






INTRODUCCIÓN
La esteatohepatitis no alcohólica (EHNA) debe considerarse integrada en una entidad de espectro más amplio, el hígado graso no alcohólico (HGNA), que abarca desde la esteatosis hepática como forma inicial, pasando por la EHNA propiamente dicha, hasta la cirrosis hepática, a menudo filiada como criptogenética por haber perdido sus características específicas, en el otro extremo del espectro. La EHNA es similar histológicamente a la hepatitis alcohólica y se caracteriza por esteatosis macrovesicular, necroinflamación, degeneración balonizante de los hepatocitos y fibrosis. Desde que fue descrita por Ludwig et al1 en 1980, el impacto epidemiológico, así como el número de publicaciones recientes sobre esta entidad, ha sido enorme2-5; la prevalencia de HGNA en la población general se estima en el 20%, y la de EHNA, en un 2-3%, por lo que el HGNA sería potencialmente la hepatopatía más común en países desarrollados6-8, y puede considerarse la manifestación hepática del síndrome metabólico. En la tabla I se mencionan las entidades relacionadas con la aparición del EHNA.

PATOGENIA
Los mecanismos patogénicos de la EHNA aún no están bien definidos, de modo que algunos de ellos son meras hipótesis. No obstante, hoy día tiende a aceptarse la teoría del «doble impacto»: en el «primer impacto», en el que la resistencia a la insulina desempeña un papel