. 57% sobre hipertermia sola26.
Tratamiento en enfermedad metastásica o recurrente
Con respecto al tratamiento de estos pacientes, Verma et al. (2008) documentó que no hay evidencia de que la tasa de respuesta objetiva y la tasa de PLE se traduzca en aumento de SG. Algunos grupos de pacientes se benefician más del manejo paliativo. Los jóvenes (< 40 años), aquellos con un ECOG de 0-1; ausencia de metástasis hepáticas, PLE largo, neoplasias de alto grado o con histología de liposarcoma y sarcoma sinovial26,32. Los pacientes sintomáticos pueden iniciar terapia sistémica, que de acuerdo a la severidad de los síntomas, puede ser en combinación o monoterapia. Mientras que los asintomáticos con enfermedad limitada van a cirugía. Los asintomáticos avanzados pueden ser sólo observados o recibir terapia sistémica. La terapia más utilizada continúa siendo el esquema basado en antraciclinas, en monoterapia o en combinación con ifosfamida, cuyas respuestas varían entre 10% y 46%26.
Entre los fármacos biológicos que se ensayan hoy en día para sarcomas de tejidos blandos están: cixutumumab, imatinib, sunitinib, sorafenib, crizotinib, pazopanib e inhibidores de mTOR26. Existen varios estudios con respecto al manejo de esta clase de pacientes, como ejemplo podríamos citar un estudio fase II donde se utilizó gemcitabina en sarcomas avanzados y metastásicos; donde se observó una tasa de sobrevida media de 15 meses, sobrevida estimada a un año de 63% y sobrevida media libre de progresión de 13 meses23.
En México, la terapia con antraciclinas es un recurso básico, ya que en muchos Centros no se realiza hipertermia ni se administra radioterapia; sin embargo, recursos como ifosfamida pueden resultar incosteables para muchos pacientes que reciben atención institucionalizada. La recomendación que se ajusta más a esta situación es el uso de esquemas basados en antraciclinas, en adyuvancia y neoadyuvancia, con apoyo en manejo multidisciplinario, con supervivencia libre de progresión, protocolos de estudio e hipertermia26.
Tratamiento con radioterapia
La radioterapia se recomienda para el manejo de la enfermedad residual, los márgenes estrechos o positivos. La radioterapia es el tratamiento adyuvante de elección en los sarcomas; sin embargo, en tronco y abdomen ésta llega a considerarse sólo cuando existe un límite negativo pero insuficiente (menor a 1 cm, o margen positivo en donde no se pueda realizar ampliación del margen)3. La radioterapia adyuvante ha demostrado beneficio en SG, y se considera indicación en tumores de alto grado33-35, mayores de 5 cm36, con localización profunda, y en los que se encuentran márgenes cercanos (< 1 cm) o positivos35,36. El uso de radioterapia adyuvante no sustituye a resección completa cuando ésta es posible36,37. Los pacientes con tumores de bajo grado y con margen negativo no requieren adyuvancia38.
La braquiterapia adyuvante se usa sola o en combinación con radioterapia externa. Se recomienda en monoterapia en resecciones completas y tumores de alto grado, siempre y cuando el volumen de tratamiento pueda ser cubierto por el implante. Se usa braquiterapia como sobreimpresión a radioterapia externa cuando no se logra R0, o si existe ulceración de la piel, en sarcomas de bajo grado o en riesgo de contaminación quirúrgica del campo. Las dosis recomendadas en monoterapia son: en LDR, de 45-50Gy/4-6 días, a una tasa de dosis de 0.45Gy/h. Cuando se usa como sobreimpresión a radioterapia externa (45-50Gy), la dosis recomendada es 15-25Gy/2-3 días. La tasa de dosis se puede incrementar39.
