




La enfermedad celíaca (EC) es una entidad mediada por fenómenos autoinmunes, que se presenta en sujetos susceptibles genéticamente. El 90% de los pacientes presenta el heterodímero HLA-DQ2, y el 10% restante suele presentar el HLA-DQ8.
ObjetivoEstudiar las características de la EC en la población pediátrica de Cantabria y en sus familiares de primer grado, fundamentalmente en los aspectos relacionados con el haplotipo, la serología y sus formas de presentación clínica.
Pacientes y métodosEstudio de 86 pacientes celíacos y 215 familiares de primer grado. Se recogieron datos clínicos, analíticos, inmunológicos, histológicos y de tipificación genómica.
ResultadosEl 95% de los caso se iniciaron con clínica clásica y el 5% eran formas monosintomáticas. Un 95% presentaba positividad a anticuerpos antigliadina (AAG) y antitransglutaminasa (AATG), y eran negativos el 5% (todos con déficit de IgA). Genotípicamente, un 71% eran portadores del DQ2 (incluidos los homocigotos y los heterocigotos), y un 9,5% del DQ8. Un 22% no presentaba heterodímero de riesgo. En el estudio familiar se hallaron 6 familiares con EC (3 AAG positivos y 4 AATG positivos). Del total, el 49% de los familiares portaba el DQ2, un 15% el DQ8, y un 40% no presentaba el heterodímero de riesgo.
ConclusionesEl HLA más prevalente en nuestra comunidad fue el DQ2 (71%), claramente menor que lo publicado en nuestro medio. La prevalencia de EC en familiares de primer grado fue similar al resto de España (2,8%). Nuestros datos apoyan la necesidad del estudio sistemático en familiars de primer grado de pacientes celíacos.
Celiac disease (CD) is an autoimmune disease that affects genetically predisposed individuals. The HLA-DQ2 heterodimer is present in nearly 90% of patients while HLA-DQ8 is found in the remaining 10%.
AimTo study the characteristics of CD in pediatric patients in Cantabria and their first-degree relatives, with special emphasis on factors related to haplotype, serology, and forms of clinical presentation.
Patients and methodsEighty-six patients with CD and 215 first-degree relatives were HLA genotyped. Clinical, laboratory, immunologic, and histological data were obtained from all patients.
ResultsClinical presentation was classical in 95% of the patients and mono-symptomatic in the remaining 5%. Anti-gliadin antibodies (AGA) and anti-transglutaminase antibodies (ATGA) were positive in 95% of the patients and negative in 5% (all with IgA deficiency). DQ2 was found in 71% of the patients (homozygotes or heterozygotes) and DQ8 was found in 9.5%. No heterodimers of risk were found in 22%. CD was found in six relatives (three were positive for AGA and four were positive for ATGA). Forty-nine percent of the relatives carried the DQ2 heterodimer and 15% the DQ8 heterodimer; no heterodimers of risk were found in 40%.
ConclusionsThe most prevalent HLA found in patients with CD in the autonomous region of Cantabria was DQ2 (71%). This prevalence is clearly lower than that reported in other Spanish regions. The prevalence of CD among firstdegree relatives was similar to that found in other studies performed in Spain (2.8%). Our data support the need for systematic study of the first-degree relatives of patients with CD.
La enfermedad celíaca (EC) es una entidad mediada por fenómenos autoinmunes, que se define como una intolerancia permanente al gluten; se caracteriza por una lesión inflamatoria en el intestino delgado que se produce en sujetos susceptibles genéticamente y con unos factores ambientales propicios1.
Las manifestaciones clínicas más frecuentes son las derivadas del cuadro malabsortivo. Cada vez con más frecuencia se describen formas oligo/monosintomáticas con determinados síntomas, como elevación de las transaminasas, alteración del esmalte dentario, estreñimiento, retraso puberal, artritis, osteoporosis, epilepsia (con o sin calcificaciones intracraneales) y ataxia, entre otras1,2.
