




Peutz–Jegher syndrome (PJS) is characterised by the presence of hamartomatous polyps in the gastrointestinal tract and mucocutaneous melanosis.1,2 Its incidence is from 8300 to 29,000 live births. Family history of PJS is required for its diagnosis.3 Secondary biliary cirrhosis is caused by the chronic interruption of bile flow,4 which ends up irreversibly damaging the liver parenchyma.
Here we report the case of a female patient diagnosed with PJS at 8 months of age, with a history of the condition diagnosed in the father, brother and paternal uncle. She was brought to hospital for abdominal pain and signs of chronic liver disease; biliary cirrhosis secondary to bile duct obstruction was diagnosed.
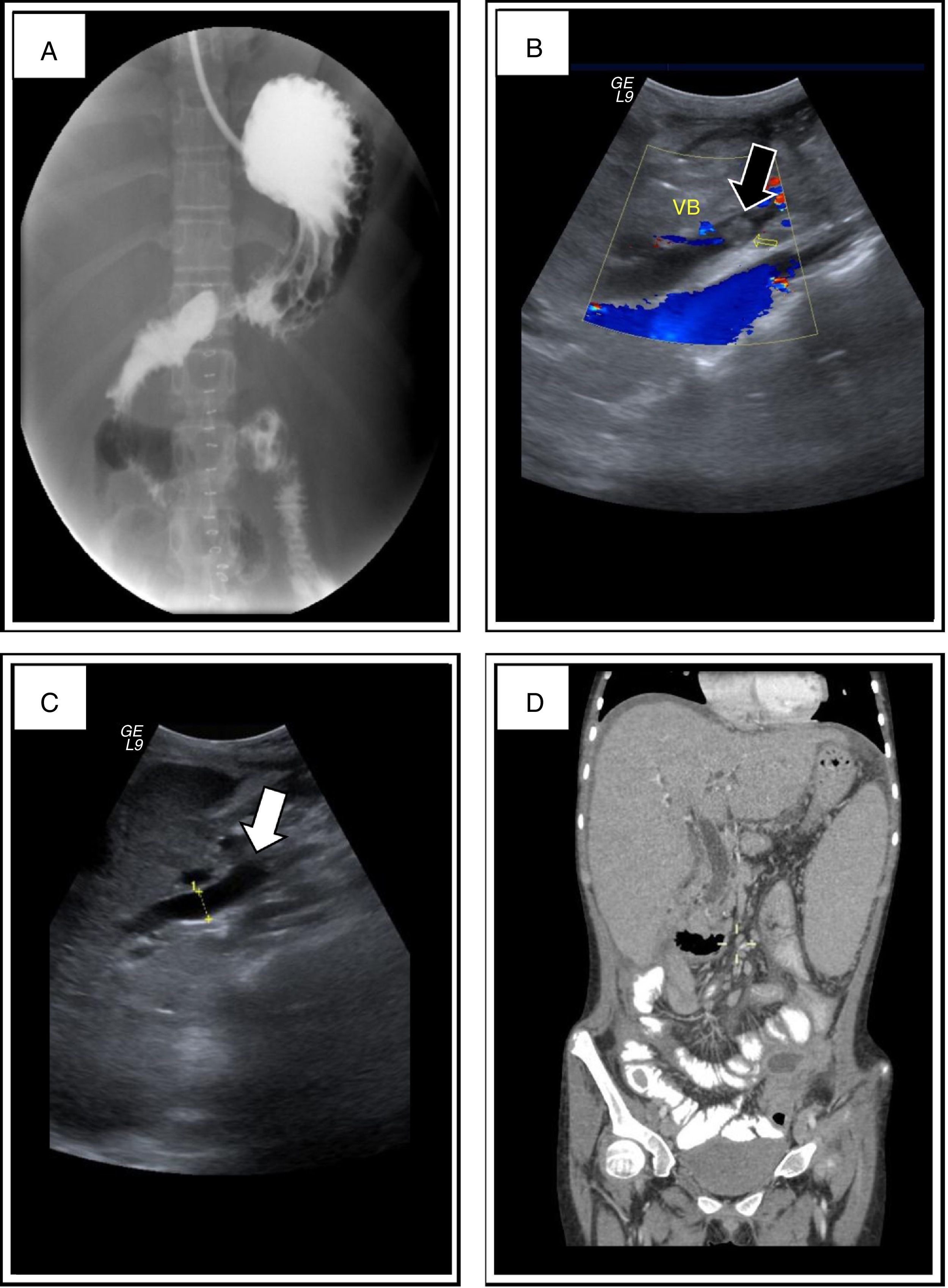
11-year-old girl who had the following symptoms for the 3 preceding days: nausea, gastric vomiting, bloating, gas not passing through properly, constipation and abdominal pain. Contrast study prior to admission: filling defects in the stomach, duodenum and jejunum (Fig. 1A). Physical examination: jaundice in conjunctivae and skin; perioral melanoplakia, oral mucosa and arms; heart and lung sounds with no compromise; ascites; collateral venous network; generalised pain on abdominal palpation; positive Blumberg's sign; decreased bowel movement frequency; and thenar and hypothenar hypertrophy. Abdominal X-ray, with evidence of intestinal occlusion: poor distribution of intestinal air and absence of gas in rectal ampulla. Abdominal ultrasound: increased echogenicity within the gallbladder and bile duct dilatation (Fig. 1B and C). Paraclinical tests showing evidence of cholestasis and leukocytosis: total bilirubin 9.31mg/dl, direct bilirubin 5.61mg/dl, GGT 221g/dl, ALT 46IU/l, AST 40IU/l, LDH 286IU/l, ALP 596IU/l, leukocytes 20103/IU, segmented neutrophils 83% and Hb 15.3g/dl.