primordial, predominaría el depósito de ácidos grasos y triglicéridos en los hepatocitos, lo que daría lugar a la esteatosis, la cual no está siempre quiescente, pues los hepatocitos cargados de lípidos podrían actuar como reservorios susceptibles a un «segundo impacto», en el que intervendrían factores adicionales que condicionarían estrés oxidativo y peroxidación lipídica, y que en algunos pacientes ocasionarían las lesiones típicas de la EHNA. Es importante recalcar que hay una amplia variabilidad individual entre los pacientes con HGNA, pues unos sólo desarrollan esteatosis, algunos EHNA, y otros cirrosis e incluso hepatocarcinoma. Por ello se necesitan estudios para comprender mejor los factores nutricionales, ambientales y genéticos que pueden modular la susceptibilidad a enfermar de estos pacientes.
Primer impacto
Resistencia a la insulina. La resistencia a la insulina parece ser un factor patogénico clave y reproducible en la EHNA9-11. Se define como la disminución de la capacidad de la insulina para ejercer sus funciones biológicas en tejidos diana típicos como el músculo esquelético, el hígado o el tejido adiposo. Este concepto se engloba dentro del denominado síndrome metabólico o síndrome X, en el que se asocian varias entidades clínicas, como la obesidad, diabetes mellitus, hiperlipidemia e hipertensión arterial, que comportan un mayor riesgo de enfermedad cardiovascular.
