




Las causas más comunes de esteatohepatitis son el consumo de alcohol y los trastornos metabólicos. Existen diversos métodos basados en determinaciones bioquímicas (transferrina deficiente en hidratos de carbono) y en la realización de cuestionarios (AUDIT, CAGE, MALT) útiles para la detección del consumo subrepticio de alcohol. A pesar de que se están desarrollando nuevos métodos no invasivos tanto basados en lipidómica (Owl-Liver®) como en determinaciones bioquímicas y parámetros antropométricos (NAFLD Fibrosis score) o en métodos de imagen (DeMILI NASH-MRi®), ninguno se ha postulado como definitivo, y la biopsia hepática constituye el método de referencia. La patogenia de la esteatohepatitis alcohólica y no alcohólica presenta elementos comunes como la resistencia a la insulina, el estrés oxidativo mediado por el citocromo CYP2E1, la adiponutrina y su gen PNPLA3 y la microbiota. Los cambios en el estilo de vida, incluido el abandono del consumo de alcohol, la dieta y el ejercicio constituyen la primera línea de tratamiento.
The most common causes of steatohepatitis are alcohol intake and metabolic disorders. Several methods based on biochemical determinations (carbohydrate deficient transferrin) and questionnaires (AUDIT, CAGE, MALE) are useful for detecting surreptitious alcohol intake. Although new non-invasive methods are under development, based both on lipidomics (Owl-Liver®) and on biochemical determinations and anthropometric parameters (NAFLD Fibrosis score) or imaging methods (DeMILI NASH-MRi®), none has been proposed as definitive and the gold standard continues to be liver biopsy. The pathogenesis of alcoholic and non-alcoholic steatohepatitis shares some elements such as insulin resistance, cytochrome CYP2E1-mediated oxidative stress, adiponutrin and its PNPLA3 gene, and the microbiota. The first-line treatment consists of lifestyle changes, including giving up alcohol, diet and exercise.
La esteatohepatitis puede tener etiologías variadas: origen metabólico, viral, debido al alcohol o bien posterior a una resección intestinal o un bypass yeyuno-ileal. La enfermedad metabólica hepática (NAFLD) es la hepatopatía más prevalente en el mundo desarrollado, originada por el cambio del estilo de vida junto con el incremento de sobrepeso/obesidad, resistencia a la insulina (RI), diabetes mellitus tipo 2 e hipertensión arterial, englobados en el síndrome metabólico. Entre los factores de riesgo se encuentra la combinación de un consumo de energía elevado, en el cual se ingieren hidratos de carbono simples procesados rápidamente y grasas saturadas, con bajos niveles de actividad física y un hospedador susceptible, que presente un polimorfismo genético, PNPLA31, junto a factores como la edad avanzada, el género masculino, la etnia hispánica y la presencia de las principales características del síndrome metabólico.
El origen de la esteatosis hepática parte de una falta de equilibrio entre la adquisición y la eliminación de triglicéridos. Los triglicéridos son formados ensamblando 3 ácidos grasos a una columna de glicerol vía enlaces éster. Estos ácidos grasos usados para la formación de triglicéridos derivan de 3 fuentes: tejido adiposo, síntesis de novo o de la dieta, sobre la cual es esencial actuar, ya que aproximadamente el 20% de la ingesta diaria va a parar al hígado2. El resultado es la acumulación de triglicéridos en el citoplasma del hepatocito, y puede generar tanto necroinflamación como fibrosis, conduciendo en último término al fallo hepático, cirrosis y carcinoma hepatocelular3.
Para detectar NAFLD es necesario evidenciar la esteatosis hepática mediante métodos de imagen y descartar causas secundarias de acumulación de grasa en el hepatocito, como consumo significativo de alcohol, uso de medicación esteatogénica, hepatitis virales, hepatotoxicidad, enfermedad de Wilson o nutrición parenteral total4. La prevalencia de NAFLD oscila entre el 2,8 y el 46%, según el método de detección utilizado (biopsia hepática [12,2%], ecografía [29,9%], espectroscopia por resonancia magnética [31%] o métodos bioquímicos [7-11%]) y la población en estudio.
La prevalencia estimada de esteatohepatitis es del 3 al 5%, con una incidencia de 2 nuevos casos/100 personas/año.
Detección de alcoholismo subrepticioExisten toda una serie de determinaciones tanto bioquímicas como cuestionarios realizados por el paciente para identificar consumo de alcohol.
CuestionariosEl cuestionario Alcohol Use Disorders Identification Test (AUDIT), desarrollado por la OMS, es un método simple de cribado para identificar pacientes con un patrón de consumo de alcohol perjudicial. El AUDIT consta de 10 preguntas sobre las consecuencias del consumo, la dependencia, la cantidad y la frecuencia del mismo. También se ha determinado un alto coeficiente de correlación (0,78) entre el AUDIT y el CAGE en pacientes ambulatorios. Presenta una sensibilidad del 80% y una especificidad del 90% para un punto de corte de 85.
El test Chronic Alcoholism General Evaluation (CAGE) es un sencillo cuestionario de 4 preguntas. Su sensibilidad oscila entre el 49 y el 100% y su especificidad entre el 79 y el 100%, en función del punto de corte propuesto y de la gravedad del problema relacionado con el alcohol. Se recomienda limitar el uso del CAGE a la detección de la dependencia alcohólica, mientras que AUDIT tiene un mejor rendimiento para la detección de bebedores de riesgo.
