




Los rasgos clínicos del síndrome de neoplasia endocrina múltiple tipo 1 (NEM-1) son: hiperplasia o adenoma de las glándulas paratiroides, adenoma hipofisario y tumores endocrinos gastroenteropancreáticos. Se debe a mutaciones del gen MEN1, localizado en la región q13 del cromosoma 11. El pronóstico de los pacientes depende del crecimiento tumoral y de su potencial metastático.
Pacientes y métodoSe revisan las historias clínicas de los miembros de esta familia (6 varones y 2 mujeres) con NEM-1 diagnosticados entre 1995 y 2007 en el Hospital Donostia de San Sebastián.
El estudio familiar de todos los pacientes y familiares (19 casos de 2 generaciones) se hizo en dos fases. La primera, mediante técnica de cribado de mutaciones y la segunda, por multiplex ligation-dependent probe amplification (MLPA) para detectar deleciones del gen.
ResultadosEl cribado de mutaciones no permitió identificar ninguna variante patogénica en el probando de esta familia. El estudio mediante MLPA reveló una deleción que afectaba al exón 1 y 2 del gen MEN1. De los 10 familiares con esta alteración molecular, 8 presentaron algún rasgo fenotipico del síndrome (8 con hiperparatiroidismo, 2 con prolactinomas y 3 con gastrinomas) tras 12 años de seguimiento.
ConclusiónSe comentan las formas clínicas del síndrome NEM-1 en esta familia y la alteración molecular encontrada. El estudio de deleciones del gen MEN1 debería incorporarse al cribado molecular sistemático.
The clinical features of multiple endocrine neoplasia type-1 (MEN-1) syndrome are hyperplasia or adenoma of the parathyroid glands, pituitary adenoma and gastroenteropancreatic endocrine tumors. This syndrome is due to mutations in the MEN1 gene, located on the q13 region of chromosome 11. Prognosis depends on tumoral growth and metastatic potential.
Patients and methodWe reviewed the medical records of the members of a family (6 men and 2 women) with MEN-1 syndrome diagnosed between 1995 and 2007 in Hospital Donostia, San Sebastian (Spain). Familial study of all patients and family members (19 cases from 2 generations) was performed in 2 phases. The first phase consisted of mutation screening and the second of multiplex ligation-dependent probe amplification (MLPA) to detect deletions.
ResultsScreening of mutations identified no pathogenic variants in the proband of this family. MLPA revealed a deletion affecting exons 1 and 2 of the MEN1 gene. Of the 10 family members with this molecular alteration, 8 had at least one phenotypic feature of this syndrome (hyperparathyroidism in 8, prolactinomas in 2, and gastrinomas in 3) after 12 years of follow-up.
ConclusionWe discuss the clinical forms of MEN-1 syndrome in this family and the molecular alteration found. Study of MEN1 gene deletions should be incorporated into routine molecular screening.
Los rasgos clínicos del síndrome de neoplasia endocrina múltiple tipo 1 (NEM-1; OMIM#131100) son: hiperplasia o adenoma de las glándulas paratiroides, adenoma hipofisario y tumores endocrinos gastroenteropancreáticos (TEGEP)1. Algunos pacientes pueden desarrollar tumores adrenales corticales, adenomas tiroideos, carcinoides en los bronquios, el timo y el estómago, angiofibromas faciales, colagenomas y lipomas. La enfermedad sigue un patrón de herencia dominante, con penetrancia casi completa entre la quinta y la sexta décadas de la vida, y expresividad variable. Esta enfermedad se debe a mutaciones del gen MEN1, localizado en la región q13 del cromosoma 11, cuya proteína se conoce como menina. El pronóstico y la calidad de vida de los pacientes dependen de la sintomatología causada por las alteraciones hormonales, del crecimiento local de los tumores y de su potencial metastásico.
Se presentan las características clínicas de una familia con síndrome de NEM-1. En 10 familiares estudiados se ha encontrado una deleción del gen MEN1 que afecta a los exones 1 y 2. Ocho de ellos han presentado alteraciones bioquímicas y/o síntomas relacionados con la presencia de tumores. Los otros 2 familiares portadores de la mutación no han desarrollado hasta la fecha ningún rasgo fenotípico del síndrome.
