




La colangiopatía portal es el conjunto de alteraciones que pueden aparecer en la vesícula y en la vía biliar en relación con la existencia de una trombosis crónica de la vena porta y el desarrollo de circulación colateral. La obstrucción crónica de la vena porta es una causa frecuente de hemorragia digestiva por varices esofágicas, pero su posible repercusión sobre la vía biliar es menos conocida. Presentamos el caso clínico de un varón con un cuadro de ictericia secundaria a colangiopatía portal, analizando posteriormente la patogenia de esta entidad, los métodos diagnósticos y las diferentes posibilidades terapéuticas disponibles.
Portal cholangiopathy encompasses a group of abnormalities of the biliary system and gallbladder that occur secondary to chronic portal vein thrombosis and collateral venous circulation. Chronic obstruction of the portal vein is a frequent cause of gastrointestinal variceal bleeding, but data on biliary tract abnormalities are limited. We report the case of a male patient with obstructive jaundice secondary to portal cholangiopathy. We describe the pathogenesis of this entity, and the various diagnostic and therapeutic options available.
La trombosis crónica de la vena porta extrahepática y la aparición de colaterales venosas periportales en forma de cavernoma pueden asociarse con anomalías estructurales de la vía biliar (estenosis, dilataciones, angulaciones), denominadas en conjunto colangiopatía portal (CP) o biliopatía portal. A pesar de que las pruebas de imagen revelan datos de CP en un porcentaje elevado de los pacientes con trombosis crónica y cavernomatosis portal (81-100%), solo una cuarta parte de éstos presentarán síntomas1,2.
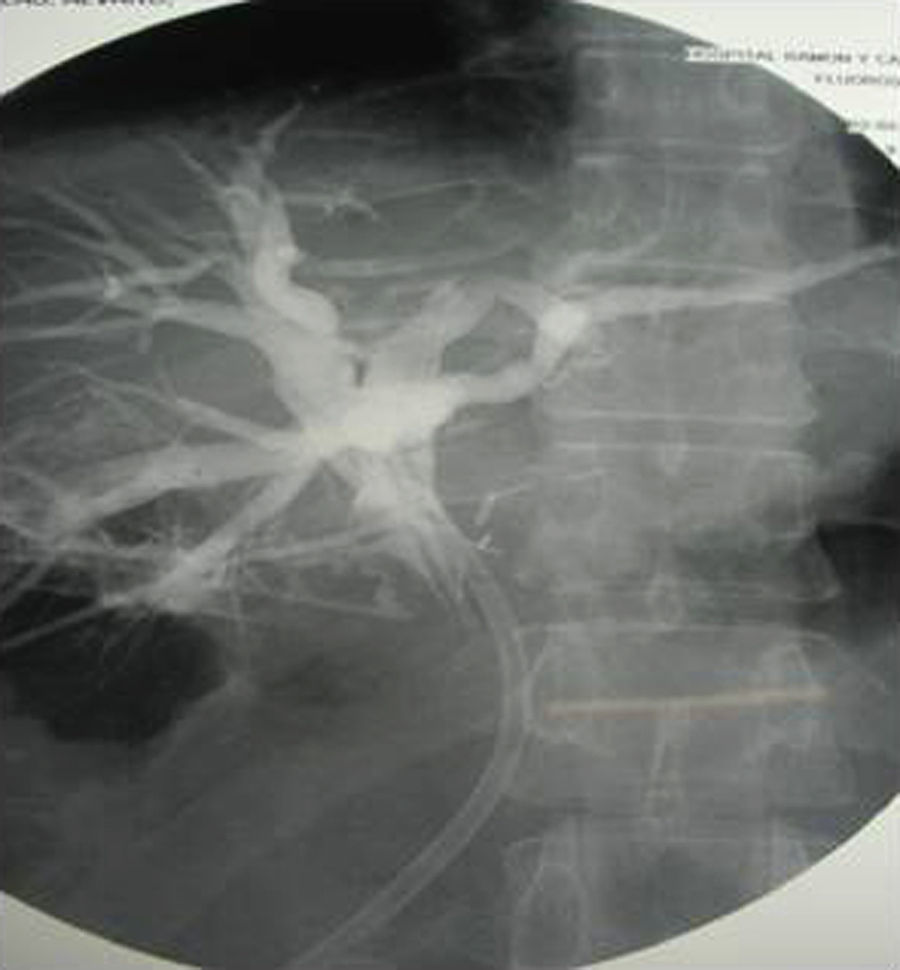
Caso clínicoVarón de 42 años sin antecedentes médicos que acudió a urgencias por ictericia de 10 días de evolución, acolia y coluria, sin dolor abdominal, pérdida de peso o fiebre. No refería ni consumo de fármacos, ni hábitos tóxicos, ni antecedentes epidemiológicos de riesgo para hepatitis virales. En la exploración física destacaba la presencia de ictericia. Presentaba bilirrubina de 5mg/dl, GOT 66 U/l, GPT 72 U/l, GGT 75 U/l y fosfatasa alcalina 287 U/l; Hb 14g/dl, VCM: 95fl, leucocitos 6,7 x 103/μl, plaquetas 189 x 103/μl e INR 1. En urgencias se realizó una ecografía abdominal y una TC, visualizando dilatación de la vía biliar intrahepática y del conducto hepático común, con un dudoso defecto de repleción en el cístico y una masa hipodensa de 25mm en el hilio hepático, de bordes mal definidos