El Munchner Alkoholismus Test (MALT) es un cuestionario muy útil en casos con dependencia encubierta. Consta de 2 partes, el MALT objetivo (MALT-O) cumplimentado por el entrevistador, y que recoge datos de anamnesis, exploración física y analítica, y el MALT subjetivo (MALT-S), que es completado por el paciente y consta de 27 ítems que exploran aspectos psicoconductuales y posibles repercusiones del consumo de alcohol.
Se ha detectado una elevada correlación entre el AUDIT y el MALT (r = 0,88) en ambos sexos, así como correlaciones de 0,47 y 0,46 para hombres y mujeres, respectivamente6.
Marcadores bioquímicosEl consumo crónico de alcohol incrementa el volumen corpuscular medio (VCM) y no constituye un buen marcador de cambios en el comportamiento del bebedor.
En las hepatopatías de origen alcohólico la relación AST/ALT suele ser superior a 2, a diferencia del resto de enfermedades hepáticas en las que predomina la alteración de la ALT.
La transferrina deficiente en hidratos de carbono (TDC) es el primer marcador bioquímico de los bebedores crónicos aprobado por la FDA. Es una forma de transferrina con bajo contenido en hidratos de carbono, cuya concentración aumenta de manera paralela al incremento de consumo de alcohol. La TDC no mide un consumo ocasional, para que aparezcan concentraciones elevadas es necesario un consumo de al menos 60-80g de etanol/día durante una semana, por lo que no se considera adecuada para detectar bebedores esporádicos. Su especificidad se sitúa entre el 70 y el 100% y la sensibilidad oscila del 69 al 100%7, llegando ambas al 100% cuando coexiste un incremento de la GGT y de la TDC. La TDC puede ser utilizada además en la monitorización de la abstinencia del consumo de alcohol, especialmente para detectar recaídas, ya que durante la abstinencia alcohólica el valor de TDC se normaliza en aproximadamente 2 semanas. A diferencia de la GGT o el VCM, no suele alterarse por otras enfermedades o por el consumo de fármacos u otras sustancias. La TDC de manera independiente puede identificar cerca del 65% de los pacientes con recaídas, pero la combinación con la GGT mejora el rango de reconocimiento alrededor de un 74%8.
La GGT se encuentra en las membranas celulares endoteliales y durante el consumo crónico de alcohol sus niveles séricos frecuentemente se elevan. Se cree que este incremento está causado por la síntesis acelerada de GGT y/o liberación rápida por parte de células hepáticas dañadas o muertas. Es un marcador muy sensible (35-85%) y específico (81-89%) en la detección de consumo de alcohol9. Se normaliza a las 2-6 semanas de abstinencia, lo que permite hacer un seguimiento de la misma.
Se han desarrollado 2 métodos para medir la concentración de acetaldehído libre: a) la cromatografía y la detección de epítopos derivados de este compuesto, asociados con proteínas plasmáticas, y b) la inmunoglobulina A contra epítopos modificados por el acetaldehído.
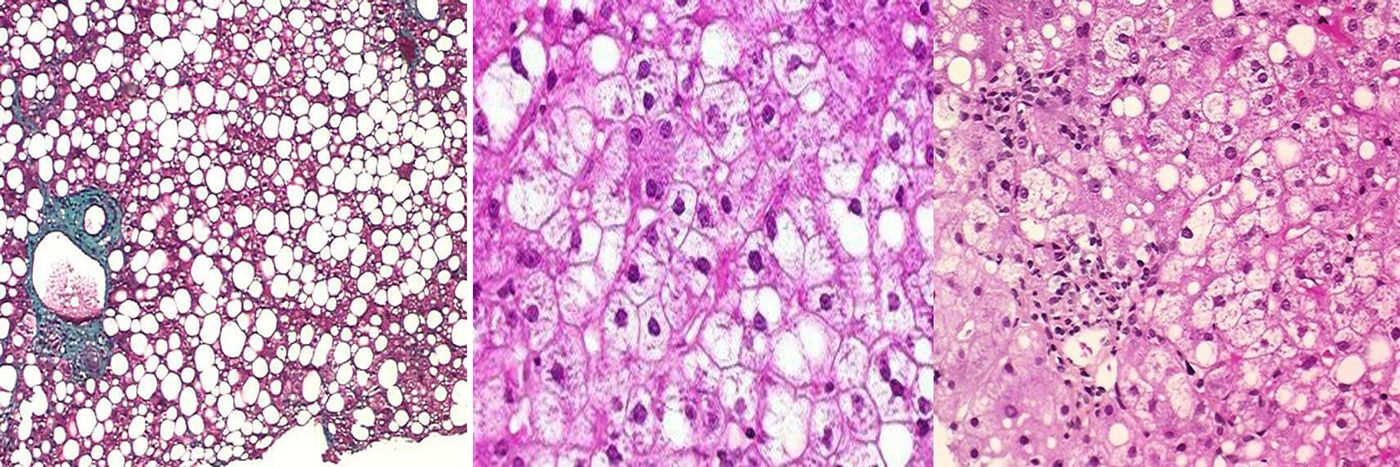
Diagnóstico y pronóstico de enfermedad metabólica hepáticaEl diagnóstico histológico de esteatohepatitis es variable y subjetivo. La valoración conjunta de la esteatosis, la inflamación y la degeneración balonizante permite al patólogo diagnosticar esteatohepatitis. El método más común es el NAS Score (NAFLD Activity Score)10 que provee una puntuación en función del grado de esteatosis (0-3), la inflamación lobulillar (0-3) y la balonización hepatocitaria (0-2). Una puntuación ≥ 5 señala esteatohepatitis, mientras que si es inferior a 3 esta lesión sería poco probable. Se considera que si el NAS está entre 3 y 4 el diagnóstico de esteatohepatitis podría ser indeterminado. En la figura 1 se pueden observar los hallazgos histopatológicos tanto de esteatosis como de degeneración lobulillar y balonización hepatocitaria. Por tanto, el cálculo del NAS Score permite mejorar la objetividad pero mantiene una zona gris en la cual el diagnóstico de esteatohepatitis no es definitivo.