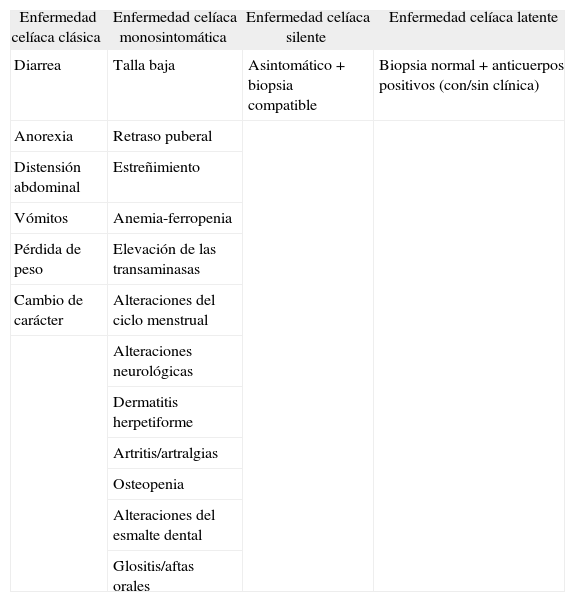
En los últimos años se han descrito formas silentes y latentes de la enfermedad (tabla I), sobre todo detectadas en familiares de primer grado de pacientes celíacos1,3,4. En la forma silente la enfermedad es asintomática, y presenta marcadores serológicos positivos y una biopsia compatible. La forma latente presenta una serología positiva y una biopsia normal que, con el tiempo, evolucionará a una atrofia de vellosidades, con el consiguiente riesgo de desarrollo de la enfermedad1. Con la descripción de estas nuevas formas de EC, su prevalencia en la población europea está en torno al 1%1.
Manifestaciones clínicas y clasificación
| Enfermedad celíaca clásica | Enfermedad celíaca monosintomática | Enfermedad celíaca silente | Enfermedad celíaca latente |
| Diarrea | Talla baja | Asintomático + biopsia compatible | Biopsia normal + anticuerpos positivos (con/sin clínica) |
| Anorexia | Retraso puberal | ||
| Distensión abdominal | Estreñimiento | ||
| Vómitos | Anemia-ferropenia | ||
| Pérdida de peso | Elevación de las transaminasas | ||
| Cambio de carácter | Alteraciones del ciclo menstrual | ||
| Alteraciones neurológicas | |||
| Dermatitis herpetiforme | |||
| Artritis/artralgias | |||
| Osteopenia | |||
| Alteraciones del esmalte dental | |||
| Glositis/aftas orales |
Se sabe que la EC es más frecuente entre los familiares de pacientes, cuyas cifras oscilan entre el 2,6 y el 16%5. Sin embargo, difícilmente estos familiares se diagnostican por las manifestaciones clínicas, ya que el 50% está asintomático o presenta formas atípicas que dificultan su identificación6.
La sospecha diagnóstica debe ser fundamentalmente clínica y reforzarse con los marcadores serológicos. Los pacientes con EC presentan positividad de los anticuerpos antigliadina (AAG), antitransglutaminasa (AATG) y antiendomisio (AAE); estos últimos cada vez se utilizan menos, y han sido desplazados por los AATG, que han demostrado una alta sensibilidad y una especificidad diagnóstica superior al 90%7,8.
Respecto al diagnóstico de certeza, la biopsia intestinal de yeyuno es el patrón de referencia. La biopsia más importante y necesaria es la primera, que se realizará en el momento del diagnóstico de sospecha y antes de la supresión del gluten; puede ser la única en niños mayores de 2 años que presentan una sintomatología clínica clásica, con una respuesta clara a la supresión del gluten9.