y aspecto inflamatorio que contactaba con la pared de la vesícula biliar que, a su vez, contenía barro biliar. Estos hallazgos radiológicos se confirmaron en una colangio-RM realizada una vez ingresado el paciente, que se interpretó inicialmente como un posible síndrome de Mirizzi (fig. 1). Se solicitó una colangiopancreatografía retrógada endoscópica (CPRE) donde se observó dilatación de la vía biliar intra y extrahepática proximal, sin defectos de repleción en su interior, con una disminución marcada del calibre del colédoco distal, de bordes regulares, no indicativa de estenosis neoplásica. Tras realizar una esfinterotomía biliar, se pasó una sonda-balón de Fogarty de 11,5mm sin encontrar salida de litiasis y, finalmente, ante el cuadro de colestasis asociado a estenosis coledoceana de origen no aclarado, se optó por colocar una prótesis plástica biliar recta de 9cm/10F. A continuación, en una ecoendoscopia se identificó una masa hipoecogénica en el hilio hepático de 25mm, de bordes mal definidos que englobaba la arteria hepática y la porta, asociada a una imagen compatible con cavernomatosis portal, con presencia de importante circulación venosa colateral en contacto estrecho con el colédoco en su porción más distal (fig. 2). Durante la ecoendoscopia se efectuó una punción aspirativa con aguja fina (PAAF) de la masa hiliar, obteniendo células de aspecto histiocitario con citoplasmas de aspecto xantomizado y otras células inflamatorias, principalmente de tipo linfoide, sin evidencia de celularidad maligna. Para completar el estudio y descartar la presencia de una neoplasia del hilio hepático se efectuó una laparotomía y se tomaron muestras de la masa hiliar. Durante el procedimiento quirúrgico se confirmó la presencia de una importante circulación colateral en el epiplón menor, se realizó una colangiografía intraoperatoria descartándose el diagnóstico de síndrome de Mirizzi al no encontrar litiasis en el conducto cístico y finalmente se extirpó la vesícula (fig. 3). El examen de las biopsias obtenidas reveló la presencia de tejido fibroadiposo-vascular sin datos de malignidad y adenitis con granulomas epitelioides no necrosantes, con tinción de Ziehl-Neelsen negativa y PCR-Mycobacterium tuberculosis negativa. Se remitió parte de la muestra para cultivo de hongos y micobacterias que fueron negativos. Así mismo se realizó una citometría de flujo donde no se detectaron células linfomatosas y se determinaron las concentraciones de la enzima conversora de la angiotensina en sangre, que resultaron normales.