El patrón de esteatosis puede ser dividido en macro y microvesicular, donde los hepatocitos pueden contener bien una gran vacuola de grasa mayor que el propio núcleo del hepatocito que desplaza el núcleo o bien varias y pequeñas inclusiones citoplasmáticas sin desplazamiento nuclear significativo11. La esteatosis macrovesicular es más común y se encuentra tanto en NAFLD como en la enfermedad hepática alcohólica, mientras que la microvesicular es generalmente una forma de esteatosis más severa y se puede presentar en un rango más amplio de enfermedades, como hepatotoxicidad, defectos en la betaoxidación de ácidos grasos y síndrome de Reye. La evaluación de la esteatosis hepática tiene también un papel crucial en la evaluación del donante candidato para el trasplante hepático, ya que en concreto el subtipo macrovesicular se ha asociado con un mayor incremento de disfunción primaria del injerto12.
Métodos bioquímicos no invasivos para el diagnóstico de esteatohepatitisOwl-Liver® es un método que consiste en la determinación de 540 metabolitos séricos combinando la cromatografía y la espectroscopia de masas, ofreciendo un perfil lipídico dependiente del índice de masa corporal, lo cual indica que los mecanismos patogénicos del NAFLD son diferentes dependiendo del nivel de obesidad individual. Usando un modelo multivariante estratificado basado en esta premisa se separan pacientes con y sin esteatohepatitis, obteniéndose un AUROC de 0,85 en la cohorte de validación teniendo en cuenta el punto de corte 0,54, con una sensibilidad de 71% y una especificidad de 92%13.
Recientemente se ha tratado de demostrar la eficacia de la combinación de varios biomarcadores como son la citoqueratina 18 (CK-18), la proteína de unión al ácido graso del adipocito (AFABP) y el factor de crecimiento 21 del fibroblasto (FGF21) para diagnosticar NAFLD y NASH. Los fragmentos de CK-18 reflejan el grado de apoptosis hepatocelular, una característica de la esteatohepatitis14. Asimismo, el AFABP está involucrado en la interacción entre adipocitos y macrófagos, lo que puede conducir a inflamación y resistencia a la insulina15. La expresión del mRNA FGF-1 en el hígado incrementa el grado de esteatosis, y sus niveles séricos se encuentran significativamente incrementados en pacientes con NAFLD16. Los resultados obtenidos para detectar esteatosis fueron una AUROC 0,91 (IC 95% 0,87-0,95) para la CK-18, 0,66 (IC95% 0,59-0,74) en el caso de AFABP y para el FGF-21 0,84 (IC 95% 0,79-0,90). En el caso de la esteatohepatitis, los resultados obtenidos fueron significativamente peores, CK-18 0,70 (IC 95% 0,61-0,78), AFABP 0,59 (IC 95% 0,50-0,68) y FGF-21 0,62 (IC 95% 0,53-0,71), todos en comparación con la biopsia17. Por ello, se plantea la combinación de CK-18 y FGF-1 como biomarcadores para la detección de esteatohepatitis.
FibroMax® comprende una serie de pruebas no invasivas (FibroTest, SteatoTest, NashTest) analizando una serie de parámetros bioquímicos (ALT, AST, GGT, colesterol total, triglicéridos, glucosa, bilirrubina total, haptoglobina, apolipoproteína-1 y alfa-2-macroglobulina) y antropométricos (edad, peso y altura). Provee información tanto del estado necroinflamatorio del paciente como de su fibrosis. Es aplicable tanto a pacientes con hepatitis virales, enfermedad hepática alcohólica y pacientes con NAFLD. El AUROC para el SteatoTest es de 0,80, para NashTest de 0,83 y para el FibroTest de 0,84 (IC 95% 0,76-0,92). Como limitación principal destaca que no calcula para índices de masa corporal (IMC) superiores a 30kg/m2, ajustándolos a este valor18.