En pacientes con tumores > 8 cm de alto grado, se reporta un porcentaje de falla a distancia > 40%, y por este motivo es importante diseñar nuevas estrategias como la quimioradioterapia preoperatoria. Se han reportado resultados alentadores con quimio-radioterapia bajo esquema MAID (mesna, adriamicina, ifosfamida y dacarbazina) concomitante con radioterapia, en dosis de 44Gy, seguido de cirugía, y finalmente 3 ciclos más de quimioterapia con el mismo esquema36,40-42.
Los pacientes tratados con radioterapia postoperatoria cursan con más fibrosis y complicaciones tardías, y los tratados con radioterapia preoperatoria tienen mayor riesgo de problemas de cicatrización en la herida. Cada vez hay más evidencia que apoya el uso de la modalidad preoperatoria, pues ha demostrado su impacto en SG y SVCE43.
Asimismo, se recomienda el uso de resonancia magnética contrastada para valorar la respuesta, sobre todo es importante evaluar el edema en T2, ya que en > 60% de los pacientes existen células tumorales en el edema44.
Los factores de mal pronóstico incluyen: localización en extremidades, edad de presentación (adultos), estadio clínico avanzado y presencia de la translocación t(2;13)16,17. El factor pronóstico principal para los sarcomas de tejidos blandos de extremidades y tronco es el tipo histológico. El factor pronóstico de control locorregional es el margen negativo y suficiente (considerado al menos 1 cm libre de tumor). Otros factores son los antecedentes de recurrencia locorregional y el tamaño tumoral. Los factores pronósticos para metástasis son: tipo histológico, tamaño y profundidad. Se ha encontrado que la tasa global de supervivencia a 15 años para sarcomas de tejidos blandos de extremidades es de 68.4%, comparada con 59.5% para los troncales, y 50% para los retroperitoneales. El grado tumoral es un factor pronóstico importante, pues la tasa de supervivencia a 12 años es de 92% para sarcomas de bajo grado, 75% para sarcomas de grado intermedio y 43% para sarcomas de alto grado. Otro factor pronóstico importante es el tamaño tumoral, debido a que los tumores < 5 cm tienen una tasa de supervivencia a 12 años del 70%, y los tumores > 5 cm tienen una tasa del 49.5%. La SG de pacientes con una cirugía con márgenes negativos fue del 67% comparada con una del 49% para la cirugía con márgenes positivos3.
La supervivencia que se puede esperar a 5 años en estadio I es de 90%; en estadio II de 70%; en estadio III de 50%, y en estadio IV es de 10% al 20%. El seguimiento de los pacientes se contempla, inicialmente para cada 3 meses dentro de los primeros 2 años, para pasar a cada 6 meses en los siguientes 3 años y una vez al año, a partir del quinto año3.
Conclusiones
Nuestro caso en particular representa el primero reportado en la cavidad torácica izquierda a nivel mundial. El rabdomiosarcoma esclerosante representa una dificultad para clasificarlo dentro del grupo de los distintos rabdomiosarcomas, por lo cual se ha tipificado como un nuevo tipo de rabdomiosarcoma20,21. También podríamos concluir que la quimioterapia adyuvante en sarcomas no es considerada estándar; su aplicación requiere individualizar el caso por parte de un grupo multidisciplinario. Si se inicia la terapia es preferible optar por esquemas basados en antraciclinas y con ifosfamida26. El pronóstico en rabdomiosarcoma de adultos es peor que en niños, no obstante se ha demostrado que con la implementación de protocolos de tratamientos adoptados de casos pediátricos trasladados a casos en adultos, se podría incrementar la sobrevida en pacientes adultos19.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Financiamiento
Los autores no recibieron patrocinio para llevar a cabo este artículo.
* Autor para correspondencia:
Av. Revolución N° 1182, esquina Barranca del Muerto, 2° piso,
Colonia San José Insurgentes, Delegación Benito Juárez, México D.F., México.
Teléfono: 5593 5300, ext. 130.
Correos electrónicos: crubeluis@yahoo.com.mx, crubeluis@gmail.com (Luis Cruz-Benítez).