Dentro de los factores genéticos, se ha demostrado una fuerte asociación entre esta enfermedad y la molécula HLA de clase II, especialmente en nuestro medio HLADQ2 y HLA-DQ8. En la mayoría de los estudios realizados hasta el momento9–15 el 90% de los pacientes presenta el heterodímero HLA-DQ2, que está codificado por los alelos DQA1*0501 y DQB1*0201. Este heterodímero, a su vez, se asocia en la mayoría de los casos al DR3, y en sujetos heterocigotos al DR7/DR5. El 10% restante suele presentar un segundo heterodímero de riesgo, HLA-DQ8, codificado por los alelos DQA1*0301 y DQB1*0302, asociado en este caso al DR4. Algunos pacientes no son portadores de estos heterodímeros de riesgo, pero se ha observado que al menos presentan uno de los dos alelos (en la mayoría de los casos uno de los alelos del DQ2, el DQA1*0501 o el DQB1*0201); se ha descrito un mínimo porcentaje de pacientes con ambos alelos ausentes1.
Aproximadamente el 30-40% de la población general presenta el heterodímero DQ2, por lo que su presencia no es condición de riesgo suficiente para padecer la enfermedad y sugiere que pueden estar implicados otros genes, tanto no DQ como no HLA14.
PACIENTES Y MÉTODOSHemos realizado un estudio retrospectivo de 86 pacientes menores de 15 años con EC confirmada, controlados en las consultas de gastroenterología infantil del Hospital Universitario Marqués de Valdecilla, de Cantabria, y 215 familiares de primer grado (78 padres, 82 madres y 55 hermanos). En todos ellos se hizo un estudio clínico, serológico y genómico. En los casos sugestivos de posible EC se realizó una biopsia intestinal.
De todos los sujetos incluidos en el estudio se revisaron las historias clínicas, recogiéndose los siguientes datos en el momento del diagnóstico: datos clínicos, parámetros analíticos (hemoglobina, hematocrito, hierro, ferritina, transaminasas, glucosa y colesterol) estudio inmunológico, histológico y tipificación genómica (HLA tipo II: DRB1, DQB1 y DQA1). La determinación de la concentración sérica de IgA total se realizó mediante nefelometría (Dade Behring, Marburg, Alemania). Los AAG y AATG se estudiaron mediante ELISA comerciales (Biosystems, Barcelona y Binding Site, Birmingham, Reino Unido). Se consideraron títulos positivos de AAG y AATG unas cifras superiores a 20 UA/l (unidades arbitrarias/ litro) y a 25 U/ml (unidades titulación/mililitro), respectivamente.
En caso de deficiencia selectiva de IgA, se estudió el isotipo IgG de los autoanticuerpos, mediante el empleo de un conjugado específico.
La tipificación HLA se realizó a partir de ADN genómico extraído de células mononucleares sanguíneas mediante DNAzol. Los alelos HLA DRB1, DQA1 y DQB1 se detectaron mediante reacción en cadena de la polimerasa (PCR)-SSO dot blot inverso, PCR-SSP y PCR-RFLP, respectivamente. La biopsia se realizó mediante cápsula de Crosby según la técnica habitual; se extrajeron 2 muestras, y se procesaron posteriormente para su estudio histológico, considerándose compatible con el diagnóstico de EC la que cumplía criterios de MARSH IIIa, IIIb o IIIc9.
RESULTADOSEstudio de los pacientesFormas de presentación clínicaDe los 86 pacientes seleccionados en el estudio, un 66% correspondía a mujeres (57 pacientes) y un 34%, a varones (29 pacientes). El inicio de los síntomas estaba entre los 7 meses y los 6 años de edad, con una media de edad de 14,93 ± 8,46 meses. El rango de edad en el momento del diagnóstico estaba entre los 10 meses y los 6 años y 10 meses, con una media de 22,39 ± 12,84 meses.