PACIENTES Y MÉTODOCaso 1 (III-1): índiceVarón de 47 años de edad, que ingresó por melenas. Antecedentes familiares de hiperparatiroidismo (HP) en 3 hermanos, y personales de cólicos nefríticos, litiasis renal derecha e hipercalcemia desde hace 12 años. No refería hábitos tóxicos ni ingesta previa de fármacos gastrolesivos. Tomaba de manera puntual omeprazol. La exploración física era normal, salvo por la presencia de lipomas en la fosa ilíaca derecha. En la bioquímica destacaba un aumento de las enzimas hepáticas: fosfatasa alcalina 556 U/l (valores normales, 40-129), GGT 387 U/l (8-61), GOT 54 U/l (6-38) y GPT 61 U/l (6-41); calcio 10,2 mg/dl (8,4-10,2), PTH intacta 80,8 pg/ml (10-65), BAO de 7,6 mEq/h y MAO de 21 mEq/h. Las serologías (virus de la hepatitis B y C, citomegalovirus, herpes simplex y virus de Epstein-Barr) y la batería de hormonas (gastrina, ACTH, T4 libre, TSH sensible, insulina, glucagón, PIV, adrenalina, noradrenalina, ácido 5-OH indolacético y cortisol en orina) resultaron negativas o normales. Se realizan una resonancia magnética (RM) y una gammagrafía de las glándulas paratiroides, una ecografía y una tomografía computarizada (TC) abdominales, que fueron normales. La gastroduodenoscopia demostró una duodenitis erosiva con presencia de Helicobacter pylori. Se hizo tratamiento erradicador y con omeprazol (20 mg/día). Se solicitó un estudio molecular del gen MEN1 y un test de secretina, que fue positivo.
Dos años más tarde, a causa de un hiperparatiroidismo, se extirpan 2 paratiroides, y un año después, por continuar con hipercalcemia, las 2 restantes con autotrasplante de tejido paratidoideo en la masa muscular del brazo izquierdo. A los 3 años de seguimiento, en una ecografía abdominal, se pone de manifiesto una masa hiperecogénica de 8,8 _ 9,1 cm en el lóbulo derecho hepático. Ante este hallazgo se realiza un estudio de los marcadores tumorales, bioquímica y hemograma, que fueron normales. La TC abdominal, la angiogammagrafía con hematíes marcados y la punción-aspiración con aguja fina (PAAF) de la lesión fueron compatibles con un hemangioma. Doce meses después, el paciente reingresa por presentar disnea de medianos esfuerzos durante el último mes, poco apetito y pérdida de 3-4 kg de peso. A la exploración se apreció un soplo sistólico I-II/VI en todos los focos, hepatomegalia de 2 traveses de dedo por debajo del reborde costal y edemas maleolares con fóvea. Se recibe el estudio genético que confirma las mutaciones en el locus 11q13. El ecocardiograma reveló una hipertrofia concéntrica del ventrículo izquierdo con fracción de eyección conservada, válvulas sigmoideas con estenosis aórtica, hipertensión arterial pulmonar moderada y trastornos de la función diastólica tipo I. Se instaura tratamiento con oxígeno y diuréticos. A los 7 días de hospitalización presentó de manera brusca dolor precordial, disnea, cianosis, shock y sudoración. El paciente falleció súbitamente a pesar de la aplicación de terapia con dopamina, cardioversión eléctrica y maniobras de resucitación.
