




La enfermedad de Wilson es una alteración que presenta una herencia autosómica recesiva caracterizada por la acumulación tóxica de cobre en el hígado y posteriormente en el cerebro y otros órganos. Su diagnóstico clínico se basa en la detección de concentraciones bajas de ceruloplasmina en suero, el incremento de excreción de cobre en orina, la presencia de anillos de Kayser-Fleisher en la córnea, y/o unos niveles altos de cobre en el tejido hepático. El diagnóstico puede ser difícil en el caso de ausencia de síntomas típicos, lo que puede resultar en la no administración de terapia profiláctica en estos pacientes. El estudio molecular ha mejorado el diagnóstico de la enfermedad, incluso en los casos dudosos. En el presente trabajo se detallan las diferentes técnicas aplicadas al diagnóstico molecular de la enfermedad y las mutaciones más comunes descritas. En la actualidad la secuenciación directa del gen ATP7B es el método más ampliamente utilizado para la detección de mutaciones. El estudio molecular y la identificación de mutaciones en el gen ATP7B ofrecen la posibilidad de diagnóstico en pacientes con enfermedad de Wilson y en sus familiares, permitiendo el asesoramiento genético, y la posibilidad de realizar diagnósticos prenatales y preimplantacionales.
Wilson¿s disease is an autosomal recessive disorder characterized by toxic copper accumulation in the liver and subsequently in the brain and other organs. Clinical diagnosis is based on the detection of low serum ceruloplasmin concentrations, increased urinary copper excretion, Kayser-Fleisher rings in the cornea, and/or high copper levels in hepatic tissue. Diagnosis can be difficult when the typical symptoms of the disease are absent, a situation that can lead to a lack of prophylactic therapy in these patients. Molecular study has improved the diagnosis of this disease, even in doubtful cases. The present article outlines the various techniques applied in the molecular diagnosis of Wilson's disease and the most commonly described mutations. Currently, direct sequencing of the ATP7B gene is the most widely used method to detect mutations. Molecular study and identification of ATP7B gene mutations allow diagnosis of individuals with Wilson's disease and their relatives, as well as the possibility of genetic counselling and prenatal and preimplantation genetic diagnosis.
La enfermedad de Wilson (EW, OMIM #277900) o degeneración hepatolenticular es un trastorno del metabolismo del cobre que se hereda siguiendo un patrón autosómico recesivo. Esta enfermedad fue descrita por primera vez en 1912 por Kinnear Wilson1. La EW puede presentar un amplio espectro clínico, con manifestaciones hepáticas, neurológicas, psiquiátricas o con una combinación de ellas, en individuos entre 3 y 60 años2. El diagnóstico se basa en la detección de concentraciones bajas de ceruloplasmina en suero, el incremento de excreción de cobre en orina, la presencia de anillos de Kayser-Fleisher en la córnea, y/o unas concentraciones altas de cobre en el tejido hepático2. El diagnóstico puede ser difícil en el caso de ausencia de síntomas típicos y en familiares asintomáticos. Sin embargo, un diagnóstico precoz es esencial ya que el tratamiento con agentes quelantes de cobre o cinc oral previene el daño hepático y neurológico causado por la acumulación de cobre3,4. El diagnóstico molecular puede determinar la presencia o ausencia de mutaciones, permitiendo el diagnóstico de EW incluso cuando los datos clínicos son equívocos3.
La incidencia de la EW varía entre las diferentes poblaciones entre 1/30.000 y 1/100.000, con una frecuencia estimada de portadores de 1/905. Existen poblaciones en las que la prevalencia es mayor, como en las Islas Canarias, donde se ha calculado en 1/2.6006. El gen ATP7B es el único gen descrito causante de la EW y codifica para una ATPasa transportadora de cobre. Este gen se halla localizado en el cromosoma 13 (13q14.3), tiene 80kb, contiene 21 exones y produce una proteína de 1.465 aminoácidos7-10. El gen ATP7B tiene un 57% de identidad con el gen ATP7A, que está implicado en la enfermedad de Menkes. La proteína ATP7B es un transportador transmembrana intracelular de cobre, que tiene un papel esencial en la incorporación de cobre a la ceruloplasmina y en el transporte de cobre fuera del hepatocito a la bilis. Se expresa principalmente en el hígado, riñón y placenta. Una función incorrecta de ATP7B da como resultado la formación de una ceruloplasmina inestable, que es rápidamente degradada. También da como resultado la acumulación de cobre en los hepatocitos11. Este exceso de cobre se acumula en tejidos extrahepáticos, incluyendo el cerebro y la córnea (donde es visible como anillos de Kayser-Fleischer), siendo la responsable de las manifestaciones clínicas de la EW12.