El 95% de los sujetos (82 pacientes) presentaba clínica malabsortiva, y el resto eran formas monosintomáticas (3 pacientes): una de ellas se detectó en el contexto de una tiroiditis al realizarse el cribado de autoinmunidad; los otros 2 casos se detectaron durante el estudio de una anemia ferropénica recurrente tras el tratamiento con suplementos de hierro. Un 2,3% (2 pacientes) presentaba síndrome de Down. Dado que es una entidad mediada por fenómenos autoinmunes, se buscaron otras enfermedades asociadas, y se detectaron en un 3,5% del total de casos estudiados otras enfermedades autoinmunes asociadas (3 pacientes): tiroiditis, diabetes mellitus y miastenia gravis.
Estudios serológicosSe detectaron 4 casos con déficit selectivo de IgA (4,6%). Los AAG se analizaron en 82 pacientes, y fueron positivos en el 95% de los casos (78 pacientes), el 5% restante que presentaba negatividad en los anticuerpos correspondía a los 4 pacientes con déficit selectivo de IgA; de estos 4 casos, todos presentaban positividad para el isotipo IgG. En el caso de los AATG, se disponía de muestras de 56 pacientes, con positividad en 51 de los niños estudiados (91%). El 9% de los pacientes con AATG negativos correspondía a 4 casos con déficit selectivo de IgA y uno con valores totales de IgA dentro de la normalidad. También en este caso todos los pacientes con déficit de IgA eran positivos para el isotipo IgG.
Estudio anatomopatológicoSe revisaron 78 biopsias duodenales, un 80% (62 pacientes) se catalogaron como atrofia subtotal (Marsh IIIb) y un 20% (16 pacientes), como atrofia parcial severa (Marsh IIIa). De los 86 pacientes, en 4 casos no se pudo realizar una biopsia, dado el deterioro de su estado general, y en otros 4 casos la biopsia fue fallida y no valorable. Hemos incluido estos casos dentro del estudio, ya que normalizaron la sintomatología clínica tras la retirada del gluten, los resultados eran normales en la histología intestinal de la segunda biopsia y todos presentaron una recaída clínica e histológica tras la sobrecarga con gluten.
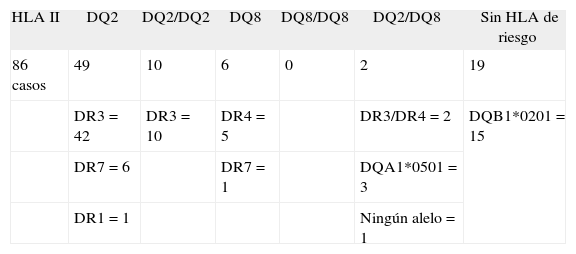
Estudio HLADentro de la tipificación genómica se analizó el HLA de clase II de los 86 pacientes (tabla II). Un 57% eran heterocigotos DQ2, un 11,6%, DQ2-DQ2, un 7%, DQ8, un 2,3%, DQ2-DQ8, y no se encontró ningún caso que fuera DQ8- DQ8. Un porcentaje elevado (22%) no era portador de HLA de riesgo. Del total de casos sin haplotipo HLA de riesgo, un 79% portaba el alelo DQB1*0201, un 16% el DQA1*0501 y sólo un 5% tenía ambos alelos de riesgo ausentes.
Distribución de alelos HLA en los pacientes celíacos
| HLA II | DQ2 | DQ2/DQ2 | DQ8 | DQ8/DQ8 | DQ2/DQ8 | Sin HLA de riesgo |
| 86 casos | 49 | 10 | 6 | 0 | 2 | 19 |
| DR3 = 42 | DR3 = 10 | DR4 = 5 | DR3/DR4 = 2 | DQB1*0201 = 15 | ||
| DR7 = 6 | DR7 = 1 | DQA1*0501 = 3 | ||||
| DR1 = 1 | Ningún alelo = 1 |
DQ2: DQA1*0501 + DQB1*0201.
DQ8: DQA1*0301 + DQB1*0302.