Métodos de imagen para el diagnóstico de esteatohepatitisLos métodos de imagen más comunes poseen una baja sensibilidad para establecer correctamente el estadio de fibrosis (FibroScan®19), detectar la presencia de esteatohepatitis (tomografía computarizada20) o bien diferenciar entre esteatosis y fibrosis (ecografía abdominal21) y tienen problemas para cuantificar de manera precisa la cantidad de grasa en el hepatocito, viéndose limitados especialmente por el grado de obesidad del paciente y por aquellos que poseen un exceso de gas intestinal o ascitis. La espectroscopia por resonancia magnética mide la cantidad grasa alojada en el hepatocito calculando la diferencia entre señales de los lípidos y del agua. Esta técnica es considerablemente más precisa y sensible que la tomografía computarizada y la ecografía22, pudiéndose detectar incluso concentraciones milimolares de ciertos metabolitos seleccionados. Es una técnica segura, rápida y no invasiva, pero actualmente su disponibilidad es limitada.
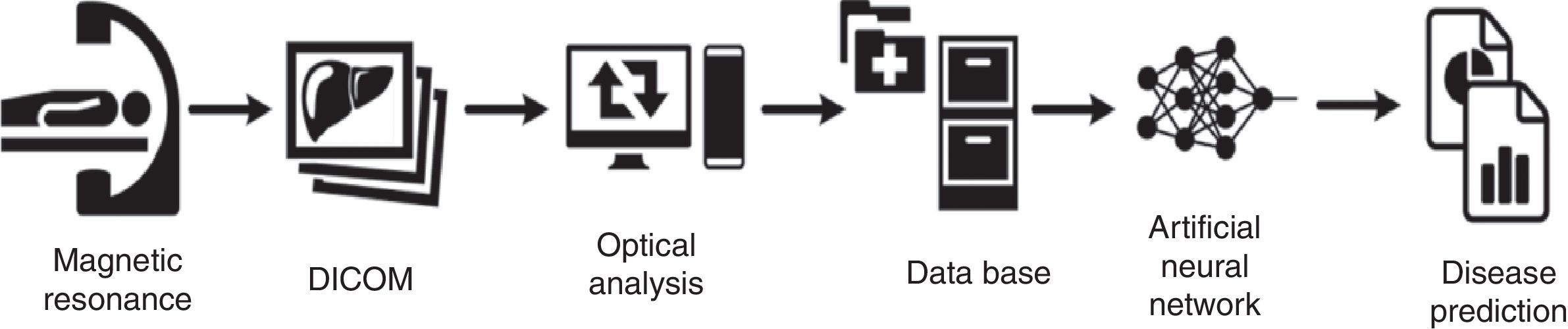
Dado que resulta crucial el uso de una herramienta precisa, fiable, no invasiva y rentable, actualmente se está desarrollando DEMILI®, un nuevo software que utiliza el procesamiento de imágenes de resonancia magnética combinando el análisis óptico con la arquitectura de redes neuronales artificiales, y permite determinar tanto el grado como la distribución de la fibrosis y la presencia de esteatohepatitis en pacientes con NAFLD23. Las AUROC obtenidas con este procedimiento son 0,91 para la fibrosis y 0,89 para detectar esteatohepatitis utilizando el mismo protocolo de resonancia magnética (fig. 2).
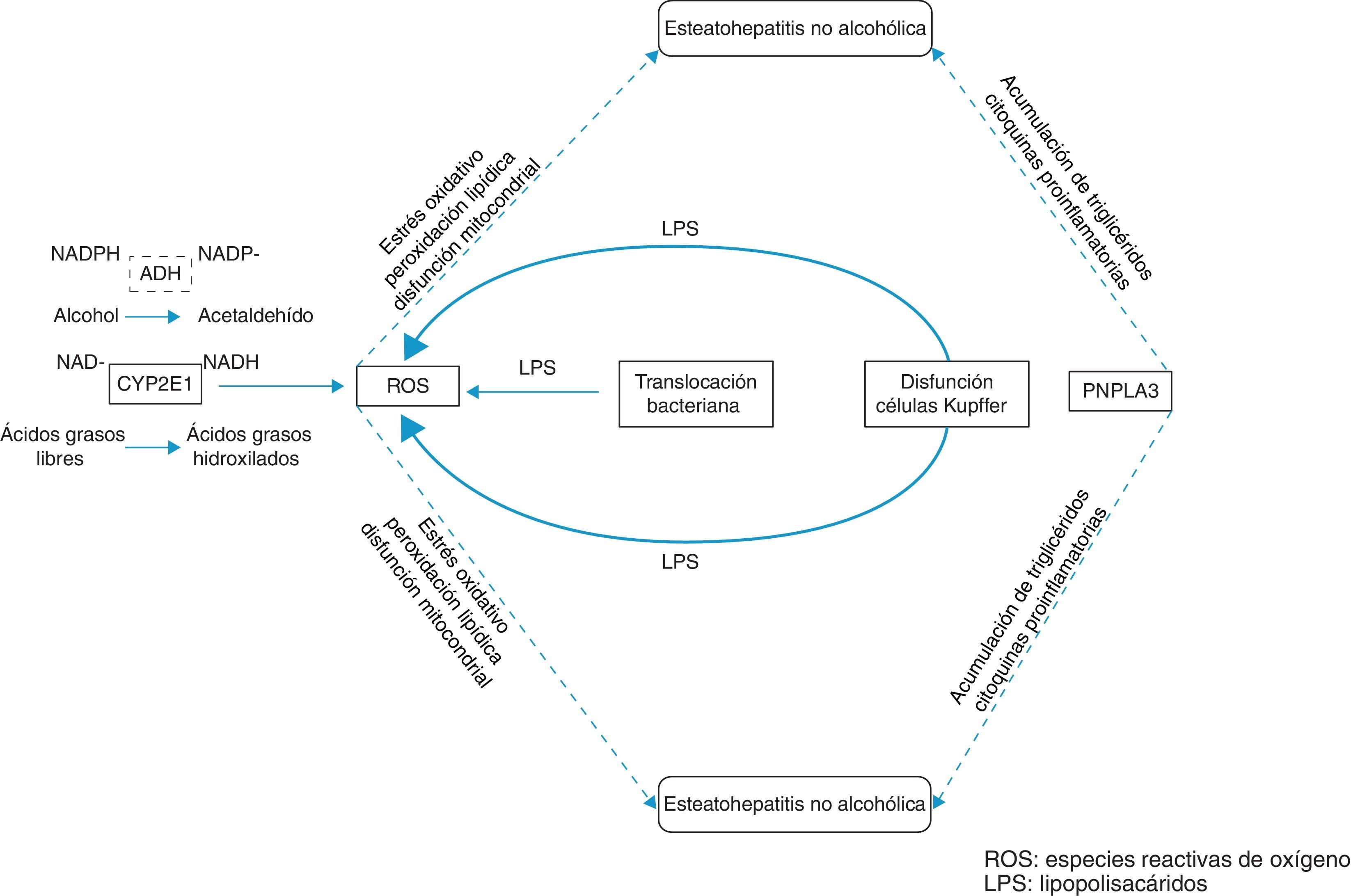
Vías patogénicas comunes de esteatohepatitis alcohólica y no alcohólica.")
La patogenia de estas 2 entidades presenta elementos comunes y otros específicos. La esteatohepatitis alcohólica muestra, como elementos más característicos, los efectos tóxicos del paso de etanol a acetaldehído, un incremento de la actividad lipogénica y una disminución de la retirada de triglicéridos del hígado. Sin embargo, en la esteatohepatitis no alcohólica destaca la resistencia a la insulina, la cual aumenta la absorción de ácidos grasos libres, la síntesis de novo de ácidos grasos y la acumulación de triglicéridos en el hígado, así como la inhibición de la conversión de estos en colesterol unido a las lipoproteínas de baja densidad (LDL) (fig. 3).

Los factores relacionados con la esteatohepatitis engloban el consumo de alcohol, estrés oxidativo, resistencia a la insulina, factores genéticos y, más recientemente, la microbiota intestinal.
