


El sarcoma histiocítico (SH) en una neoplasia especialmente rara en niños. Habitualmente afecta piel, ganglios linfáticos, tracto gastrointestinal y partes blandas. En la mayoría de los casos se presenta en estadios de la enfermedad muy avanzados, con pronóstico malo a pesar del tratamiento.
Se presenta el caso de un niño de 4 años con enfermedad localizada en tejido celular subcutáneo, el diagnóstico se hizo por inmunohistoquímica con CD68 positivo. El paciente permanece vivo y sin datos de actividad de la enfermedad después de 6 ciclos de quimioterapia con ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP), a 18 meses del diagnóstico.
Histiocytic sarcoma is a special uncommon neoplasm in childhood. Affect skin, lymphatic nodes, gastrointestinal tract and soft tissues. Frequently is present like advanced disease with poor prognostic in spite of treatment.
We present a four-year-old boy with local histiocytic sarcoma in soft tissues; the diagnostic was made by immunohistochemistry with CD68+. He received 6 cycles of cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) chemotherapy and is alive and healthy to 18 months of diagnostic.
Introducción
El sarcoma histiocítico (SH) es una neoplasia rara en todos los grupos de edad, pero es particularmente rara en niños. La etiología es desconocida, pero se ha asociado a la pérdida del supresor tumoral p16 INK4A. La deleción del locus CDKN2A que codifica p16INK4A se ha visto también en leucemia linfoblástica de células B y T1.
Los SH son neoplasias malignas de precursores histiocíticos que habitualmente afectan piel, ganglios linfáticos, tracto gastrointestinal y partes blandas. Pueden presentarse en niños y adultos, sin embargo la mayoría de casos se ve en adultos (media de edad 46 años), con predilección por hombres. El pronóstico es malo, presentándose en la mayoría de los casos en estadios de la enfermedad muy avanzados. El diagnóstico combina las características morfológicas con marcadores inmunohistoquímicos: positividad de marcadores histiocitarios (CD68, lisozima) junto con ausencia de marcadores de células T o B, células mieloides y células histiocíticas diferenciadas (células de Langerhans, células dendríticas foliculares y células dendríticas interdigitantes)2.
El diagnóstico de los tumores histiocitarios y de células dendríticas no puede ser hecho únicamente por la información clínica. Microscópicamente, estos tumores abarcan un amplio espectro de características histológicas, con patrones epitelioides fusocelulares, de células redondas indiferenciadas, o con gran componente inflamatorio, que enmascara la población tumoral, por lo que la inmunohistoquímica (CD68, CD21, CD23, CD35, CD1A y S-100) y la microscopía electrónica son esenciales para llegar al diagnóstico2.
El examen anatomopatológico suele revelar la desestructuración de la arquitectura normal del órgano, debido a una infiltración difusa de células neoplásicas polimórficas de gran tamaño, con contorno redondo u ovalado, y de citoplasma amplio y eosinófilo, con núcleos grandes y excéntricos. Con frecuencia se observan células gigantes multinucleadas. Los fenómenos de hemofagocitosis son escasos y suele existir un número variable de células reactivas acompañando a los linfocitos.
Para establecer el diagnóstico definitivo es necesario el estudio inmunofenotípico, y por definición debe existir expresión de uno o más marcadores histiocitarios (CD68, alfa 1-antitripsina, lisozima, CD11c, CD14), con ausencia de marcadores de células dendríticas/Langerhans (CD1a, CD21, CD35), de los linfocitos B y T (CD2, CD3, CD8, CD19, CD20, CD22, CD79a), y de los marcadores mieloides específicos (mieloperoxidasas, CD13, CD33, CD34). La lisozima suele presentar un patrón granular intracitoplasmático con una mayor acentuación habitualmente a nivel de aparato de Golgi (patrón Golgi positivo).
El SH en una neoplasia maligna que muestra características morfológicas e inmunofenotípicas de histiocitos maduros3. El diagnóstico diferencial morfológico del SH incluye seudotumor inflamatorio, sarcoma folicular de células dendríticas, sarcoma de células dendríticas interdigitantes, histiocitosis maligna de células de Langerhans, linfoma anaplásico de células grandes, melanoma y otros sarcomas2,4.