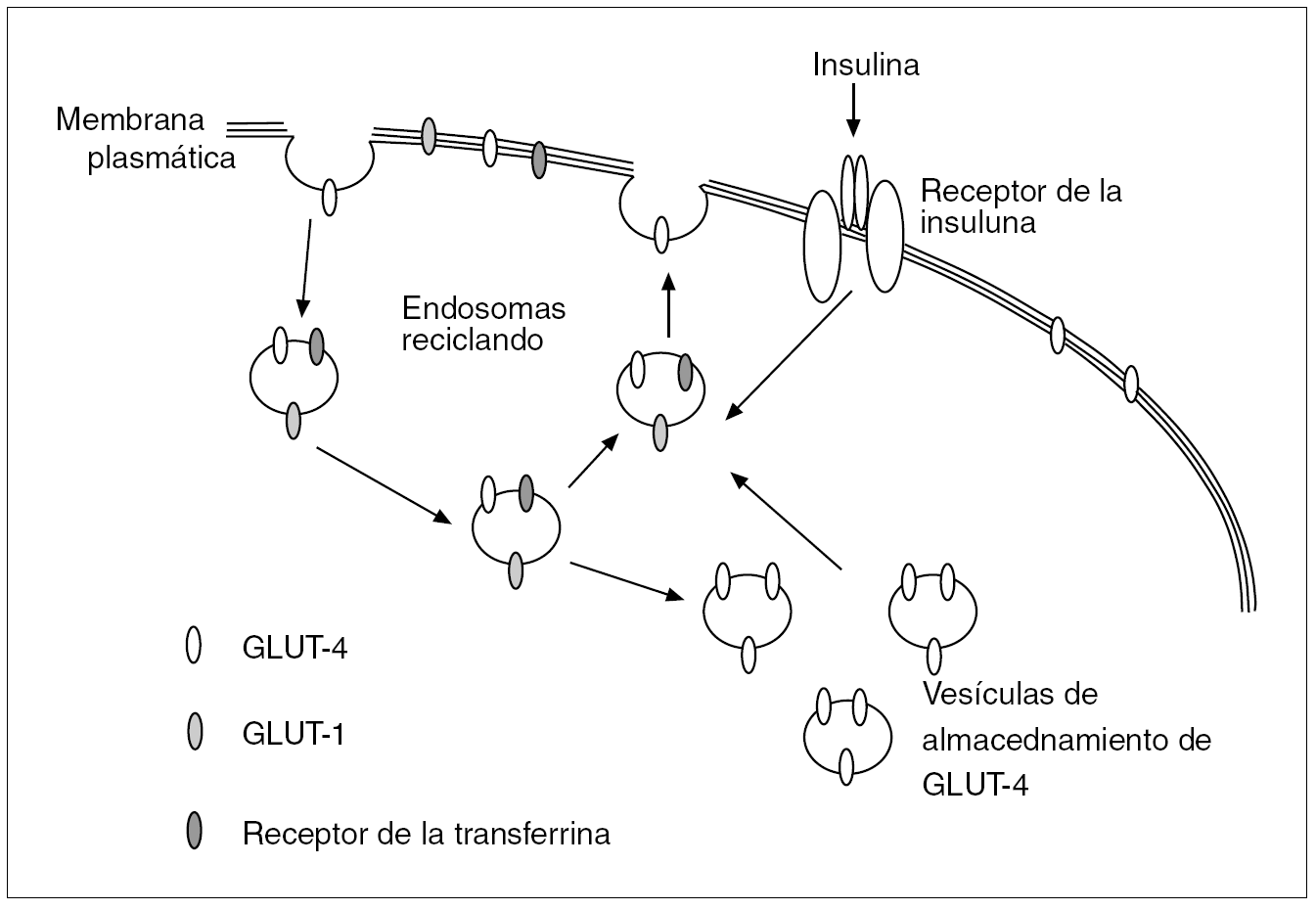
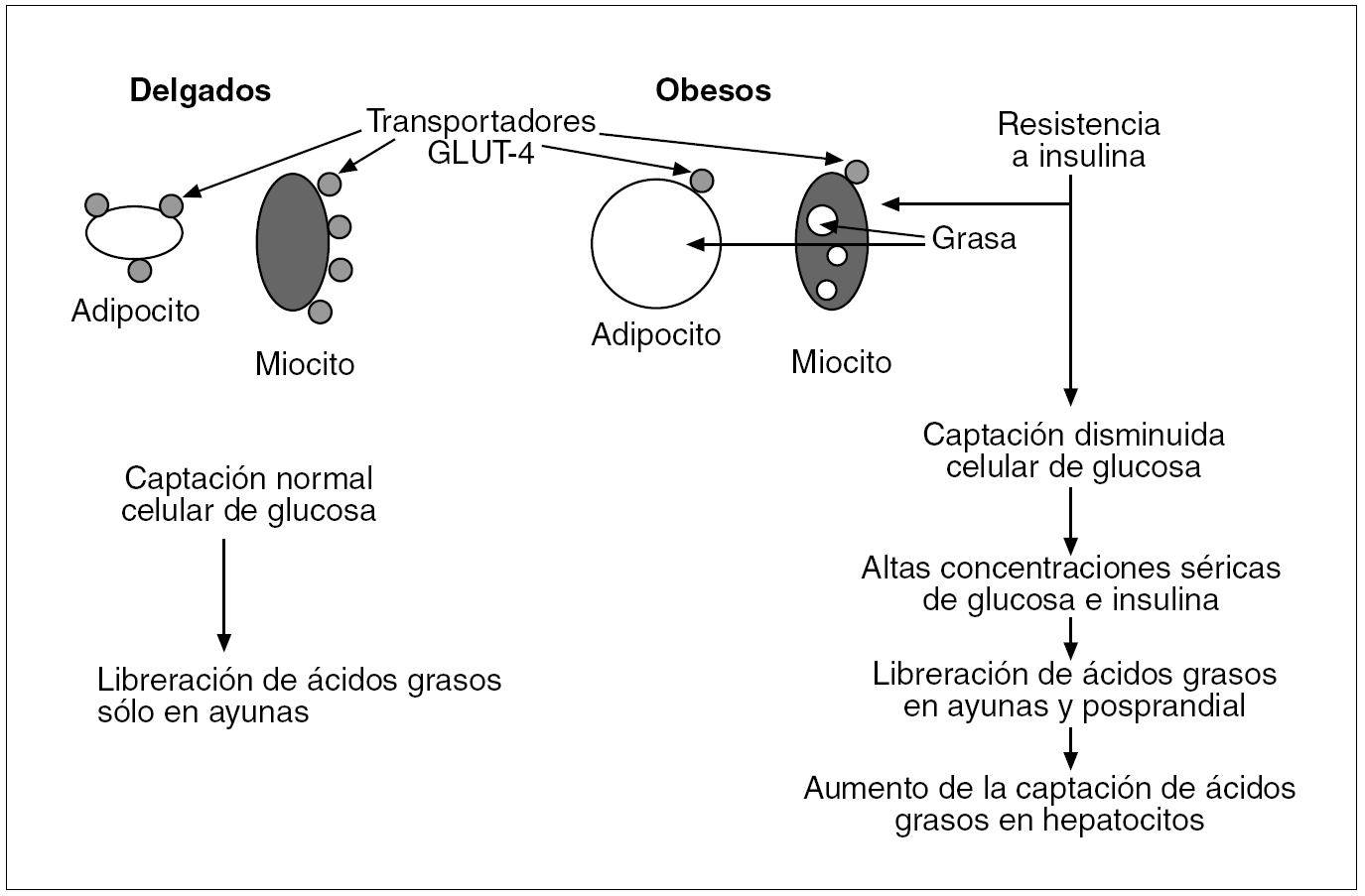
El receptor de la insulina está constituido por 2 subunidades alfa de localización extracelular donde se acopla la insulina, y 2 subunidades beta que penetran a través de la membrana celular en el citoplasma. Actúa como una tirosincinasa que induce, al acoplársele la insulina, una fosforilización de varias proteínas intracelulares denominadas sustratos del receptor de la insulina (IRS), de los cuales el mejor estudiado es el ISR-1, que induce la translocación del transportador de glucosa GLUT-4 a la membrana celular y así incrementa la captación de glucosa. El transporte intracelular de glucosa estimulado por la insulina está pues regido preferentemente por la translocación del GLUT-4 a la membrana celular desde las vesículas de almacenamiento o desde los endosomas del interior de la célula; en estos endosomas también se localizan los receptores de la transferrina y el GLUT-1. La insulina a su vez aumenta la frecuencia del reciclaje del sistema endosómico12 (fig. 1). Se ha observado una expresión disminuida del gen de GLUT-4 en la membrana plasmática de los adipocitos y miocitos de obesos que causa una insuficiente captación celular de glucosa y unas concentraciones altas de glucosa/insulina, reflejo de la resistencia a la insulina en estos pacientes (fig. 2).