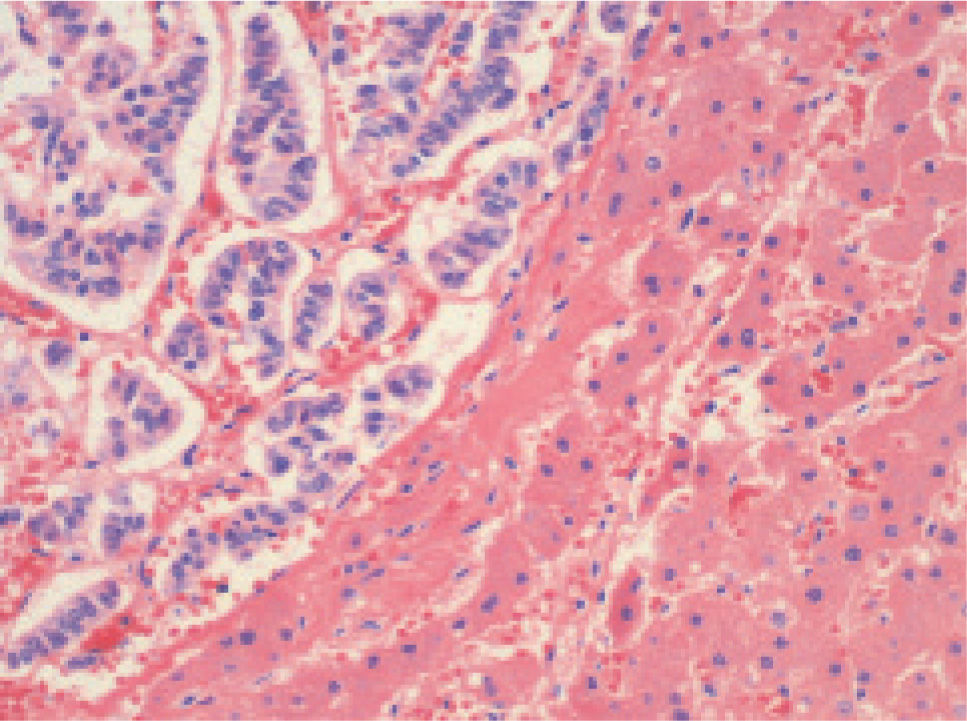
El estudio necrópsico confirmó la existencia de cardiomegalia con hipertrofia leve concéntrica del ventrículo izquierdo, congestión vascular difusa en los pulmones y fibrosis pretraqueal secundaria a una intervención quirúrgica previa, esófago con acantosis glucogénica y tumor con diferenciación neuroendocrina en el lóbulo derecho hepático de 12,5 cm de diámetro, con extensa necrosis hemorrágica (un 70% de la tumoración aproximadamente) (fig. 1), así como una congestión pasiva crónica y aguda del hígado con focos de necrosis centrolobulillar, y en el riñón derecho un hematoma en grasa perirenal del polo inferior y espermatocele del testículo derecho.
Caso 2 (III-8): primo segundo.")
Varón de 40 años de edad, con varios familiares de segundo grado portadores del gen MEN1, que ingresó en el servicio de digestivo por melenas. Entre sus antecedentes personales destacaban una pancreatitis aguda presentada hacía 13 años y una intervención por HP hace 11. En los últimos años, había presentado varios cólicos nefríticos por nefrolitiasis y pérdida de agudeza visual del ojo derecho por telangiectasias yuxtafoveales. Era fumador de un paquete de cigarrillos/día y bebedor de 100 g alcohol/día.
En la exploración física tenía las constantes vitales normales, presentaba una intensa palidez mucocutánea, la auscultación cardiopulmonar era normal y en el abdomen no había hallazgos patológicos. Entre las pruebas bioquímicas complementarias y determinaciones hormonales cabe resaltar los siguientes parámetros: urea 83 mg/dl, hemoglobina 7,5 g/dl, hematocrito del 21%, proteínas totales 4,7 g/dl, glucemia 118 mg/dl, fósforo inorgánico 2,4 mg/dl (2,5-4,5), PTH intacta 73,6 pg/ml, gastrina 305 pg/ml (28-115) y calciuria 530 mg/24 h (100-300). La endoscopia con biopsias gástricas demostró una úlcera duodenal, Helicobacter pylori positiva, con sangrado activo. Se hizo una esclerosis con adrenali- na. En la RM de páncreas se constató una atrofia del parénquima, una dilatación del Wirsung con litiasis intraluminal y calcificaciones, junto con una masa de 3 cm, heterogénea, en la cara posterior de la cola pancreática. La RM de la hipófisis fue normal y la ecografía de tiroides mostró un nódulo de 17 _ 14 _ 12 mm, de consistencia media y bien delimitado, en el lóbulo izquierdo.