El pronóstico de los SH y de células dendríticas intedigitantes es malo. Hasta la fecha, no se ha llegado a un consenso sobre el régimen quimioterapéutico estándar a seguir en este grupo de pacientes, se ha usado quimioterapia para linfoma no Hodgkin, con respuestas variables al tratamiento, pero sin obtenerse resultados favorables en la mayoría de los casos. Se ha probado tratamiento, también sin éxito, con poliquimioterapia incluyendo doxorrubicina, ciclofosfamida, vincristina y prednisona. Hay un caso descrito en el que se obtuvo mejoría tras régimen quimioterapéutico con doxorrubicina, bleomicina, vinblastina y dacarbazina (ABVD). Son tumores con conducta muy agresiva, y con respuesta quimioterapéutica variable y poco establecida.
El SH (la contraparte maligna de histiocitos/macrófagos) se ha reportado en asociación, y algunas veces clonalmente relacionado a linfoma/leucemia linfoblástica aguda de células B o T, o a otras neoplasias hematológicas como leucemia mielomonocítica crónica1,5.
La Organización Mundial de la Salud (OMS) clasifica a las neoplasias hematopoyéticas basada en la línea celular de la cual derivan6. La rareza de los SH hace difícil formular guías de tratamiento basadas en la evidencia. La cirugía, radioterapia, quimioterapia, trasplante