 Oesophagogastroduodenal series and bowel transit in which filling defects in the stomach, duodenum and jejunum compatible with polyps were found. (B) and (C) Ultrasound in which increased echogenicity in the gallbladder was documented, which was not mobilised with gravity manoeuvres, with no posterior acoustic shadowing, compatible with a polyp (black arrow), and bile duct dilatation (white arrow). (D) Computed axial tomography (CT) in which there was bile duct dilatation, hepatomegaly and splenomegaly.")
(A) Oesophagogastroduodenal series and bowel transit in which filling defects in the stomach, duodenum and jejunum compatible with polyps were found. (B) and (C) Ultrasound in which increased echogenicity in the gallbladder was documented, which was not mobilised with gravity manoeuvres, with no posterior acoustic shadowing, compatible with a polyp (black arrow), and bile duct dilatation (white arrow). (D) Computed axial tomography (CT) in which there was bile duct dilatation, hepatomegaly and splenomegaly.
Exploratory laparotomy: icteric free fluid, dilated loops of bowel, ileocolic intussusception and volvulated right colon; liver greyish, hard and granular; and large, indurated gallbladder with irregular borders. The intussuscepted and volvulated segment was resected, and the following were also performed: right colectomy, ileostomy, distal closure in Hartmann's pouch of the transverse colon, liver biopsy, gallbladder resection and Kehr's T-tube placement. Histopathology: hamartomatous polyps with necrosis in the area of intussusception; gallbladder with hamartomatous arboriform polyp; liver: partially conserved architecture, irregular porto-portal fibrosis, hepatocytes with icteric cytoplasmic membrane dye, Kupffer cells with lipofuscin, Glisson's capsule with lymphoplasmacytic infiltrate and reactive mesothelial hyperplasia. Diagnosis: macronodular and micronodular biliary cirrhosis, with no neoplasms, and dense deposit disease ruled out.