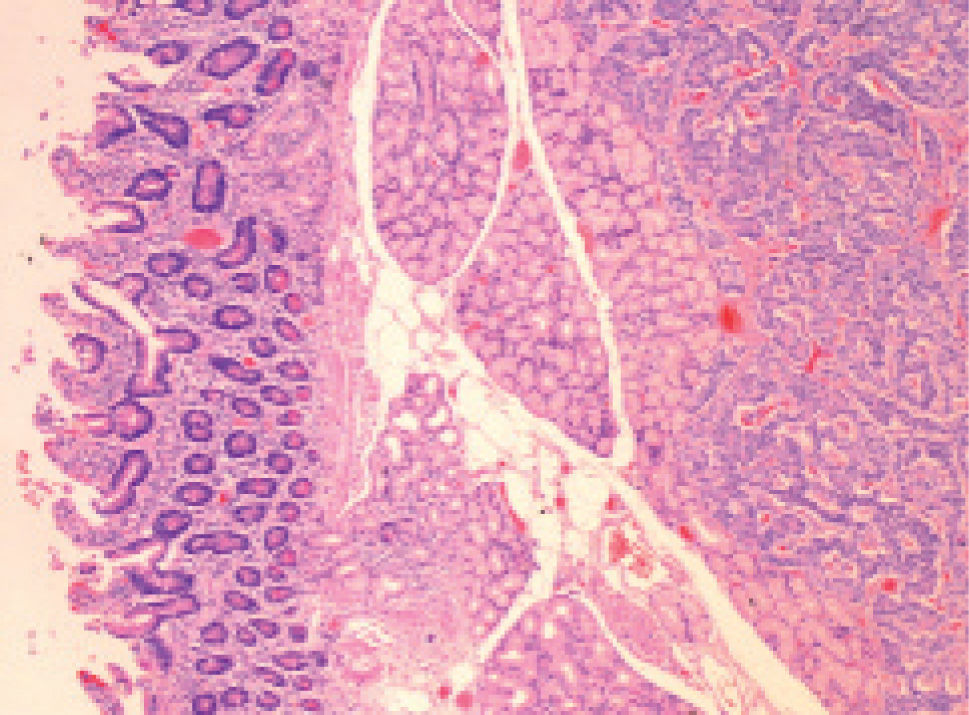
Ante la sospecha clínica de NEM-1 se solicitó un estudio genético, una gammagrafía con octreótida y una ecoendoscopia. Se inició tratamiento con esomeprazol (80 mg/día) y se llevó a cabo tratamiento erradicador de H. pylori. El estudio molecular confirmó que era portador de la mutación de MEN1. Tras encontrar y confirmar mediante octreoscán y PAAF dos nódulos pancreáticos con diferenciación neuroendocrina, se procedió a la resección total del páncreas (fig. 2). Se trata con insulina y enzimas pancreáticas. Los controles posteriores (laboratorio, TC y octreoscán) fueron normales. En la actualidad, transcurridos 4 años de la intervención, no ha presentado ninguna incidencia, salvo algún episodio de hipoglucemia.
Caso 3 (IV-1): hijo.")
Varón de 25 años de edad, con antecedentes familiares de HP (padre y tres tíos paternos) y personales de pirosis, así como una epigastralgia de 3 años de evolución. Tratado con omeprazol y triple terapia para H. pylori, continúa con molestias digestivas. Refiere desde hace 2 años deposiciones líquidas (6-8 al día). Ingresa en el servicio de urgencias por una úlcera yeyunal perforada y es remitido al servicio de aparato digestivo para su estudio. La exploración física fue normal. En la analítica resaltaban los siguientes datos: calcio plasmático 11,2 mg/dl, fósforo 2,8 mg/dl, calciuria 6.642 mg/24 h, fosfaturia 1,9 mg/24 h (0,4-1,3), PTH intacta 137,8 pg/ml y gastrina 1.921 pg/ml. Las determinaciones plasmáticas de TSH sensible, T4 libre, testosterona, prolactina, GH, insulina, glucagón, factores de crecimiento análogos a la insulina, ACTH, PIV, polipéptido pancreático, cortisol, calcitonina, serotonina, vitamina D-25 hidroxi y catecolaminas en orina de 24 h fueron normales.
La fibrogastroscopia con biopsias, tránsito intestinal, RM de silla turca y de abdomen, y octreoscán no revelaron la presencia de lesiones. El test de secretina fue positivo. A pesar de no haberse detectado gastrinoma ni hiperplasia paratiroidea, se instauró tratamiento con omeprazol (80 mg/día) seguido de control periódico por el servicio de endocrinología. A los 30 meses del diagnóstico, la gastrina era de 5.049 pg/ml, la PTH de 136,7 y la cromogranina A de 1.090 ng/ml (19,4-98,1). Se confirmó la presencia de 4 nódulos hipervasculares de 15-20 mm, peripancreáticos y en la cola pancreática. Se añadió octreótida mensual (hasta 30 mg). Diez meses después se extirparon la paratiroides y el timo. En la actualidad el paciente sigue asintomático. La gastrina ha bajado a 900 pg/ml y la cromogranina A a 160,4 pg/ml. El estudio molecular del gen MEN1 confirmó su condición de portador de la alteración.