Consumo de alcoholRecientemente se ha publicado un estudio realizado en ratones ob/ob a los cuales se les proporcionó 2,5 g de alcohol/kg al día durante 6 semanas versus ratones control. Los resultados ofrecidos por este trabajo indican que el alcohol disminuyó la esteatosis macrovesicular, la apoptosis y ciertos parámetros inflamatorios, como las transaminasas y la infiltración neutrofílica, así como aumentaron los niveles de adiponectina y Akt24. Asimismo, de manera reciente se ha propuesto que un consumo moderado de alcohol podría tener un efecto protector contra la NAFLD. Se analizaron 4.957 hombres y 2.155 mujeres sin enfermedades hepáticas previas, de los cuales el 40% de los hombres no bebedores resultó tener NAFLD y entre aquellos que sí consumían la cifra disminuyó al 28%, por lo que el consumo de alcohol se asoció de manera inversa con la posibilidad de presentar NAFLD con un OR de 0,54. Comparando la proporción de pacientes con NAFLD con la frecuencia de consumo se detectó que el 38% de los pacientes bebían de 1 a 3 días a la semana, el 29% de 4 a 6 días a la semana y el 24% bebían a diario25.
Para descartar el alcohol como causa principal de la esteatohepatitis, y que por tanto, ésta sea englobada dentro de la etiología no alcohólica, habría que establecer un límite a partir del cual el consumo de alcohol sería el origen de la hepatitis, que actualmente en mujeres se encuentra en 60 g/día y en hombres en 80 g/día. El problema estriba en que no existe un umbral de ingesta etílica para definir no alcohólica. A esto se añade que los métodos para conocer la cantidad y la frecuencia del consumo de alcohol son muy subjetivos y poco precisos, y que existe cierta dificultad para establecer la etiología cuando se solapan factores metabólicos y consumo de alcohol, ya que además las vías metabólicas que conducen al estrés oxidativo son compartidas, tanto en esteatohepatitis alcohólica como no alcohólica.
Estrés oxidativoLa esteatohepatitis alcohólica es el resultado del incremento de esteatosis, inflamación, estrés oxidativo y disfunción mitocondrial, debido a que el etanol es metabolizado a acetaldehído por la alcohol deshidrogenasa a través del citocromo P450 2E1 (CYP2E1) en los microsomas y a través de la catalasa en los peroxisomas26. El metabolismo del etanol por la primera vía (sistema MEOS) provoca una disminución de NADH, lo que genera esteatosis por la estimulación de síntesis de ácidos grasos, y la sobreexpresión del CYP2E1 contribuye al desarrollo de esteatohepatitis por la generación de especies reactivas de oxígeno27. Estas son tóxicas para las células, ya que reaccionan con las macromoléculas, desnaturalizan las proteínas y dañan el ADN.