de células tallo y una combinación de estas modalidades, han sido usadas con resultados pobres7,8.
Presentación del caso
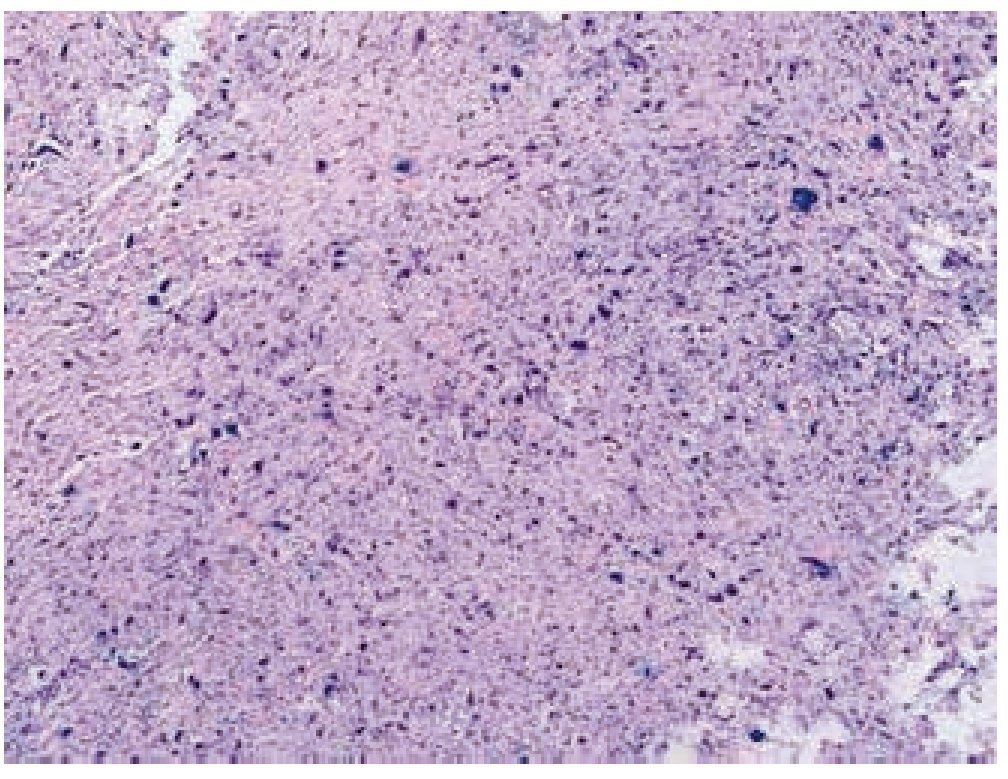
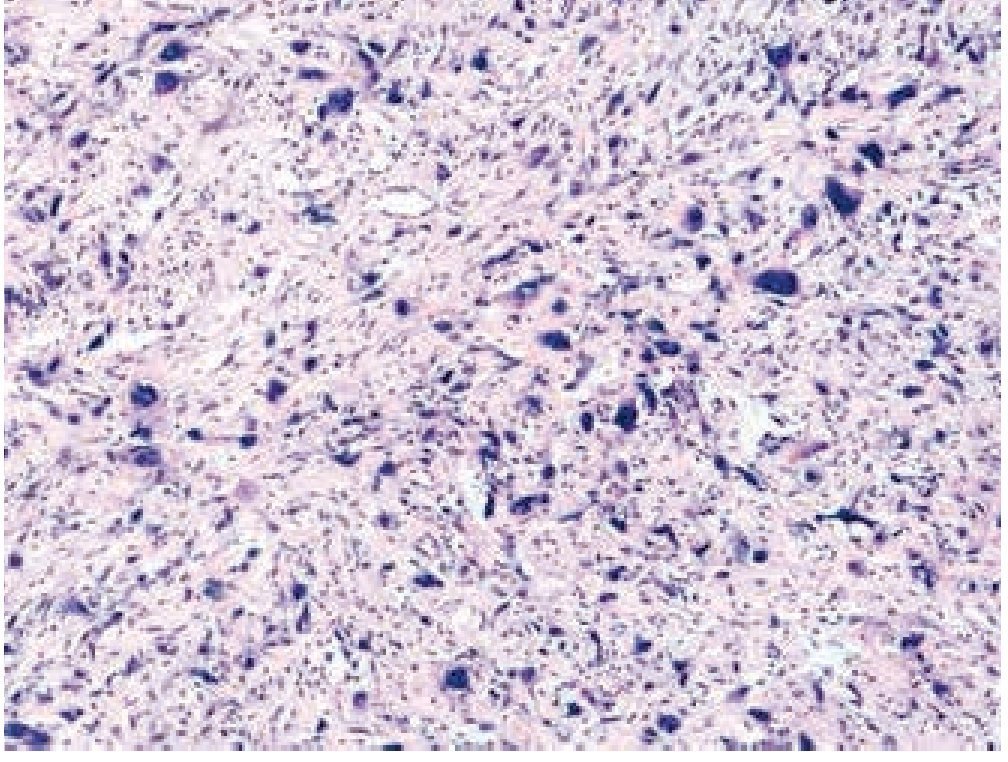
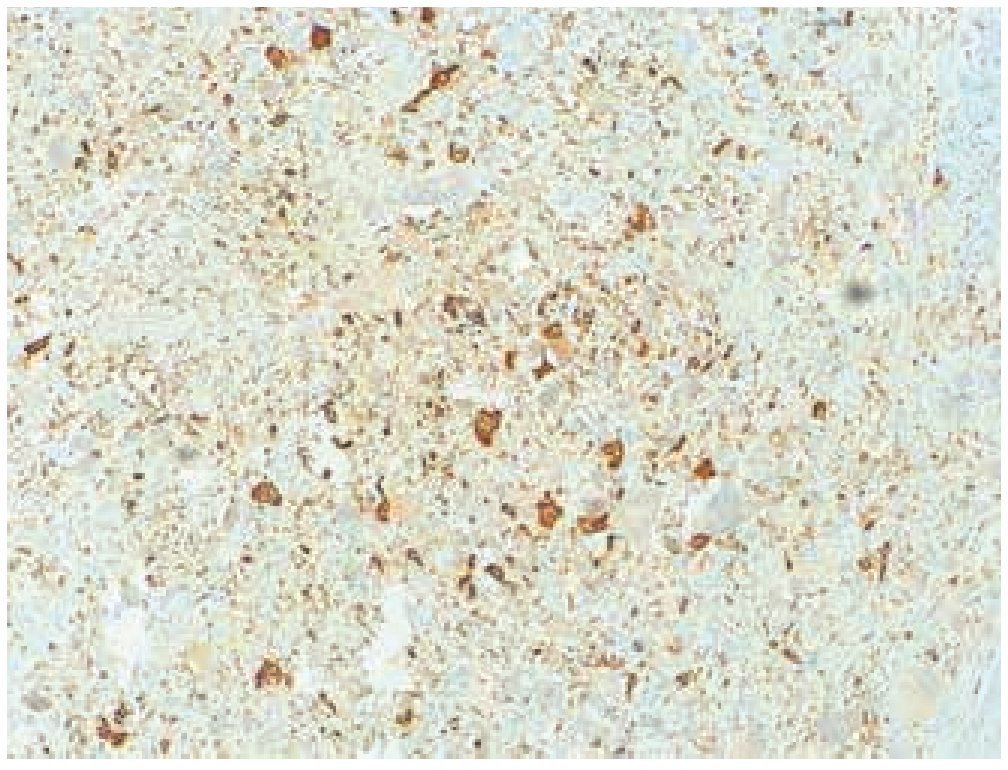
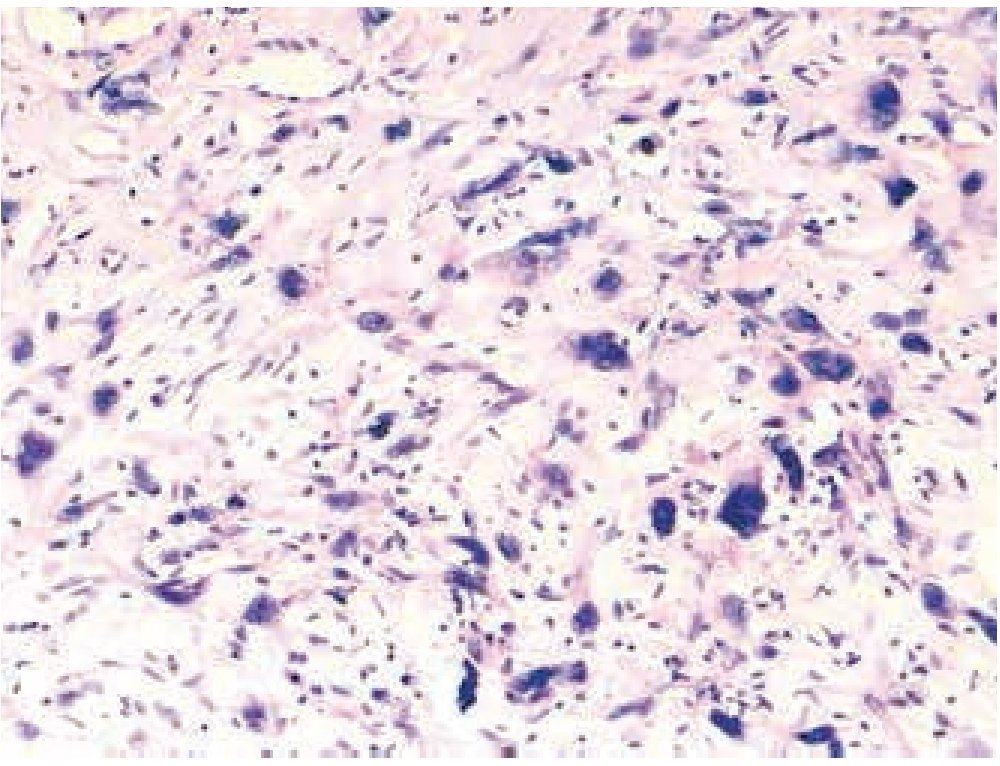
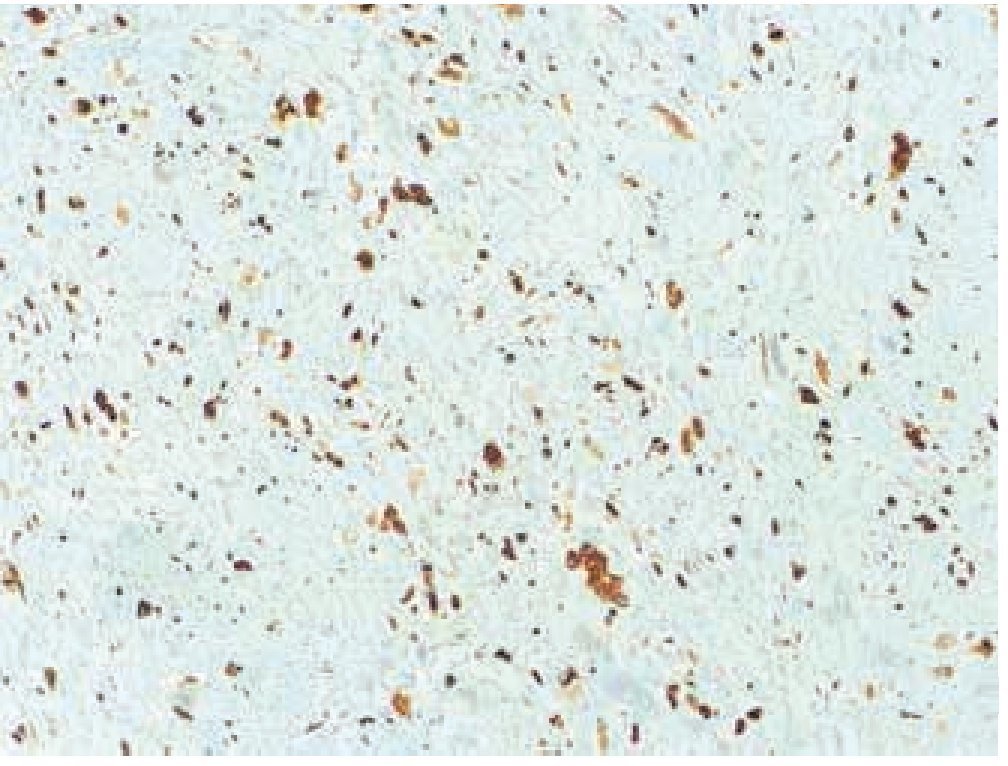
Masculino de 4 años de edad, antecedente de abuela paterna fallecida por cáncer de mama. Cuadro clínico de 6 meses de evolución con nodulación en brazo izquierdo de 1 cm de diámetro, móvil, no dolorosa, dura, no adherida a planos superficiales, ni profundos. Resto de la exploración