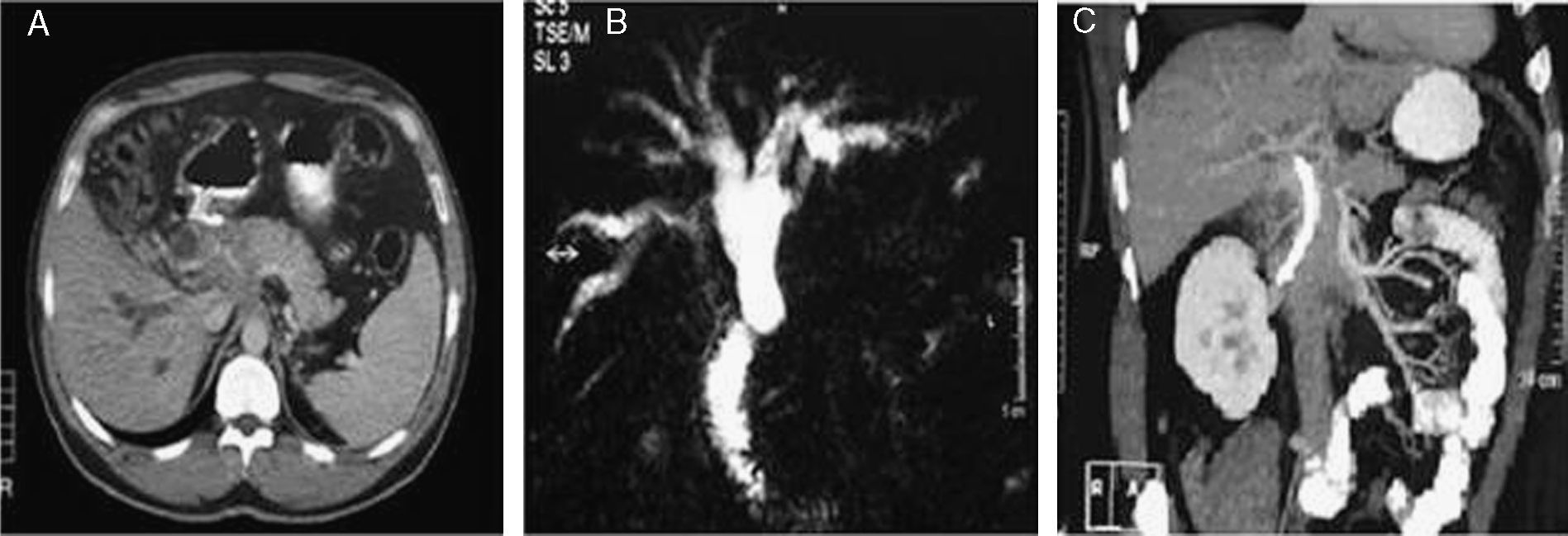
 TC que muestra una masa hipodensa en el hilio hepático. B) Colangiografía con RM: se observa dilatación de la vía biliar intrahepática y colédoco proximal que se amputa de forma brusca pero regular. C) Proyección MIP coronal en fase portal: se encuentra un aumento de partes blandas en torno a 1/3 proximal a la prótesis con densidad similar a la del contenido de los vasos venosos que no permite diferenciar si se trata de una masa con captación de contraste o una cavernomatosis portal.")
TC y colangio-RM. A) TC que muestra una masa hipodensa en el hilio hepático. B) Colangiografía con RM: se observa dilatación de la vía biliar intrahepática y colédoco proximal que se amputa de forma brusca pero regular. C) Proyección MIP coronal en fase portal: se encuentra un aumento de partes blandas en torno a 1/3 proximal a la prótesis con densidad similar a la del contenido de los vasos venosos que no permite diferenciar si se trata de una masa con captación de contraste o una cavernomatosis portal.