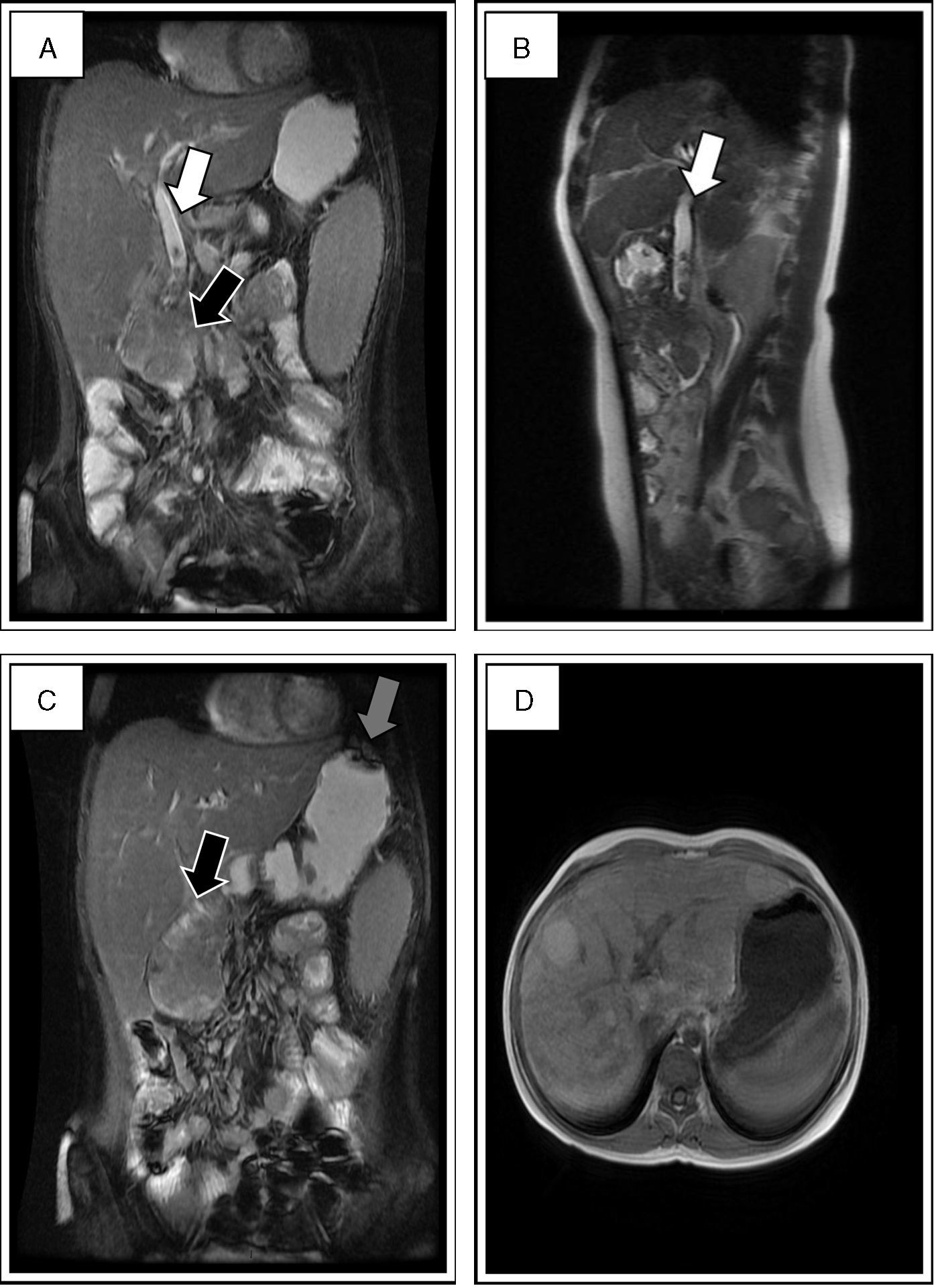
Approach for liver disease: serology for CMV, EBV, HIV; parvovirus B19, HAV, HBV and HCV negative; autoimmune aetiology: ANA, ANCA, anti-SM, anti-LKM1 tests negative; normal expanded metabolic screen and alpha 1-antitrypsin 121mg/dl. Panendoscopy: polyp in ampulla of Vater that obstructs its lumen; resection of 60% of lesion; bypass tube placed. Histopathology: hamartomatous polyp. Computed tomography: bile duct dilatation (Fig. 1D). Magnetic resonance cholangiopancreatography (MRCP): dilated bile duct, signalling defects in the interior, 6.7×4.8cm lesion in the second part of the duodenum and hyperintense nodular lesions in the parenchyma suggestive of hepatic cirrhosis (Fig. 2). Multiple endoscopic procedures were performed with resections of polyps and attempts to permeabilise the bile duct. These were unsuccessful because of the size of the lesion in the ampulla of Vater; the liver condition had deteriorated and the patient was therefore not a candidate for surgery. In the end the liver damage led to death.