negativa. Un ultrasonido mostró lesión sólida en tejido celular subcutáneo, sin afectación de estructuras óseas ni musculares. Se realizó biopsia excisional de la lesión, la microscopía de luz con tinción de hematoxilina & eosina (figs. 1, 2 y 3) reportó neoplasia maligna de estirpe histiocítica. La inmunohistoquímica fue: CD68 +++ (fig. 4), EMA reacción focal, CD4 +, S-100 +, CD45 negativa (fig. 5), citoqueratina negativa (fig. 6); CD1A, HMB45, CD30 y CD2 negativos; y Ki67 con índice de proliferación de 60% (fig. 7).
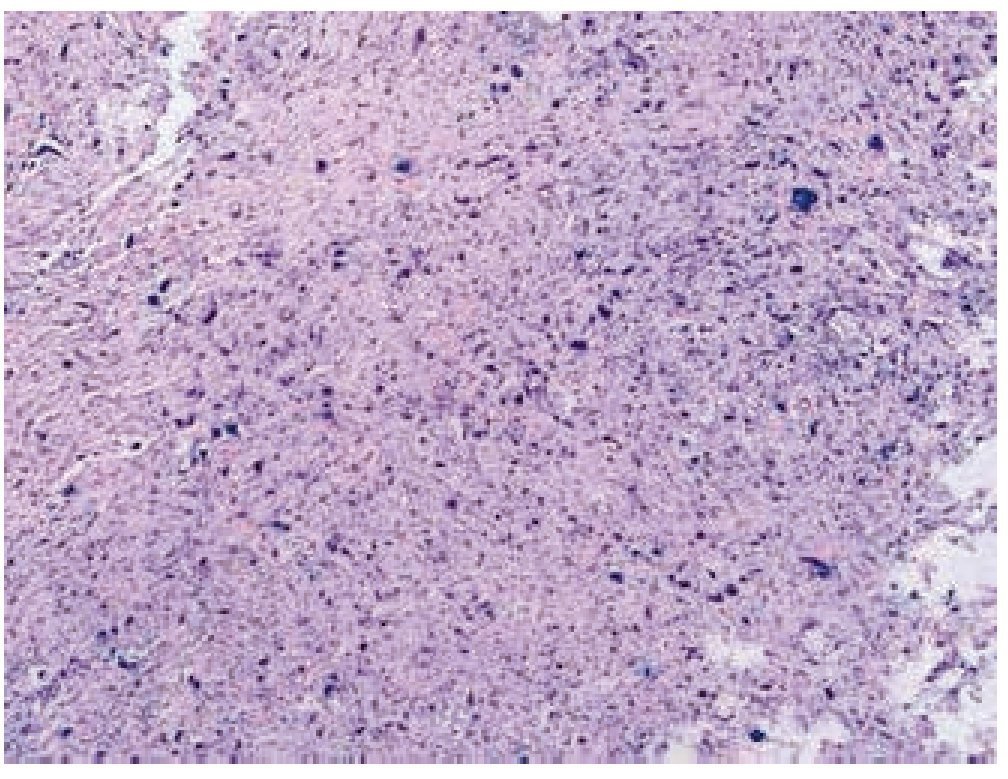
Figura 1 Microfotografía 1 (tinción de hematoxilina & eosina, 10x).