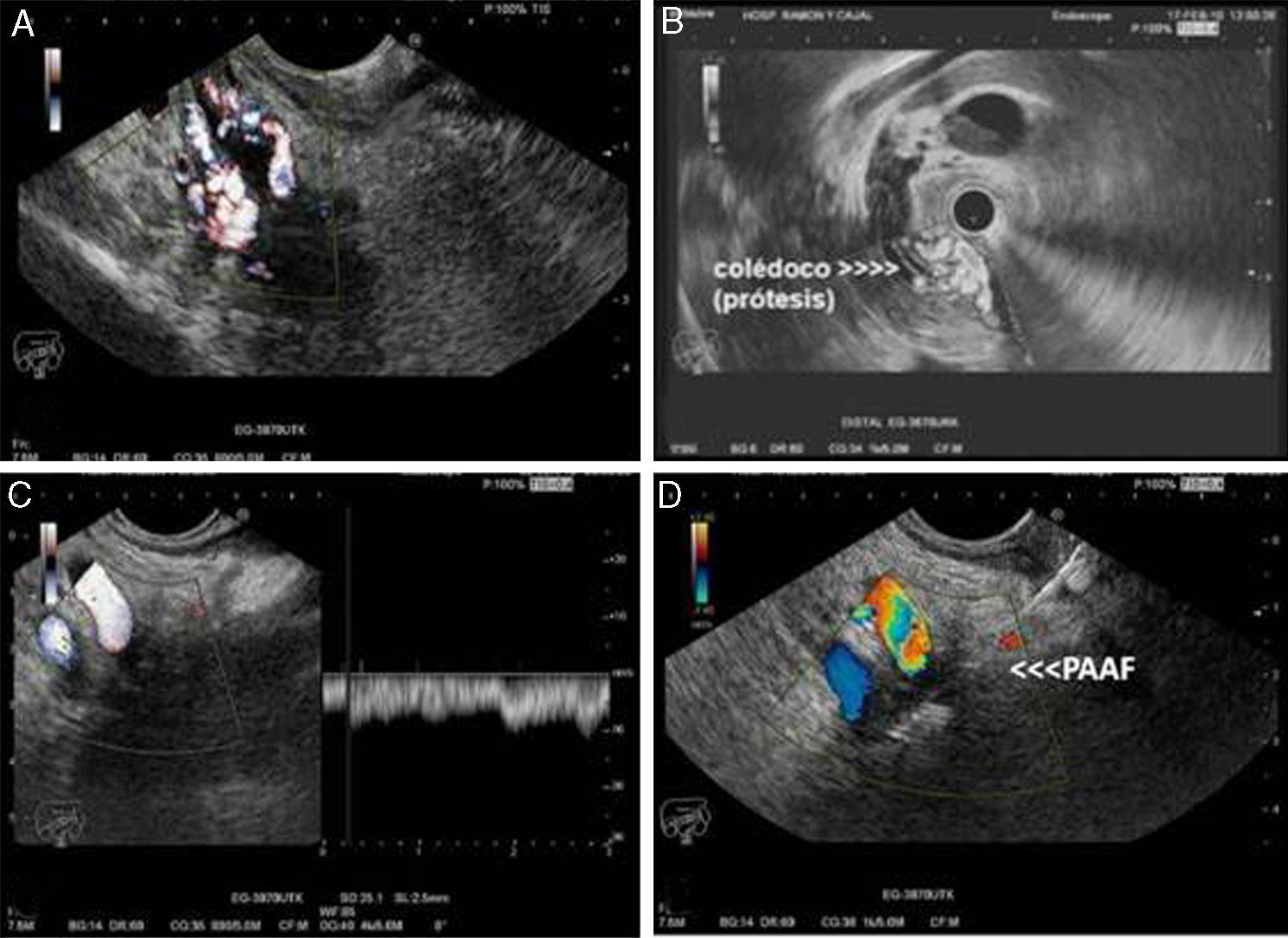
 Ecoendoscopia lineal desde el bulbo duodenal donde se identifica el colédoco con marcada circulación colateral pericoledoceana observada mediante Doppler. B) Ecoendoscopia radial del colédoco distal con prótesis plástica en su interior y circulación venosa colateral pericoledoceana. C) Ecoendoscopia lineal desde la cavidad gástrica donde se identifica una masa hipoecogénica de bordes mal definidos que engloba la porta (Doppler-pulsado) y la arteria hepatica. D) PAAF guiada por ecoendoscopia de la masa descrita en C.")