En todos los pacientes se analizó también el alelo DRB1, y se encontró la siguiente asociación (tabla II): de los heterocigotos DQ2, el 86% se asociaba a DR3, un 12%, al DR7, y un 2%, al DR1; entre los homocigotos DQ2-DQ2, el 100% se asociaba a DR3-DR3; entre los heterocigotos DQ8, un 85% estaba asociado al DR4 y un 15% al DR7, y entre los heterocigotos DQ2-DQ8, el 100% era DR3-DR4.
Estudio de familiaresEstudio serológicoSe encontró positividad a AAG tipo IgA en 9 de los 215 familiares (4,2%). Del 95,8% restante con AAG negativos, 2 de ellos correspondían a familiares con déficit de IgA. En ellos se determinó el isotipo de anticuerpos IgG, que fue positivo en los 2 sujetos. Los AATG IgA fueron positivos en 11 (5,1%) de los 215 casos, y entre los que presentaban negatividad a éstos se encontraban también los 2 familiares con déficit selectivo de IgA referidos previamente, en los que la detección de los AATG tipo IgG fue positiva. Así, si unificamos a todos los pacientes positivos (IgA + IgG), encontramos 11 familiares (5,1%) con AAG y 13 familiares (6%) con AATG.
Por último, en 2 familiares (incluido uno dentro de los 9 con positividad para AAG y el otro dentro de los 11 con positividad para los AATG) se detectó simultáneamente positividad para ambos anticuerpos, AAG y AATG tipo IgA, y ambos se encontraban asintomáticos.
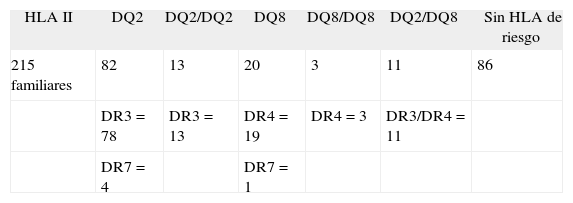
Estudio HLALa tipificación genómica (tabla III) detectó 82 familiares heterocigotos para el HLA-DQ2 (38,1%), de los cuales 4 se asociaban al DR7 y 78, al DR3. Trece familiares (6%) eran homocigotos DQ2-DQ2, todos asociados al DR3- DR3; se encontraron 20 familiares (9,3%) heterocigotos DQ8, 19 asociados al DR4 y 1 al DR7; 3 (1,4%) eran homocigotos DQ8-DQ8, todos ellos asociados al DR4-DR4; 11 familiares (5,1%) eran heterocigotos DQ2-DQ8, todos asociados al DR3-DR4; por último, 86 (40%) familiares no eran portadores de ningún heterodímero de riesgo.
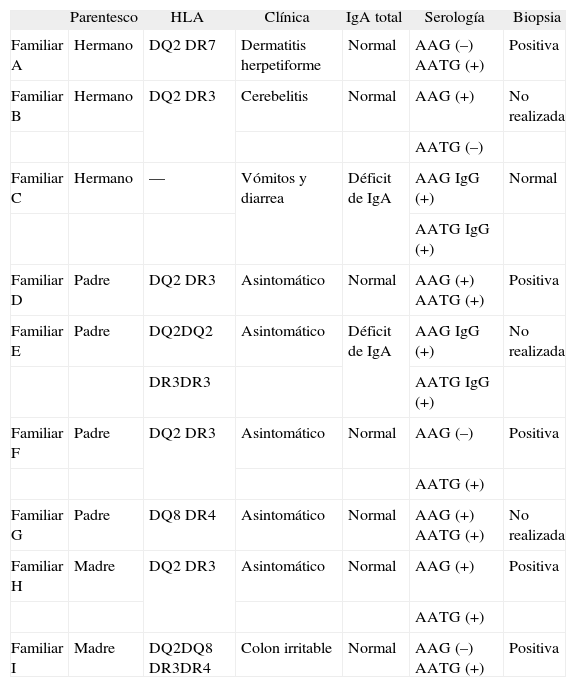
ClínicaLos resultados obtenidos del análisis de la sintomatología clínica de los familiares se recogen en la tabla IV. Clínicamente, presentaron algún tipo de síntoma 4 familiares (3 hermanos y una madre) de los 215 casos: un hermano presentaba una lesión cutánea y fue diagnosticado de dermatitis herpetiforme; un hermano con ataxia cerebelosa fue diagnosticado de cerebelitis; un hermano con cuadro de vómitos y diarrea de pocos meses de evolución y una madre con alteraciones gastrointestinales inespecíficas fueron diagnosticados de síndrome del intestino