Figura 2 Microfotografía 2 (tinción de hematoxilina & eosina, 40x).
Figura 3 Microfotografía 3 (tinción de hematoxilina & eosina, 40x).
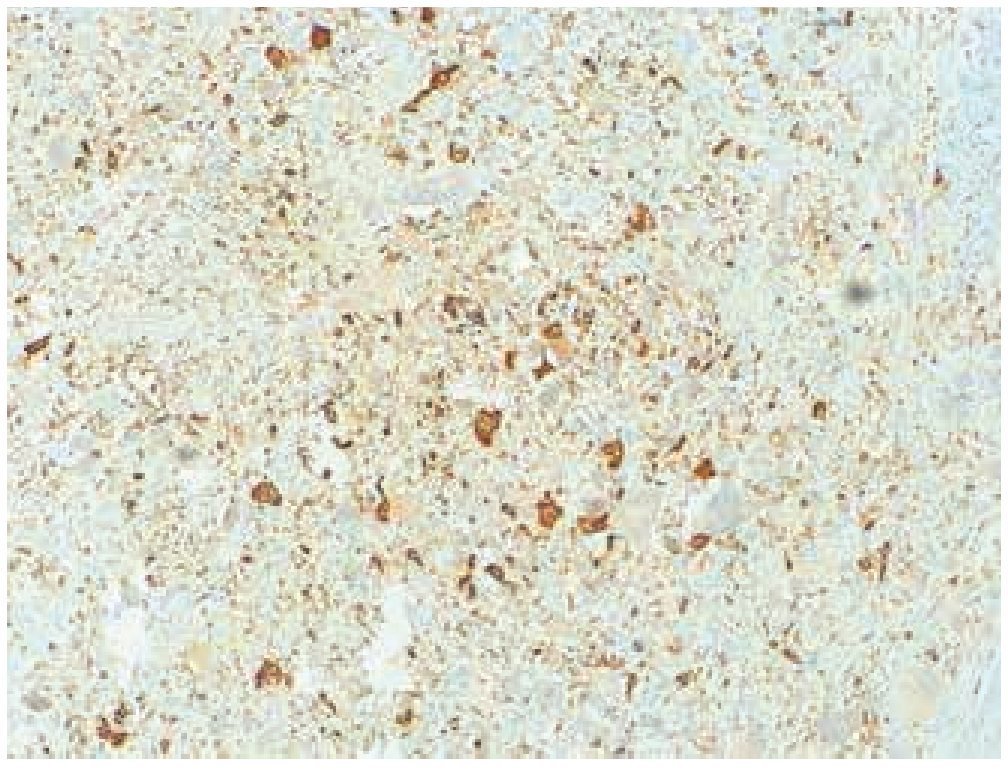
Figura 4 Microfotografía 4 (tinción de inmunohistoquímica CD68 positivo, 10x).
Figura 5 Microfotografía 5 (tinción de inmunohistoquímica CD45 positivo, 10x).
Figura 6 Microfotografía 6 (tinción de inmunohistoquímica citoqueratina negativa, 10x).