El consumo crónico de etanol provoca varias situaciones: a) altera la capacidad fagocítica y la activación de las células de Kupffer (sistema reticuloendotelial que destoxifica endotoxinas); b) incrementa la actividad de CYP2E1, y c) altera la permeabilidad de la barrera gastrointestinal, lo que hace más fácil la difusión al torrente sanguíneo de las endotoxinas (translocación bacteriana de estos componentes de la pared celular de las bacterias gramnegativas)28. Estas toxinas, por tanto, inducen la formación de citocinas proinflamatorias, como TNF-α e IL-6 por las células de Kupffer (vía TLR-4), que se añaden a las especies reactivas del oxígeno para incrementar el estrés oxidativo29. En definitiva, el etanol induce la peroxidación lipídica por la formación de radicales libres y por el descenso de agentes antioxidantes (fundamentalmente glutatión)30.
La esteatohepatitis no alcohólica está estrechamente relacionada con la obesidad, la diabetes mellitus tipo 2 y la hiperlipidemia, situaciones en las que CYP2E1 es inducido, ya que tanto los ácidos grasos libres como las cetonas son sustratos de la reacción31. El segundo «impacto» de la patogenia de esta enfermedad32 es la peroxidación de ácidos grasos debido al incremento del estrés oxidativo que, al igual que en la esteatohepatitis alcohólica, es provocada por la inducción de CYP2E1 y la disfunción mitocondrial. En concreto, este citocromo es capaz de metabolizar ácidos grasos poliinsaturados en ácidos grasos ω-hidroxilados, que son citotóxicos a altas concentraciones33. Se ha observado una correlación entre el CYP2E1 y la peroxidación lipídica en pacientes obesos. En el paso de esteatosis a esteatohepatitis las especies reactivas del oxígeno se muestran decisivas; se ha observado un aumento de CYP2E1 en ratas alimentadas con dietas ricas en grasa34, lo que indica que la cantidad de grasa hepática es importante en la patogenia de la esteatohepatitis por incremento de CYP2E1. Otro aspecto importante es la interferencia que ejerce la esteatosis sobre la microcirculación sinusoidal y el aclaramiento hepatocelular de señales endógenas y exógenas (endotoxinas35, ácidos grasos libres), provocando que las células de Kupffer potencien su acción, es decir, una producción excesiva de especies reactivas del oxígeno y citocinas proinflamatorias36.
Resistencia a la insulinaLa insulina ejerce su efecto sobre el crecimiento y la supervivencia del hepatocito a través de una serie de señales en las que tienen gran importancia los receptores de insulina 1 (IRS-1) y 2 (IRS-2) y el factor de crecimiento insulínico tipo 1 (IGF-1)37. En dicha cascada de señalización existen otros componentes importantes como PI3K que, tras diversos procesos, activan Akt que se encarga de potenciar la proliferación y supervivencia celular, además de incrementar el metabolismo energético y la utilización de glucosa. Por tanto, la vía de la señalización de la insulina a través de PI3K inhibe la apoptosis activando Akt, destacando en ella PTEN como inhibidor de dicho proceso38. La exposición crónica al alcohol altera los mecanismos normales de supervivencia celular mediante la inhibición de la actividad de PI3K (efecto mediado por el incremento de niveles de PTEN)39. Además, la RI hepática es consecuencia de una reducción de los receptores de insulina, así como de la alteración de la fosforilación de las vías IRS-1, IRS-2 y IGF-1. En otros estudios se apunta a la alteración de la unión de la insulina con su receptor como el desencadenante fundamental40. Por tanto, los efectos combinados sobre la vía de señalización de la insulina conllevan, junto con el estrés oxidativo, una alteración del metabolismo energético y de la síntesis de ADN que comprometen la supervivencia celular. Mientras que en la esteatohepatitis alcohólica la RI es consecuencia del consumo de etanol, en la no alcohólica es causa fundamental (primer paso de la teoría del «doble impacto»). La RI suprime la sensibilidad del receptor de insulina situado sobre la membrana del hepatocito, lo que implica la síntesis de ácidos grasos libres y la captación de grasa por los hepatocitos. Además, conlleva la síntesis de novo de ácidos grasos libres vía SREBP-1c (mediante Akt y mTOR) y ChREBP. Posteriormente pueden ser oxidados en la mitocondria, incorporados a los triglicéridos o almacenados y secretados en forma de vesículas de colesterol LDL41. Sin embargo, la activación de SREBP-1c inhibe la oxidación de ácidos grasos libres y contribuye a la menor formación de LDL, por lo que se dificulta el transporte del exceso de triglicéridos hacia el torrente sanguíneo42. Asimismo, la RI disminuye el efecto inhibitorio de la propia insulina sobre la producción de glucosa, mientras que el efecto estimulador sobre la lipogénesis es mantenido43. El incremento del estrés oxidativo propio de la RI es lo que termina de favorecer el paso a esteatohepatitis.