Estudio de mutacionesEl análisis molecular se llevó a cabo en dos etapas. En la primera fase se eligió la técnica de cribado utilizando cebadores específicos de ADN que flanqueaban cada uno de los exones del gen, de modo que se analizó la secuencia codificante completa. Esta técnica permite analizar la existencia de mutaciones del tipo cambio de aminoácido, pequeñas deleciones o inserciones, o variantes que alteren el proceso de splicing. En una segunda fase del estudio se analizó la existencia de grandes deleciones del gen, dado que se trata de un tipo de alteración que no puede ser detectada por secuenciación, y está presente en un 5-10% de las familias NEM-1 negativas para la primera fase del estudio. Para ello, se eligió la técnica multiplex ligation-dependent probe amplification (MLPA). Se llevó a cabo una hibridación con sondas marcadas con fluoresceína y específicas de los exones 1, 2, 3, 7 y 10, así como para regiones cromosómicas próximas y lejanas al gen que se utilizaron como controles internos del ensayo. En todos los casos se comparó con el patrón obtenido a partir de ADN de un individuo sano, utilizado como control negativo.
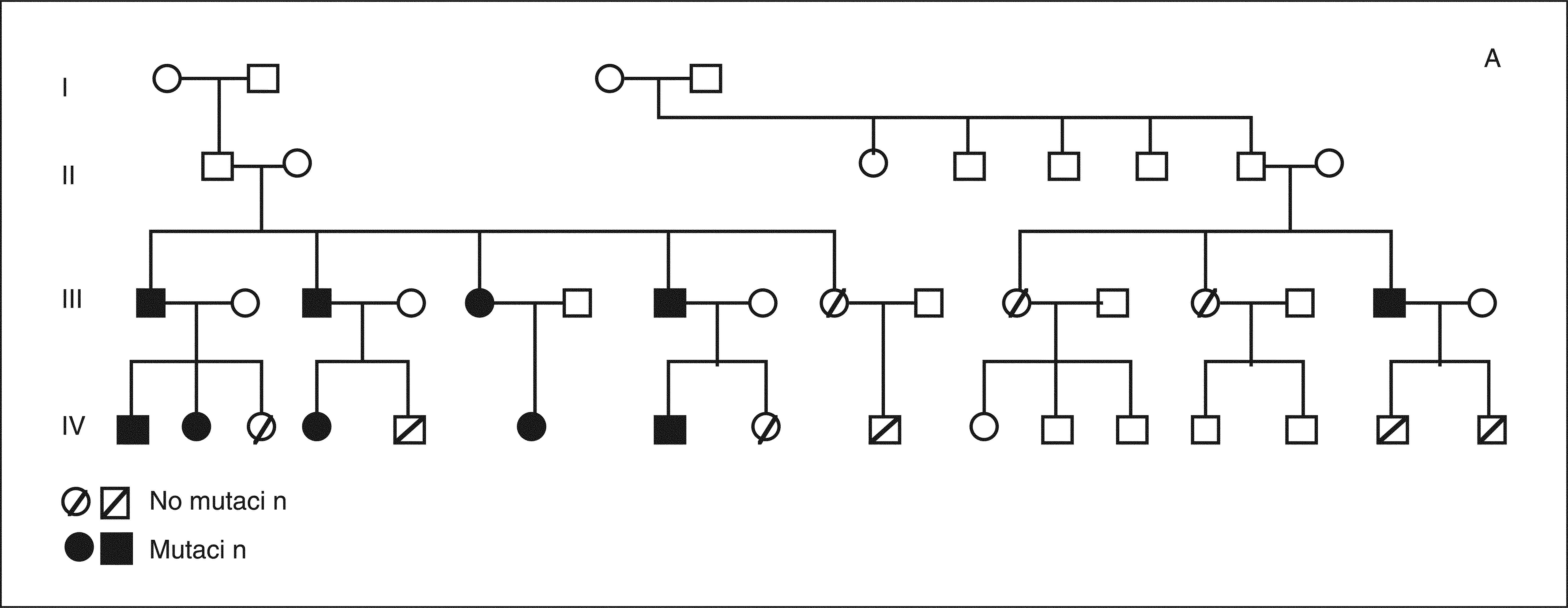
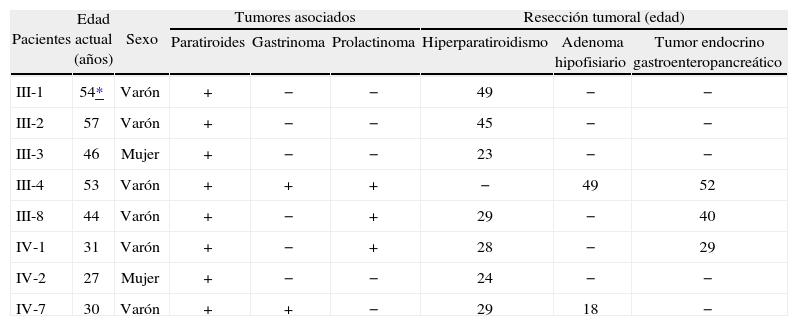
RESULTADOSEl árbol genealógico se muestra en la figura 3A, y se detalla la mutación detectada en los distintos miembros de la familia (fig. 3B). El cribado de mutaciones realizado a partir de secuenciación (primera etapa) no permitió identificar ninguna variante patogénica en el probandus de esta familia. El estudio mediante MLPA reveló una deleción que afectaba al exón 1 y 2 del gen MEN1 (fig. 3B). Esta alteración molecular estaba presente en 10 de los familiares estudiados correspondientes a 2 generaciones. De estos 10 miembros, 8 presentaron algún rasgo fenotípico del síndrome tras 12 años de seguimiento. Sus datos demográficos (edad, sexo, neoplasias asociadas y resección tumoral) se recogen en la tabla I. Otros 2 familiares (casos IV-4 y IV-6), con mutación del gen, aún no tienen ningún rasgo fenotípico del síndrome.