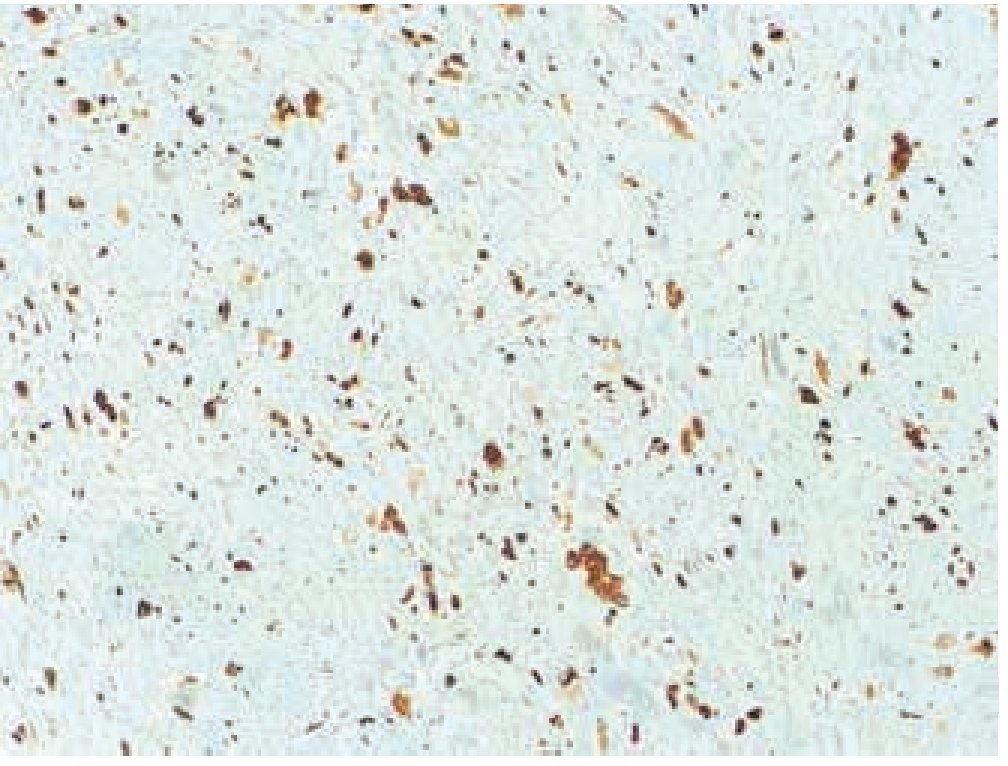
Figura 7 Microfotografía 7 (tinción de inmunohistoquímica Ki67 positivo, 10x).
Los estudios de laboratorio fueron normales, la radiografía de tórax con ensanchamiento mediastinal superior (timo hiperplásico), las tomografías de tórax y abdomen normales, la imagen de resonancia magnética de cráneo y médula espinal normales, el aspirado de médula ósea, el citoquímico y citológico de líquido cefalorraquídeo fueron negativos. Se manejo con 6 ciclos de ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) con buena tolerancia, sin presentar datos de actividad durante todo el tiempo de tratamiento y pasando a vigilancia con todos los estudios de extensión negativos. A la fecha tiene una sobrevida de 17 meses a partir del diagnóstico en remisión completa continua.
Discusión
El SH es raro en niños, se conoce poco acerca de su comportamiento clínico, aunque en general se reporta como una enfermedad agresiva que se presenta en estadios avanzados de la enfermedad, con un mal pronóstico a pesar de las diversas modalidades de tratamiento, en este caso llama la atención la presencia de enfermedad localizada sólo en el tejido celular, no se documentó enfermedad a otros niveles, lo que explica posiblemente la buena evolución que hasta el momento ha tenido el paciente, quien tiene a la fecha una sobrevida libre de enfermedad de 17 meses. Se ha considerado que la expresión de CD163 puede ser un marcador de resistencia a quimio y radioterapia, y la ausencia de su expresión puede ser reactiva para el tratamiento9. Algunos datos, como el tamaño del tumor y el estadio de la enfermedad pueden tener valor pronóstico, mientras que las mitosis y el índice de proliferación celular medido por el Ki67 suelen ser muy variables y no han mostrado relación con el curso clínico10,11.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Financiamiento
Los autores no recibieron patrocinio para llevar a cabo este artículo.
* Autor para correspondencia:
Av. Félix Cuevas N° 540, Colonia Del Valle,
Delegación Benito Juárez,
C.P. 03000, México D.F., México.
Teléfono: 5200 5003, ext. 14338.
Correo electrónico: sandra_fpa@hotmail.com (Sandra Flor Páez-Aguirre).