irritable en la infancia. De estos 4 familiares con clínica, 2 tenían los AATG positivos (> 100 U) y uno los AAG positivos. En el cuarto familiar con síntomas se detectó un déficit selectivo de IgA, con AATG y AAG de clase IgG positivos. Se diagnosticaron 6 casos de EC (familiares A, B, D, F, H e I) de 215 familiares estudiados, lo que supone una prevalencia del 2,8%. En el caso del familiar B no se practicó una biopsia intestinal por negativa de la familia, y el gluten de la dieta se retiró de forma unilateral por parte de los padres. Hemos incluido este familiar dentro del grupo de pacientes celíacos, ya que los datos clínicos que presentaba, junto con la mejoría éstos tras la supresión del gluten de la dieta y la negativización de los anticuerpos, orientan claramente el diagnóstico. En 2 casos (familiares E y G) no se pudo confirmar el diagnóstico porque, a pesar de tener HLA y serología compatibles, se negaron a la realización de una biopsia. En la actualidad realizan controles clínicos evolutivos, siguen una dieta normal y se encuentran asintomáticos.
Resultados obtenidos del estudio de los familiares con serología sospecha
| Parentesco | HLA | Clínica | IgA total | Serología | Biopsia | |
| Familiar A | Hermano | DQ2 DR7 | Dermatitis herpetiforme | Normal | AAG (–) AATG (+) | Positiva |
| Familiar B | Hermano | DQ2 DR3 | Cerebelitis | Normal | AAG (+) | No realizada |
| AATG (–) | ||||||
| Familiar C | Hermano | — | Vómitos y diarrea | Déficit de IgA | AAG IgG (+) | Normal |
| AATG IgG (+) | ||||||
| Familiar D | Padre | DQ2 DR3 | Asintomático | Normal | AAG (+) AATG (+) | Positiva |
| Familiar E | Padre | DQ2DQ2 | Asintomático | Déficit de IgA | AAG IgG (+) | No realizada |
| DR3DR3 | AATG IgG (+) | |||||
| Familiar F | Padre | DQ2 DR3 | Asintomático | Normal | AAG (–) | Positiva |
| AATG (+) | ||||||
| Familiar G | Padre | DQ8 DR4 | Asintomático | Normal | AAG (+) AATG (+) | No realizada |
| Familiar H | Madre | DQ2 DR3 | Asintomático | Normal | AAG (+) | Positiva |
| AATG (+) | ||||||
| Familiar I | Madre | DQ2DQ8 DR3DR4 | Colon irritable | Normal | AAG (–) AATG (+) | Positiva |
Ig: inmunoglobulina.
El caso del familiar C, que presentaba anticuerpos de clase IgG positivos con biopsia normal (indicativo de una EC latente), no lo hemos incluido entre los 6 con casos con EC, ya que está siendo objeto de seguimiento clínico en la actualidad.
Por último, los 6 nuevos casos diagnosticados siguen actualmente una dieta sin gluten, con una evolución favorable de los títulos de anticuerpos y la clínica.