 árbol genealógico con las mutaciones detectadas en los miembros de la familia. B) imagen del análisis del fragmento correspondiente a un individuo afectado de esta familia. En azul aparece el resultado obtenido de un control normal, y en rojo el correspondiente al ADN del caso de estudio. Las flechas señalan los 2 fragmentos que mostraron amplificación para uno solo de los alelos y, por tanto, deleción de esas regiones.")
 árbol genealógico con las mutaciones detectadas en los miembros de la familia. B) imagen del análisis del fragmento correspondiente a un individuo afectado de esta familia. En azul aparece el resultado obtenido de un control normal, y en rojo el correspondiente al ADN del caso de estudio. Las flechas señalan los 2 fragmentos que mostraron amplificación para uno solo de los alelos y, por tanto, deleción de esas regiones.")
A ) árbol genealógico con las mutaciones detectadas en los miembros de la familia. B) imagen del análisis del fragmento correspondiente a un individuo afectado de esta familia. En azul aparece el resultado obtenido de un control normal, y en rojo el correspondiente al ADN del caso de estudio. Las flechas señalan los 2 fragmentos que mostraron amplificación para uno solo de los alelos y, por tanto, deleción de esas regiones.
Datos demográficos de los pacientes con neoplasia endocrina múltiple tipo 1
| Pacientes | Edad actual (años) | Sexo | Tumores asociados | Resección tumoral (edad) | ||||
| Paratiroides | Gastrinoma | Prolactinoma | Hiperparatiroidismo | Adenoma hipofisiario | Tumor endocrino gastroenteropancreático | |||
| III-1 | 54* | Varón | + | − | − | 49 | − | − |
| III-2 | 57 | Varón | + | − | − | 45 | − | − |
| III-3 | 46 | Mujer | + | − | − | 23 | − | − |
| III-4 | 53 | Varón | + | + | + | − | 49 | 52 |
| III-8 | 44 | Varón | + | − | + | 29 | − | 40 |
| IV-1 | 31 | Varón | + | − | + | 28 | − | 29 |
| IV-2 | 27 | Mujer | + | − | − | 24 | − | − |
| IV-7 | 30 | Varón | + | + | − | 29 | 18 | − |
+: presencia de tumor; —: ausencia de tumor.