Ecoendoscopia. A) Ecoendoscopia lineal desde el bulbo duodenal donde se identifica el colédoco con marcada circulación colateral pericoledoceana observada mediante Doppler. B) Ecoendoscopia radial del colédoco distal con prótesis plástica en su interior y circulación venosa colateral pericoledoceana. C) Ecoendoscopia lineal desde la cavidad gástrica donde se identifica una masa hipoecogénica de bordes mal definidos que engloba la porta (Doppler-pulsado) y la arteria hepatica. D) PAAF guiada por ecoendoscopia de la masa descrita en C.

Una vez descartado el origen neoplásico e infeccioso de la masa hiliar y ante la ausencia de un proceso litiásico, se planteó la posibilidad de que el cuadro de ictericia fuera debido a la compresión extrínseca de la vía biliar por la circulación colateral desarrollada a raíz de la trombosis portal (TP). El paciente no presentaba datos de cirrosis hepática, se realizó estudio de hipercoagulabilidad que resultó negativo y finalmente fue diagnosticado de CP idiopática (grado III)3 y se inició tratamiento con ácido ursodesoxicólico. Fue dado de alta tras 40 días de hospitalización, presentando durante el seguimiento en consultas externas una evolución clínica y analítica satisfactoria con desaparición de la colestasis. Transcurridos 4 meses, volvió a ingresar por un cuadro de hemorragia digestiva alta con inestabilidad hemodinámica, realizándose con carácter urgente una gastroscopia, en la que se encontraron varices esofágicas de pequeño tamaño sin puntos rojos ni estigmas de sangrado y una hemorragia activa procedente de la vía biliar. El cuadro cedió con el tratamiento médico (somatostatina), siendo necesaria la transfusión de 2 concentrados de hematíes. El origen de la hemorragia se atribuyó a una posible lesión por decúbito de la prótesis biliar en la pared del colédoco. Una vez superado el episodio de hemorragia aguda, se retiró la prótesis biliar por vía endoscópica sin complicaciones. En ese momento, considerando la presencia de varices esofágicas y para intentar disminuir la presión en la circulación colateral periportal se añadió propanolol al tratamiento. También se planteó la realización de cirugía derivativa portosistémica, que no fue posible al presentar una vena esplénica de pequeño calibre. Actualmente, transcurridos 7 meses desde el episodio hemorrágico, el paciente continúa asintomático, con una bioquímica hepática dentro de parámetros normales.
DiscusiónEl término colangiopatía portal fue utilizado por primera vez por Sarin et al en 19924. Desde entonces solo se han descrito unos 200 casos en la literatura médica, probablemente debido a que, entre los gastroenterólogos, existe un bajo nivel de sospecha clínica de esta entidad4–7. La patogenia de la CP no está completamente aclarada. Algunos autores señalan que la TP crónica y la transformación cavernomatosa ocasionan isquemia en los conductos biliares8, si bien la teoría más aceptada defiende que las colaterales periportales que componen el cavernoma pueden ejercer suficiente presión sobre la vía biliar como para provocar cuadros de colestasis y en ocasiones verdaderas estenosis de la vía biliar9,10 La mayoría de los casos de CP se asocian a TP crónica extrahepática (81-100%), si bien esta entidad también puede aparecer en pacientes con marcada hipertensión portal sin trombosis, ya sea de origen cirrótica (0-33%) o idiopática (9-40%)1,10.
Respecto a la presentación clínica, la mayoría de los pacientes están asintomáticos5,11. En la serie de casos de CP más numerosa publicada hasta la fecha, que incluye 67 pacientes, solo el 21% presentaron síntomas, siendo los más frecuentes el dolor abdominal, la ictericia y la colangitis aguda3. Los autores de este trabajo proponen una clasificación de la CP en función de los hallazgos radiológicos (grado I: angulaciones en el tracto biliar; grado II: estenosis sin dilatación; grado III, estenosis con dilatación) y observan que solo los casos con estenosis y dilatación biliar (grado III) presentan sintomatología. El intervalo de tiempo medio transcurrido entre el episodio de TP y la aparición de síntomas relacionados con la CP fue de 42 y 118 meses en los casos de TP aguda y crónica, respectivamente3. Existen casos descritos en la literatura médica con periodos de latencia superiores a 10 años6,12.