Factores genéticosPatatin-like phospholipase domain-containing 3 (PNPLA3) es una proteína transmembrana que tiene actividad hidrolasa frente a los triglicéridos. Es expresada en el tejido adiposo y en los hepatocitos, siendo regulada por la insulina a través de LXR y SREBP-1c44. Se ha descrito una variación (PNPLA3-148M) asociada a esteatosis hepática que confiere un riesgo 2 veces mayor de acumulación de triglicéridos a aquellos pacientes homocigotos45, por lo que se sitúa como uno de los determinantes más potentes y hace que estos pacientes sean más susceptibles a presentar NAFLD. Más recientemente, se ha publicado que el genotipo PNPLA3-GG (PNPLA3-148M) se asocia además con un incremento de riesgo en la progresión de esteatohepatitis alcohólica leve a avanzada, con una fracción atribuible del 26,6% y una OR de 11,646. Resultados similares han sido obtenidos por Trépo et al.47, ya que en europeos caucásicos PNPLA3-148M se asoció a esteatohepatitis alcohólica, fibrosis avanzada y riesgo de cirrosis. La relación entre PNPLA3 y la enfermedad hepática alcohólica podría explicarse porque la presencia de grasa es el primer paso en la lesión hepática, causando inflamación (peroxidación lipídica y liberación de citocinas). Todos estos datos indican que existen factores genéticos comunes entre ambas formas de esteatohepatitis.
Microbiota intestinalEn la etapa fetal comienza el desarrollo de una estrecha relación entre el hígado y el tracto gastrointestinal que perdurará toda la vida. Esto es debido a la localización central del tracto gastrointestinal que, junto con su vascularización única, le confiere al hígado un papel crítico a varios niveles: metabólico, inmunológico y destoxificante. Se ha descrito que la microbiota intestinal puede ejercer su influencia tanto en la etiología como en la progresión de la esteatohepatitis. Esto es debido a que promueve tanto la obesidad como el desarrollo de resistencia a la insulina, altera el metabolismo de la colina y activa la liberación de citocinas proinflamatorias48. Los tipos bacterianos más comunes pertenecen a los filos Firmicutes y Bacteroideos, los cuales de manera conjunta representan casi el 90% del microbioma tanto en ratones como en humanos49. Trasplantes experimentales de microbioma en ratones señalan que este puede influir tanto en la absorción como en la digestión de nutrientes, así como en la producción de hormonas intestinales, como el glucagón, ejerciendo su efecto sobre el metabolismo general del hospedador. Además, se ha detectado una posible relación entre el microbioma intestinal y la producción endógena de alcohol. En una cohorte de 63 niños a los que se les practicó biopsia hepática percutánea, se tomó una muestra de heces y se realizó una medición de la cantidad de etanol en sangre periférica. Se determinaron los enterotipos descritos por Arumugam et al.50: enterotipo 1 (abundancia de Bacteroides (22-39%) y menor cantidad de Prevotella (0-1%)), enterotipo 2 (abundante Prevotella (6-36%) y menos cantidad de Bacteroides (2-17%)) y enterotipo 3 (menor abundancia de ambos géneros, Bacteroides (3-16%) y Prevotella (0-8%), y mayor abundancia de Ruminococcus (0,1-6,5%). Se encontró que cada enterotipo se asociaba con un único fenotipo en concreto: esteatohepatitis, obesidad e individuos control. En este estudio se indica un posible papel por parte de la microbiota intestinal de producción endógena de alcohol con posible relación en el desarrollo de esteatohepatitis51.
TratamientoLas modificaciones del estilo de vida, que comprenden tanto la dieta, la actividad física y los comportamientos relacionados con el ejercicio, se consideran la primera terapia recomendada para la esteatohepatitis no alcohólica52. Las intervenciones del estilo de vida, que conllevan una reducción del peso corporal del paciente y el incremento de la actividad física, reducen de manera consistente la grasa hepática y mejoran el control de la glucemia y la resistencia a la insulina53. El problema fundamental de estas intervenciones estriba en la adherencia a la dieta o al ejercicio por parte del paciente. La monitorización de la adherencia se realiza mediante cuestionarios autocumplimentados, con las limitaciones inherentes a este procedimiento. Además, cuanto más compleja sea la intervención (dieta, incremento de actividad física y cambios comportamentales), más difícil es la monitorización. El proceso terapéutico debería comenzar con una intervención educativa, seguida de un plan dietético con disminución de la ingesta calórica54, y realización de ejercicio físico55. Una pérdida de peso mayor del 5% se acompaña de una mejora de la esteatosis (también mejora el riesgo cardiovascular) y más del 7% mejora la necroinflamación y la esteatohepatitis56.
En la esteatohepatitis alcohólica, es fundamental la completa abstinencia alcohólica, la cual se ha observado que reduce la lesión hepática en unos 3 meses, mejora la histología y disminuye la progresión a cirrosis57. La malnutrición suele estar asociada al consumo crónico de alcohol, por lo que es decisivo asegurar un buen aporte nutricional (1,5 g/kg de proteínas y 35-40 kcal/kg de energía)58.
- 1.
Cambios en la dieta. Generalmente se realizan recomendando dietas restrictivas en grasas (< 25%) e hidratos de carbono y ricas en proteínas, de entre 1.200 y 1.400 kcal. Esta intervención debe durar de 3 a 6 meses inicialmente, y luego se pautará una dieta menos estricta como seguimiento. A la hora de la monitorización lo adecuado sería realizar, al menos trimestralmente, una analítica bioquímica, para determinar la normalización de las transaminasas, acompañado de un cálculo de parámetros antropométricos, como el peso corporal, la medida del pliegue braquial y del perímetro abdominal, y en el caso en que sea posible, realizar al paciente una bioimpedancia para determinar los porcentajes de masa magra y grasa, así como la cantidad de agua que acumula el individuo.