El síndrome de NEM-1, o síndrome de Wermer, descrito en 19541, es una enfermedad hereditaria que se transmite de forma autosómica dominante, con penetrancia casi completa dependiente de la edad y expresividad variable. Su incidencia es del 0,25% en series de autopsias seleccionadas al azar2,3, y la prevalencia de 2 casos por 105 individuos/ año4. Estaremos ante un caso de NEM-1 esporádico si coincide en el mismo individuo el desarrollo de al menos 2 tumores endocrinos relacionados con el síndrome (hiperplasia o adenoma de paratiroides, adenoma hipofisario y TEGEP). Si además del caso índice hay otro pariente de primer grado con una de las neoplasias propias del síndrome, se considera una forma familiar de la enfermedad.
La enfermedad está causada por mutaciones germinales del gen MEN1, que contiene 10 exones distribuidos a lo largo de 9 kb de ADN genómico. El MEN1 es un gen su- presor de tumores5–7, de modo que los dos alelos deben estar inactivados para que se desarrolle el tumor. El primer evento mutacional ocurrirá en la línea germinal heredada y el segundo en la célula somática8,9. MEN1 codifica la síntesis de una proteína de 610 aminoácidos (menina), que se expresa de forma ubicua y se localiza en el núcleo en células en reposo10, fundamentalmente en el citoplasma de células en división11. La menina interactúa hasta con 12 proteínas y su función se ha relacionado con la regulación transcripcional, la estabilidad genómica y el control de la proliferación y división celular12,13.
Hasta ahora se han descrito más de 550 mutaciones distintas, distribuidas a lo largo de toda la secuencia codificante del gen. Una mayor proporción de pacientes clínicamente diagnosticados como NEM-1 esporádicos son portadores de mutaciones germinales que afectan al exón 2, frente a los diagnosticados como familiares14. En torno a un 23% de las alteraciones genera una señal de parada, el 9% afecta al splicing, un 41% provoca un cambio en el marco de lectura, un 6% consiste en deleciones o inserciones que no cambian la pauta de lectura, un 20% genera cambios de aminoácido y un 1% consiste en grandes deleciones parciales o totales del gen15, si bien el estudio de estas últimas no se realiza de forma sistemática en todos los casos. Hasta el momento hay indicios de la existencia de 4 puntos calientes de mutación, que aglutinarían algo más de un 12% de todas las mutaciones descritas hasta la fecha. Es importante recalcar que se ha descrito entre un 5 y un 10% de individuos NEM-1 sin mutaciones en el gen MEN1. Esta observación se puede explicar porque estos pacientes sean portadores de mutaciones en una región promotora del gen no analizada, o bien porque se traten de fenocopias.
Desde la identificación del gen en 19978, se ha intentado establecer una relación fenotipo-genotipo. Merece la pena destacar que el 38% de las mutaciones en MEN1 detectadas en familias con HP primario aislado consisten en cambios de aminoácido, que contrasta significativamente con la proporción de cambios de aminoácido detectada en pacientes NEM-1 (20%). Esto podría sugerir que este tipo de alteración se asocie con una inactivación menor de la proteína y, por tanto, con un efecto fenotípico más suave, pero lo cierto es que las mutaciones descritas en familias con HP están distribuidas a lo largo de todo el gen. Es más, se han descrito familias con HP portadoras de mutaciones que generan proteínas truncadas, hecho que hace difícil establecer una relación fenotipo-genotipo inequívoca. Todos estos resultados complican el consejo genético que se ofrece a estas familias, dado que por el momento se desconocen otros factores genéticos implicados en la expresividad de la enfermedad que ayuden a predecir el fenotipo más probable desarrollado por un individuo portador de mutación en el gen.