El diagnóstico de CP se basa en los hallazgos obtenidos en las diferentes pruebas de imagen9,10. La ecografía abdominal con estudio Doppler del eje esplenoportal es la exploración en la que inicialmente suele detectarse la TP y la cavernomatosis, pero no suele proporcionar un estudio óptimo de la vía biliar extrahepática1,13. En este sentido, la colangio-RM actualmente se considera la exploración complementaria más rentable para llegar al diagnóstico de CP, ya que por una parte permite confirmar el diagnóstico de TP y la presencia de circulación venosa colateral periportal alrededor del colédoco y, por otra, demuestra la repercusión del cavernoma del árbol biliar en forma de estenosis de contornos regulares, angulaciones o dilataciones segmentarias6. La CPRE, a pesar de que permite encontrar alteraciones en la vía biliar similares a las descritas en la colangio-RM, nunca debe realizarse con intención exclusivamente diagnóstica2,14. Como puede comprobarse en el caso expuesto, en ocasiones los pacientes con CP pueden presentar en el hilio hepático masas detectables mediante TC o RM, generalmente de aspecto sólido, constituidas por tejido inflamatorio y estructuras venosas colaterales de pequeño calibre, que pueden simular lesiones neoplásicas conocidas como pseudocolangiocarcinomas6,14 (fig. 1).
La ecoendoscopia, aunque es una técnica diagnóstica invasiva y requiere sedación para su realización, puede resultar útil en el diagnóstico de esta entidad, ya que al igual que la RM permite la evaluación conjunta del cavernoma y de la vía biliar. En presencia de lesiones sólidas detectadas en el hilio hepático, la posibilidad de realizar una PAAF para realizar el diagnóstico diferencial con procesos neoplásicos, puede ser un argumento a favor de la utilización de esta técnica15,16 (fig. 2).
En los pacientes diagnosticados de CP es conveniente realizar un estudio de hipercoagulabilidad para intentar aclarar el origen de la TP, así como una endoscopia oral para detectar varices esofágicas. Aunque no es infrecuente el desarrollo de varices esofágicas en pacientes con CP, la mortalidad de estos enfermos está condicionada en mayor medida por la evolución de las enfermedades asociadas a la TP más que por las complicaciones propias de la hipertensión portal17.
Aunque no existe consenso acerca del tratamiento de esta entidad, la mayoría de los autores coinciden en afirmar que los pacientes asintomáticos no requieren medidas terapéuticas. La administración de ácido ursodesoxicólico (10-15mg/kg/día) se recomienda en casos de elevación de las enzimas de colestasis2. En pacientes con cuadros de ictericia obstructiva, especialmente cuando existe sospecha de colangitis y sepsis, debe plantearse la realización de CPRE con esfinterotomía, extracción de coledocolitiasis si estuviese presente y colocación de prótesis con recambios periódicos en función de la evolución clínica y analítica7. Cabe señalar que la esfinterotomía debe ser de pequeño tamaño por el alto riesgo de hemorragia debido a la importante vascularización que puede haber alrededor de la papila2,11. Si el tratamiento endoscópico fracasa, debe plantearse la intervención quirúrgica, mediante una derivación portosistémica. La técnica quirúrgica más empleada en adultos es la derivación esplenorrenal, mientras que en la población pediátrica se recomienda la derivación mesentérico-cava18. Existen casos anecdóticos de CP tratados satisfactoriamente mediante una derivación percutánea portosistémica intrahepática19. En caso de no existir vasos derivables o cuando los síntomas persisten tras la derivación portosistémica, cabe la posibilidad de realizar una derivación bilioentérica, teniendo presente que asocia una morbimortalidad elevada18. Finalmente, el trasplante hepático puede plantearse ante pacientes con enfermedad avanzada intratable endoscópica y quirúrgicamente20.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