. In the second part of the duodenum there is a hypointense lesion of 6.7×4.8cm, with probable involvement of the ampulla of Vater (black arrow in A and C). Signalling defects in the fundus and gastric antrum suggestive of polyps (grey arrow in C). Multiple hyperintense nodular lesions were found in the liver parenchyma, which shows cirrhosis (D).")
MRCP in which the dilated bile duct was documented with signalling defects in it that stand out with the contrast agent, compatible with polyps (white arrows in A and B). In the second part of the duodenum there is a hypointense lesion of 6.7×4.8cm, with probable involvement of the ampulla of Vater (black arrow in A and C). Signalling defects in the fundus and gastric antrum suggestive of polyps (grey arrow in C). Multiple hyperintense nodular lesions were found in the liver parenchyma, which shows cirrhosis (D).
PJS is an autosomal dominant disorder with a variable and incomplete penetrance related to the mutation of the STK11/LKB1 gene, characterised by hamartomatous polyps from the gastroesophageal junction to the anal canal, mainly in the ileum.5 Melanoplakia usually occurs on the lips and oral mucosa,6 a sine qua non sign.
The manifestations begin in the first 2 decades of life; the initial reason for consultation usually involves gastrointestinal manifestations. The most common symptom is recurrent abdominal pain due to intussusception, where polyps are the “head” of the intussusception.7
There have been reports of hamartomatous polyps in uncommon areas such as the gallbladder, common bile duct and ampulla of Vater.2,8 There are limited reports of the presence of tumours in the biliary tract in the literature,9 with a higher prevalence in adults. These locations entail a high risk of bile duct obstruction, and in some cases, varying degrees of liver failure.2,9 In this case the presence of hamartomatous polyps in the gallbladder, bile duct and ampulla of Vater was documented, which is an exceptional finding in the field of paediatrics.9 These lesions caused the bile duct obstruction and, finally, micro- and macronodular biliary cirrhosis, a situation not published in the literature.
Cirrhosis manifests clinically when 80% of the liver parenchyma is affected.10 In advanced stages, as in this case, there is jaundice, pruritus, gastrointestinal bleeding, coagulopathy and ascites.10 Recognising the compromised bile flow early on can lead to a better prognosis for liver function.
Patients with PJS require follow-up and observation to prevent gastrointestinal complications. We also consider it important to actively look for hamartomatous polyps that may obstruct bile flow, a potential cause of irreversible liver damage. Close follow-up with liver function tests and early detection of clinical signs of liver disease may allow for the timely identification of cholestasis, i.e., before liver damage occurs, and thus provide the appropriate management and prevent progression to cirrhosis, thereby reducing morbidity and mortality.
FundingNo funding of any kind was received to write this article.
Please cite this article as: Vichido-Luna MA, Zárate-Mondragón F, Mora-Tiscareño MA, Cervantes-Bustamante R, Ramírez-Mayans JA. Cirrosis biliar secundaria a obstrucción de la vía biliar por pólipos hamartomatosos en una paciente con síndrome de Peutz-Jeghers. Reporte de Caso. Gastroenterol Hepatol. 2017;40:459–462.