El 1-2% de todos los casos de HP primario4, el 20-30% de todos los gastrinomas16 y menos del 3% de los tumores hipofisarios17, respectivamente, se deben al síndrome de NEM-1. La manifestación clínica más habitual de los pacientes con NEM-1 es el HP, con una penetrancia del 90- 95% a los 40-50 años. La edad de comienzo de las alteraciones bioquímicas y de los síntomas relacionados con HP en pacientes NEM-1 es entre los 15 y los 25 años, y precede en dos décadas a la de los casos de HP esporádico. No muestra predilección por varones o mujeres. La afectación suele ser pluriglandular, por lo que en más del 60% de los pacientes con paratiroidectomía subtotal hay recurrencia de la clínica, transcurridos 8 años18. En el caso índice de esta familia, el antecedente de HP en 3 hermanos nos indujo a descartar en primer lugar un HP familiar aislado. De los 8 parientes con mutaciones en el locus 11q13 que tuvieron HP, en 5 se hizo una paratiroidectomía secuencial de las 4 glándulas con autotrasplante de tejido paratiroideo en el brazo, antes de los 30 años (tabla I).
La prevalencia de los adenomas pituitarios en el síndrome oscila entre el 15 y el 30%19,20. La mayoría segregan prolactina (el 60% de los casos), GH (25%) o ACTH (5%), y los tumores no funcionantes son los menos frecuentes. El prolactinoma puede ser la primera manifestación del síndrome en el 10% de los pacientes21. Los síntomas y el tratamiento son superponibles a los de los tumores hipofisarios esporádicos, aunque suelen ser de mayor tamaño (macroadenomas) y más agresivos22. El tratamiento y el pronóstico dependen del tamaño y de la compresión sobre las estructuras contiguas. Si hay hemianopsia bitemporal o hipopituitarismo, se realiza una hipofisectomía selectiva por vía transesfenoidal o radioterapia, cuando no es posible la resección completa.
La incidencia de tumores endocrinos pancreáticos es del 40% en los individuos con NEM-111 Predominan los tumores funcionantes: gastrina (un 40% de los casos), insulina (10%), glucagón (2%) u otras hormonas (polipéptido pancreático, polipéptido intestinal vasoactivo) respecto a los adenomas no funcionantes (20%)23. La sintomatología depende de la hormona secretada. El gastrinoma junto con la enfermedad ulcerosa y la hipersecreción gástrica constituyen el síndrome de Zöllinger-Ellison (SZE), que es la manifestación inicial en el 40% del subgrupo de pacientes con NEM-1/SZE24. En ellos, el comienzo de los síntomas precede al diagnóstico de HP en el 45% de los casos24. El diagnóstico de SZE se sospecha por la aparición de múltiples úlceras pépticas, atípicas o recurrentes, o diarrea, como sucedió en el caso 3. La confirmación se hace mediante la demostración de gastrina sérica alta y de una elevada producción basal de ácido gástrico. Los gastrinomas en el síndrome de NEM-1 suelen ser múltiples, extrapancreáticos (duodeno) y varían entre hiperplasia, adenoma y carcinoma25. La mayoría responde de forma adecuada a los inhibidores de la bomba de protones. El tratamiento quirúrgico es controvertido, ya que son múltiples y a veces tienen un componente maligno y metástasis en el momento del diagnóstico26. Diversos grupos propugnan la realización de cirugía en los mayores de 3 cm, hepatectomía y/o radiofrecuencia, embolización o quimioembolización arterial si hay metástasis hepáticas, y quimioterapia sistémica (estreptozocina más doxorrubicina), interferón o análogos de la somatostatina en la enfermedad avanzada4,27,28,29.
El estudio de la mutación en MEN1 se debe hacer en el caso índice con criterios de NEM-1 (esporádico o familiar) o sospecha de éste (HP familiar aislado, gastrinoma o tumor endocrino pancreático a cualquier edad, HP primario recurrente y en los pacientes con hiperplasia paratiroidea menores de 30 años) y en los parientes asintomáticos de una familia con un caso índice de NEM-126,27,29. El algoritmo diagnóstico recomendado en los portadores asintomáticos de la mutación incluye la determinación sérica de calcio, prolactina y hormonas gastrointestinales, anualmente y la realización de técnicas de imagen (RM de hipófisis, ecoendoscopia y octreoscán) cada 3 o 5 años30,31. Si alguno de los parámetros está elevado, se procede a su confirmación por las técnicas apropiadas. La finalidad del cribado en los pacientes asintomáticos de una familia con NEM-1 es reducir la morbilidad y la mortalidad de la enfermedad. Este objetivo a veces no se consigue, pues la penetrancia del gen MEN1 es variable y la presentación de la enfermedad muy diferente en los miembros de una misma familia en cuanto a la edad y a la gravedad de los síntomas.