Hasta el momento se han descrito más de 500 mutaciones causantes de la enfermedad, localizadas a lo largo de todo el gen (The Human Gene Mutation Database in Cardiff, http://www.hgmd.cf.ac.uk/ac/index.php). Se han llevado a cabo numerosos estudios en pacientes con EW, y estos han revelado que el espectro de mutaciones en el gen ATP7B es específico de poblaciones. Además, existe una alta heterogeneidad, lo que dificulta el estudio molecular en los individuos con EW. Por otro lado, más del 50% de las mutaciones descritas son del tipo missense (o de cambio de sentido), lo que añade un nuevo problema al determinar la posible implicación de este tipo de mutaciones en la enfermedad. La distribución de mutaciones difiere entre los grupos étnicos, así como la presencia de mutaciones prevalentes13-20. La mutación más frecuente en poblaciones caucasianas es la H1069Q. Esta mutación se encuentra en un 50-80% de los casos en Europa, principalmente en la Europa del Este, mientras que su frecuencia decrece hacia el Oeste y el Sur de Europa, donde la frecuencia se sitúa entre 10 y el 30%12,14. Existen otras mutaciones comunes en Europa, pero sus frecuencias son inferiores al 10%. Por otro lado, ciertas mutaciones se encuentran preferentemente en determinadas regiones geográficas, por ejemplo, una deleción de la región 5’UTR (c.-447_427del) en la población de Cerdeña16. En las poblaciones asiáticas (japonesa, china, coreana) la mutación más frecuente es la R778L (14-50% de los casos)14.
En la población española, existen escasos estudios6,21-23. En ella, la mutación M645R representa el 27% de los cromosomas mutados22, mientras que la mutación H1069Q sólo se ha detectado en el 5% de los pacientes. En la población canaria, la mutación L708P se ha detectado en el 82% de familias, no detectándose la mutación europea más común H1069Q23. En nuestra casuística de 32 pacientes con EW molecularmente confirmada, la mutación M645R representó el 30% de los cromosomas mutados, la H1069Q el 9%, pero es de destacar que el 31% de las mutaciones eran únicas (detectadas en un único paciente).
Existen varios estudios que han intentado establecer una correlación genotipo-fenotipo valorando la edad de inicio, la presentación hepática o neurológica, la presencia de anillos de Kayser-Fleicher o los niveles de ceruloplasmina en suero. Sin embargo, no se ha podido demostrar que dicha correlación exista, lo que indica la existencia de otros factores ambientales y genéticos que afectan a la expresión fenotípica6,13,24,25.
Búsqueda de mutaciones en el gen ATP7BEl análisis molecular del gen ATP7B es la forma más precisa para realizar el diagnóstico de la EW, pero su estudio es complejo, debido tanto a la variabilidad poblacional como al gran número de mutaciones detectadas. Se han utilizado diferentes estrategias para la detección de mutaciones. Desde la clonación del gen hasta hace pocos años la técnica de SSCP (Single Strand Conformational Polymorphism) ha sido la más ampliamente utilizada, sobre todo cuando se trata de identificar mutaciones en una nueva población13,15,21,26-28. Otras tecnologías similares (DHPLC, DGGE, HRM) también han sido utilizadas, pero en menor proporción29-32.
En el momento actual, la secuenciación directa es la técnica más utilizada en el diagnóstico molecular de la EW4,33-38. Ello es debido a que es capaz de detectar el cambio específico de la secuencia de ADN y además detecta el 98% de los cromosomas mutados. En contrapartida, requiere un estudio largo, costoso y laborioso si se necesita secuenciar el gen entero.
Todas las técnicas anteriores permiten detectar cambios puntuales o pequeñas deleciones o inserciones en la secuencia de ADN. Una estrategia seguida por muchos grupos consiste en la selección de determinados exones reduciendo así costes y el tiempo del análisis. La selección de estas regiones va a depender de la población, y es necesario basarse en estudios anteriores. Así, el análisis de los exones 8, 13, 14 y 15 cubre el 60-70% de las mutaciones en Europa del Este o Central. Según nuestra casuística y la de Margarit et al22 en la población española, este estudio detectaría el 26% de los cromosomas mutados, mientras que el análisis de los exones 2, 5, 6 y 14 detectaría el 66%.