DISCUSIÓNLos datos obtenidos del estudio de nuestros pacientes confirman la mayor prevalencia de casos en niñas que en niños, de manera similar a lo registrado en la literatura médica1. De los 3 pacientes con formas de presentación oligosintomáticas, 2 de ellos (2,3%) presentaban anemia ferropénica recurrente, cifra muy inferior a lo registrado en otras series, ya que la prevalencia de EC en este grupo varía entre un 10 y un 15% en los pacientes que además asociaban algún síntoma gastrointestinal16, y hasta un 30% en los que únicamente presentaban una anemia ferropénica17. El tercero, con tiroiditis autoinmune, fue diagnosticado de EC en el curso del estudio de su enfermedad de base. La tiroiditis puede considerarse más como una enfermedad asociada que como una manifestación oligosintomática en sí misma. Este tipo de enfermedades asociadas fueron inicialmente descritas sobre todo en los adultos, pero los avances en los conocimientos sobre ella ha hecho posible detectarlas en la infancia18. Sin embargo, nuestros datos difieren de lo reflejado por otros autores, que cifran la presencia de estas enfermedades asociadas entre un 10%1 y un 15%19. Quizá la diferencia puede estar en que ellos observan un desplazamiento de los grupos de pacientes hacia edades mas tardías que las encontradas en nuestra serie.
Varias enfermedades de base genética se han asociado a la EC (síndrome de Down, síndrome de Turner, síndrome de Williams, déficit selectivo de IgA)20, y entre todas ellas el síndrome de Down ha sido el más estudiado. Un 2,5% de nuestros pacientes presentó síndrome de Down, cifra que contrasta con el 6,6% publicado en otras áreas de nuestro país21, pero es más cercana al 3,2% de Estados Unidos22 o al 3,9% de Suecia23.
En todos nuestros pacientes se realizó un estudio de HLA, y se halló una prevalencia de DQ2 en el 71%, cifra muy inferior a la referida en grupos de pacientes celíacos de nuestro país (93%)12 y en series europeas (86-94%)24. Éste es un dato que cabe destacar, dado que la población estudiada fue homogénea y en nuestro entorno no hay diferencias étnicas destacables.
En el estudio de los familiares encontramos que el 2,8% presentaba una EC, cifra idéntica a la observada en el País Vasco en un grupo de 642 familiares de primer grado25; en otras series de familiares estas cifras oscilan entre el 2,6 y el 16%5. Se ha sugerido que esta amplitud de rango podría atribuirse a errores en la selección de las familias, a una relación con el caso índice, a criterios diagnósticos o al diseño del estudio16. Incluso es posible que la edad de las personas estudiadas pueda considerarse un factor determinante, ya que se ha observado en varios estudios prospectivos que los marcadores serológicos e histológicos de EC pueden desarrollarse en personas predispuestas genéticamente tras haberse demostrado su negatividad en estudios previos12. Esto corrobora los hallazgos de nuestra serie, en la que el número de adultos es el doble que el de niños detectados. Este hecho puede sugerir la existencia de una correlación entre la duración de la exposición al gluten y el desarrollo de una respuesta inmune en sujetos predispuestos genéticamente. Aunque, por otro lado, se ha sugerido que es la edad del paciente en sí y no el tiempo de exposición al gluten el factor que condiciona la aparición de autoinmunidad26. Una prevalencia entre adultos/niños similar a la nuestra se ha descrito en un amplio estudio multicéntrico llevado a cabo en Estados Unidos27.
Hemos encontrado que el 5,1% de los familiares estudiados presentaba AAG (IgA/IgG) positivos, cifra muy similar al 6% encontrada por Corazza et al28 en su estudio de 328 familiares. Estos datos contrastan con los referidos por otros grupos en nuestro país, que reflejan cifras del 10- 12,5% de familiares con AAG de clase IgA25,29, y claramente superiores a los referido por Farré et al12 en su estudio de 669 familiares, que encontraron sólo un 1,9% de ellos con positividad a los mismos isotipos de dichos anticuerpos. En el único estudio publicado en este sentido en Cantabria autónoma, realizado en población adulta5, se encuentra un 3,8% de familiares con anticuerpos antigliadina (IgA/IgG) positivos. La explicación para estas variaciones en las cifras no está clara, y puede deberse a que los familiares de primer grado tienen una mayor predisposición a la producción de estos anticuerpos30. Complementamos el estudio con la determinación de los AATG, que han demostrado tener por sí solos un valor predictivo positivo del 75-80% en relación con la confirmación por biopsia de la EC31. Sin embargo, hemos encontrado muy pocos estudios en familiares utilizando estos anticuerpos como técnica de cribado. En nuestra serie un 6% de familiares (13 casos) presentaba AATG (IgA/IgG). Otros autores han referido cifras muy discordantes, como las recogidas en el estudio de Mustalahti et al32, en el que un 12,9% de 466 familiares estudiados presentaban positividad de estos anticuerpos (IgA); en Cantabria, en el estudio realizado en adultos, referido previamente5, se encontraron AATG (IgA/IgG) positivos sólo en un 2,7% de los familiares estudiados.