- 2.
Intervención solo a nivel de actividad física. Suele realizarse un moderado incremento en la actividad aeróbica respecto a la basal, por ejemplo, siguiendo las recomendación de caminar al menos una hora diaria. Tres meses de ejercicio aeróbico conlleva una reducción del 47 y del 48% en los niveles de ALT y AST, respectivamente59.
- 3.
Combinación de dieta y ejercicio. El objetivo de esta intervención es disminuir la ingesta calórica e incrementar la actividad física combinando las 2 anteriores en un período estimado de entre 3 y 12 meses60,61. El objetivo de estas intervenciones sería reducir al menos un 10% el peso corporal del paciente.
Las terapias farmacológicas más comunes son la vitamina E, los agonistas de la PPAR α/δ y la pioglitazona en pacientes no diabéticos. No obstante, ninguna se ha postulado como la terapia definitiva aún y se necesitan más estudios y ensayos clínicos para determinar la mejor opción. En NAFLD existen varios estudios que han investigado los efectos de la metformina sobre la histología hepática y el nivel de aminotransferasas62. Aunque la metformina mejora la resistencia a la insulina y desciende las cifras de AST/ALT, no produce mejoría histológica63. Haukeland et al. llevaron a cabo un ensayo clínico en el que hubo una eficacia similar entre el grupo metformina y el grupo placebo64. Un metaanálisis realizado por Vernon et al.65 concluyó que 6 meses de tratamiento de metformina y modificación del hábito de vida no eran superiores a la modificación de vida únicamente, en cuanto a histología y nivel de aminotransferasas. En la última guía de práctica clínica de la AASLD, la metformina no se recomienda para el tratamiento de pacientes con esteatohepatitis no alcohólica66.
El uso de agonistas de los receptores PPAR ha sido experimentado en ambos tipos de esteatohepatitis, siendo su uso en la esteatohepatitis alcohólica más limitado y reciente. Ramírez et al.67 examinaron los efectos de los agonistas de PPAR-δ (en ratas alimentadas con líquido con 37% etanol) sobre la señalización de la insulina (vía Akt), el estrés oxidativo y la enfermedad ultraestructural, concluyendo que estos fármacos reducen la resistencia a la insulina y la esteatosis hepática, pero con resultados inciertos en cuanto al estrés oxidativo y la fibrosis.
En 22 sujetos con NAFLD, la rosiglitazona se mostró más efectiva en la mejora de aminotransferasas, esteatosis hepática, balonización y escalas de inflamación68. Estos resultados fueron similares a los obtenidos por Belfort et al.69 con 45 mg/día de pioglitazona en pacientes con glucemia basal alterada o diabéticos tipo 2. Por su parte, Aithal et al.70 añadieron al estudio sobre pioglitazona la modificación del estilo de vida frente a placebo durante 12 meses, consiguiendo una mejoría considerable de la lesión hepatocelular y de la fibrosis hepática. El inconveniente de estos estudios es que tienen poco seguimiento, por lo que la efectividad de las glitazonas a largo plazo no está establecida. Además, la seguridad de estos fármacos está cuestionada por el incremento del riesgo de cáncer de vejiga y el posible incremento de riesgo cardiovascular.
La vitamina E es un antioxidante que estabiliza los electrones desparejados y previene la peroxidación lipídica71. En modelos animales se ha observado una correlación inversa entre los niveles de vitamina E y la presencia de peroxidación lipídica72. Los estudios son contradictorios, ya que en ensayos clínicos no se ha visto una mejoría clínica o bioquímica en pacientes con cirrosis alcohólica73. El uso de antioxidantes como la N-acetilcisteína, cuya función en el hígado es incrementar los niveles de glutatión, tampoco aportó resultados positivos.
La vitamina E en la esteatohepatitis no alcohólica produjo una mejoría en los niveles de aminotransferasas, esteatosis, balonización e inflamación (aunque no se ha observado efecto sobre la fibrosis hepática). En el estudio PIVENS se administró vitamina E, 800 mg/día, o pioglitazona, 45 mg/día, frente a placebo durante 96 semanas74. El tratamiento con agonistas PPARγ consigue resultados contradictorios, con mejoría de la esteatosis y de la sensibilidad hepática a la insulina pero sin efecto sobre la fibrosis o mejoría de la fibrosis sin cambios en la esteatosis o la resistencia a la insulina. El efecto beneficioso de la vitamina E en pacientes con esteatohepatitis no alcohólica demostrada por biopsia y sin diabetes mellitus tipo 2 queda en un segundo plano debido a los problemas de seguridad a largo plazo. El tratamiento con vitamina E a largo plazo incrementa la mortalidad75. Por último, la N-acetilcisteína en monoterapia en esteatohepatitis alcohólica mejoró los niveles de aminotransferasas pero no la histología hepática76.
En definitiva, tanto la elevada frecuencia de la enfermedad como el conocimiento de su posible evolución a estadios avanzados de fibrosis refuerzan la necesidad de un diagnóstico precoz y un tratamiento individualizado del paciente con enfermedad hepática metabólica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