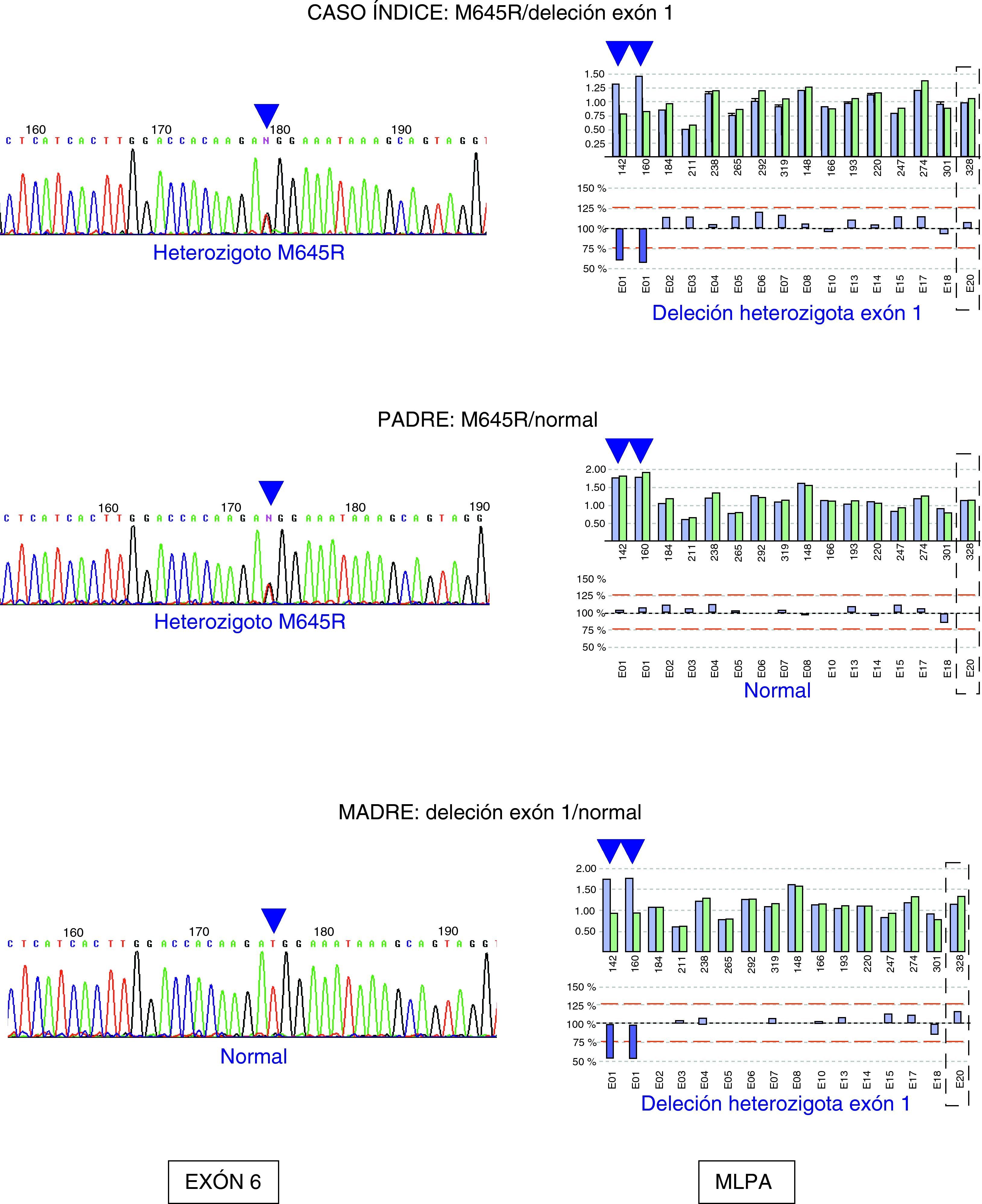
Existe otra técnica denominada MLPA (Multiple Ligation-dependent Probe Amplification) que permite detectar deleciones y duplicaciones parciales o totales de un gen. Para la mayoría de enfermedades hereditarias, las deleciones o duplicaciones parciales suponen menos del 10% de mutaciones detectadas. No existen publicaciones referentes a la utilización de esta tecnología aplicadas a la EW en la actualidad. No obstante, en nuestra casuística de 32 pacientes molecularmente diagnosticados, un paciente presentó una deleción del exón 1 en heterocigosis que fue detectada por MLPA. El otro alelo era portador de la mutación M645R. El estudio familiar permitió comprobar la segregación de ambas mutaciones (fig. 1). La utilización de esta metodología en pacientes en los cuales sólo se ha detectado una mutación puede mejorar el diagnóstico de esta enfermedad.