Si analizamos los 6 casos de enfermedad celíaca detectados en nuestro estudio, 5 de ellos presentaban AATG positivos (IgA/IgG), todos de clase IgA, en títulos superiores a 100 U/ml. Estos datos confirman la eficacia de este marcador para detectar la enfermedad y apoyan su uso como técnica de cribado muy útil en nuestro medio. De hecho, recientemente se ha postulado que las cifras superiores a 100 U/ml son muy indicativas de una EC, y en determinadas circunstancias incluso podrían evitar la realización de una biopsia intestinal para confirmar la sospecha diagnóstica de EC7.
En nuestro estudio la determinación del HLA en los familiares refleja una prevalencia de DQ2 del 49,2%, ligeramente superior al 39,5% que presenta la población general, pero muy alejadas del 64% referido en Cataluña por Farré et al12. Sin embargo, y en concordancia con estos últimos autores, en todos los familiares con EC de nuestro estudio encontramos la presencia de DQ2. Por último, cabe destacar que hemos encontrado otro haplotipo de riesgo, el DR3, en todos los pacientes diagnosticados de EC. Esto se debe a que el DR3 está en desequilibrio de ligamiento con el DQ2. Del conjunto de los 6 casos diagnosticados, 3 de ellos presentaban algún tipo de sintomatología, en concreto uno con clínica gastrointestinal. El único adulto sintomático había sido diagnosticado de síndrome de intestino irritable. Algunos estudios recientes sugieren la necesidad de un cribado de la EC en este grupo de pacientes, ya que hasta un 5% de los pacientes que cumplen los criterios Roma II para el diagnóstico de colon irritable presentaba una EC de base33. El hecho de que en nuestra serie la mayoría de los pacientes sintomáticos detectados entre los familiares sean niños apoya lo publicado, en relación con la mayor presencia de sintomatología en la infancia y la posibilidad de permanecer indetectable hasta la edad adulta en muchos casos27. El familiar E está en la actualidad en seguimiento, ya que la presencia de DQ2 en homocigosis junto con el déficit de IgA (altamente prevalente en la EC) y la presencia de AAG y AATG (clase IgG) orientan claramente el diagnóstico de EC. No obstante, debido a que se encuentra asintomático y a la negativa de éste a someterse a la realización de una biopsia intestinal, es imposible, por el momento, confirmar la sospecha diagnóstica.
En la actualidad no hay evidencias de que la realización de un cribado de la EC en la población general sea útil; pero sí se ha demostrado su utilidad cuando se realiza en poblaciones de riesgo (familiares de primer grado, diabetes mellitus tipo 1, anemia ferropénica, etc.), tanto en adultos como en niños, si bien en estos últimos se recomienda realizarlo en edades superiores los 3 años de edad, ya que en edades más tempranas los resultados son poco representativos25,31. En este sentido, la técnica ideal para el cribado de la EC –aspecto confirmado además por nuestros datos– es la detección de autoanticuerpos, especialmente los AATG, por su alta sensibilidad y especificidad. El estudio de HLA es una ayuda importante para determinar los grupos de riesgo genético, pero no sirve para definir la enfermedad.
CONFLICTO DE INTERESESLos autores declaran no tener ningún conflicto de intereses.