Estudio molecular del gen ATP7B en un paciente con enfermedad de Wilson y sus progenitores. A la derecha se muestra el electroferograma de parte de la secuencia exón 6 del gen ATP7B donde se encuentra localizada la mutación M645R: el caso índice y su padre son portadores de la mutación. A la izquierda se muestran los resultados del análisis por MLPA de los diferentes exones del gen, donde se detecta una deleción heterozigota del exón 1. El caso índice y su madre son portadores de dicha mutación.
Se han descrito técnicas para estudiar una mutación determinada, como la H1069Q24,39, o la deleción de 15 pares de bases en el promotor del gen ATP7B que identifica el 60,5% de cromosomas mutados en la población sarda16. La identificación de la mutación H1069Q únicamente puede ser útil en poblaciones con un gran porcentaje de esta mutación, pero no así en otras, como la española, donde su prevalencia es baja (sobre el 10%).
Desarrollo de paneles para detectar un determinado número de mutacionesDiversos grupos han desarrollado paneles para detectar las mutaciones más frecuentes en su población. Para ello se ha de seleccionar un panel que represente la población en estudio. En estos casos, la utilidad es alta para la población específica, pero acostumbra a ser limitada cuando se estudian otras poblaciones.
Lovicu et al40 describen una PCR específica que permite la amplificación conjunta de 6 fragmentos donde se localizan las mutaciones que identifican el 85% de cromosomas en la población sarda. La misma técnica fue utilizada por Huster et al41, en la que se incluyen 4 mutaciones europeas comunes. Dichas mutaciones (H1069Q, P1134P-fs, M769V y W779X) identifican el 61,3% de los cromosomas mutados en pacientes con EW del este de Alemania. Este panel de mutaciones detectaría únicamente un 10% de los cromosomas mutados si se estudiara la población española, y un 2,4% de la población canaria. Recientemente, el grupo de Mak et al42 ha diseñado un panel para la detección de las 28 mutaciones más prevalentes en la población china de Hong Kong.
Gojová et al43 desarrollaron un microarray de ADN para detectar 87 mutaciones y 17 polimorfismos en el gen ATP7B. En este array se han incorporado las mutaciones descritas para la población checa, pero también se han introducido mutaciones prevalentes de otras poblaciones (como la española). La utilización de este chip hubiese detectado el 95,6% de cromosomas mutados en la población canaria, y con resultados contradictorios para la población española: 94,4% según Margarit et al22, pero únicamente el 68% en nuestra casuística. La ventaja principal de esta técnica es que permite detectar un número elevado de mutaciones en un solo experimento, pero existen limitaciones en cuanto a la posible presencia de polimorfismos en los lugares donde se encuentran situados los oligonucleótidos, la utilización de maquinaria especializada y su coste. En un futuro, cuando los costes de producción y procesamiento del chip sean más baratos y las mutaciones en la EW estén más delineadas se podría incrementar su sensibilidad. De todas maneras, puede no ser competitiva con las nuevas tecnologías de secuenciación masiva, capaces de detectar mutaciones no preidentificadas.
Interpretación del diagnóstico molecular de la enfermedad de WilsonEl fenotipo de la EW es diverso a nivel clínico y bioquímico, por lo que establecer un diagnóstico puede ser difícil en ciertos pacientes3. El análisis molecular puede determinar la presencia o ausencia de mutaciones en el gen ATP7B, ayudando así al diagnóstico de la enfermedad, sobre todo en los casos en que el diagnóstico clínico es dudoso.
El estudio molecular del gen ATP7B puede dar 3 tipos de resultados: 1) detección de 2 mutaciones; 2) detección de una única mutación, o 3) no detección de mutaciones.
- 1.
El resultado molecular puede ayudar al diagnóstico clínico cuando se detectan 2 mutaciones causativas, que o bien hayan sido previamente descritas o a las que se les pueda atribuir un efecto en la proteína (por ejemplo, mutaciones tipo nonsense o deleciones o inserciones que cambien la pauta de lectura). Para ayudar a la interpretación de resultados existen bases de datos donde se recogen las mutaciones descritas en el gen ATP7B como las de la Universidad de Alberta (http://www.wilsondisease.med.ualberta.ca/database.asp) o la de Cardiff (http://www.hgmd.cf.ac.uk/ac/index.php).
- 2.
Una dificultad mayor la tenemos en el caso de un paciente en el cual únicamente se detecta una mutación en el gen ATP7B. En el caso de que existan síntomas clínicos indicativos de EW (evidencia de daño en el tejido hepático, síntomas neuropsiquiátricos asociados a anillos de Kayser-Fleycher, o imágenes cerebrales que sugieran EW) se tiene que iniciar el tratamiento4. Una respuesta al mismo en estos individuos reforzará la idea de que el diagnóstico es correcto. En cambio en pacientes en que la enfermedad progrese a pesar de la terapia será necesario reconsiderarlo. La mejora en el diagnóstico molecular de la EW hace pensar que este tipo de pacientes será cada vez menos frecuente.
- 3.
En el caso de no detectar mutaciones, el resultado no descarta completamente el diagnóstico de EW, ya que se ha descrito que el estudio mutacional identifica la presencia de mutaciones en un 65% de todos los alelos en pacientes con sospecha clínica de EW36. En estos casos se puede ofrecer un estudio indirecto del gen ATP7B. El estudio indirecto (o análisis de ligamiento) no diagnostica el caso índice pero permite identificar portadores y pacientes asintomáticos en la familia del mismo. En este caso es indispensable que el diagnóstico clínico no sea dudoso44. Para realizar este tipo de estudio además del caso índice es necesaria una muestra de los padres y hermanos. El resultado dependerá de que los marcadores genéticos estudiados sean informativos en esa familia. Se ha descrito que la utilización de marcadores flanqueantes del gen ATP7B resulta informativa en un 90% de las familias44. En estos casos es posible el diagnóstico de otros individuos de riesgo, detección de portadores y diagnóstico prenatal y preimplantacional.
Dado que es una enfermedad autosómica recesiva, los padres de un paciente con EW son portadores obligados, y tienen una probabilidad de tener descendencia afectada del 25%. Este hecho hace necesario realizar un cridado en cascada de los familiares del caso índice.
En función del resultado del caso índice, se puede hacer el estudio de familiares de riesgo. Es necesario confirmar el estudio de portadores en ambos progenitores, ya que se han descrito casos muy poco frecuentes de mutaciones localizadas en el mismo alelo45. La búsqueda de mutaciones en hermanos del caso índice requiere únicamente del estudio de las mutaciones detectadas en él, lo que hace que sea más barato y rápido. La presencia de mutaciones idénticas al caso índice identificará a pacientes afectados de EW (ya que pueden ser asintomáticos). La identificación de mutaciones permite además establecer diagnósticos prenatales y preimplantacionales.
Por otro lado, un enfermo de Wilson tiene un riesgo incrementado de tener descendencia afectada, ya que la probabilidad de que el enfermo pase el alelo mutado es del 100%, y la probabilidad de que su pareja sea portadora de una mutación es la de la población general (1/90)5. Por lo tanto, su probabilidad de tener descendencia con EW es de 1/180, mientras que la de la población general es de 1/32.400 (1/90 x 1/90 x 1/4). Se puede reducir dicha probabilidad haciendo un cribado de las mutaciones más frecuentes en la población de la pareja. Por ejemplo, un estudio de parte del gen que detecte el 70% de las mutaciones reduciría el riesgo a 1/600. En la población española esto podría conseguirse estudiando los exones 2, 5, 6 y 14 que detectan el 66% de los cromosomas mutados reportados (según nuestra casuística y Margarit et al22). En el caso de identificar una mutación en la pareja, es posible ofrecer entonces un diagnóstico prenatal o preimplantacional.
Esta misma argumentación se puede hacer con los hermanos de un caso índice, ya que su riesgo inicial de ser portadores es del 50%. En el caso de ser portadores de una mutación, se recomienda el cribado de mutaciones frecuentes en la pareja, al igual que en el caso de enfermos.
En la actualidad el diagnóstico molecular de la EW se lleva a cabo en la mayoría de los casos por secuenciación directa. El estudio molecular y la identificación de mutaciones en el gen ATP7B ayudan al diagnóstico clínico confirmando la enfermedad y ofrecen la posibilidad del diagnóstico a familiares, el asesoramiento genético, y poder realizar diagnósticos prenatales y preimplantacionales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