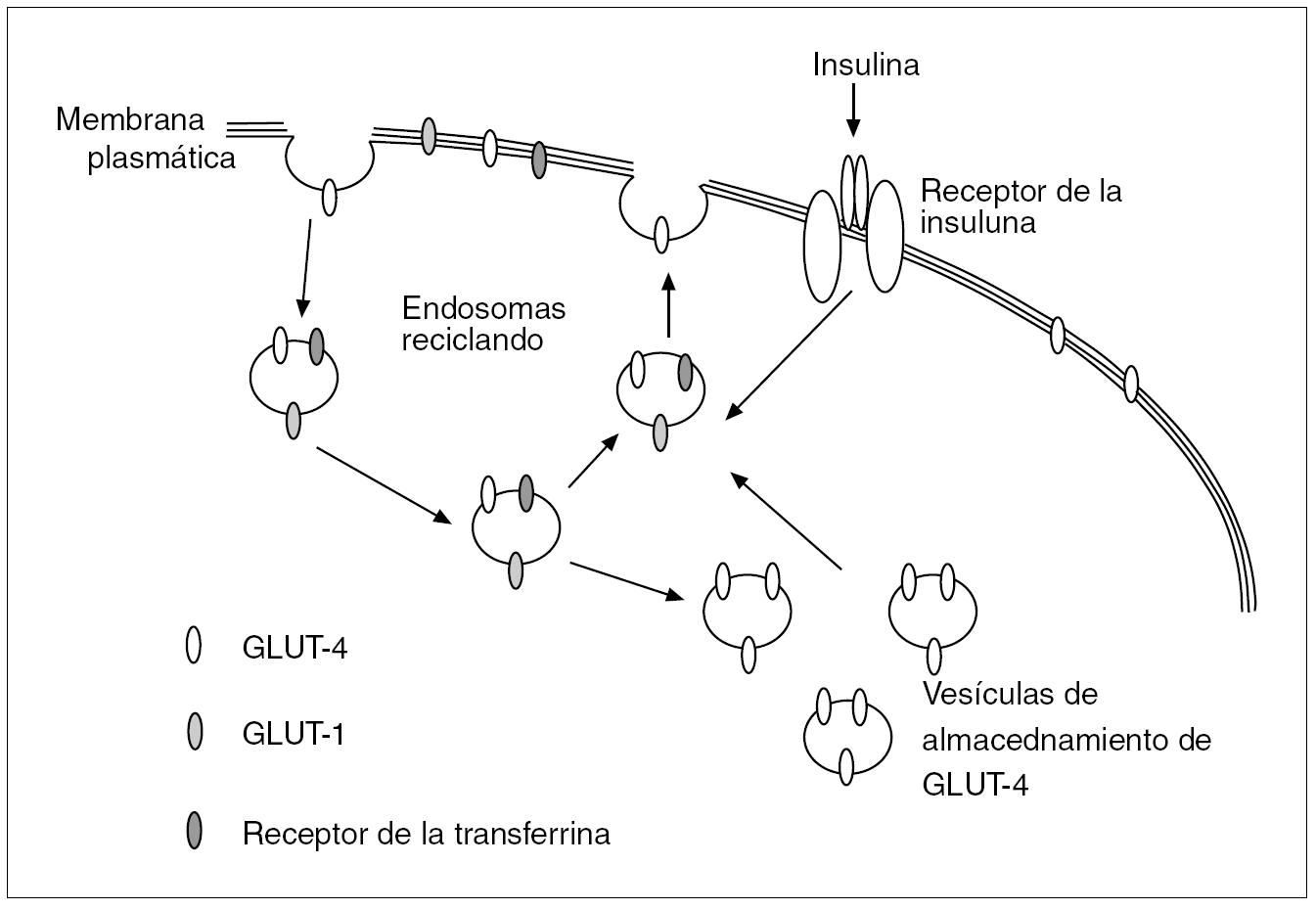
Fig. 1. Posible modelo que sirve para estimular la translocación del transportador de glucosa GLUT-4 y la captación celular de glucosa en adipocitos y miocitos. El GLUT-4 se encuentra en vesículas de almacenamiento o reciclando en endosomas; en estos últimos también se localizan los receptores de la transferrina y GLUT-1. La insulina estimula a las vesículas de almacenamiento a moverse hacia la superficie celular y allí aumentar la captación de glucosa. Asimismo aumenta la frecuencia del reciclaje del sistema endosómico. En la resistencia a la insulina, la escasez de transportadores GLUT-4 en la membrana plasmática ocasiona una captación insuficiente de glucosa y unas concentraciones séricas elevadas de glucosa e insulina.
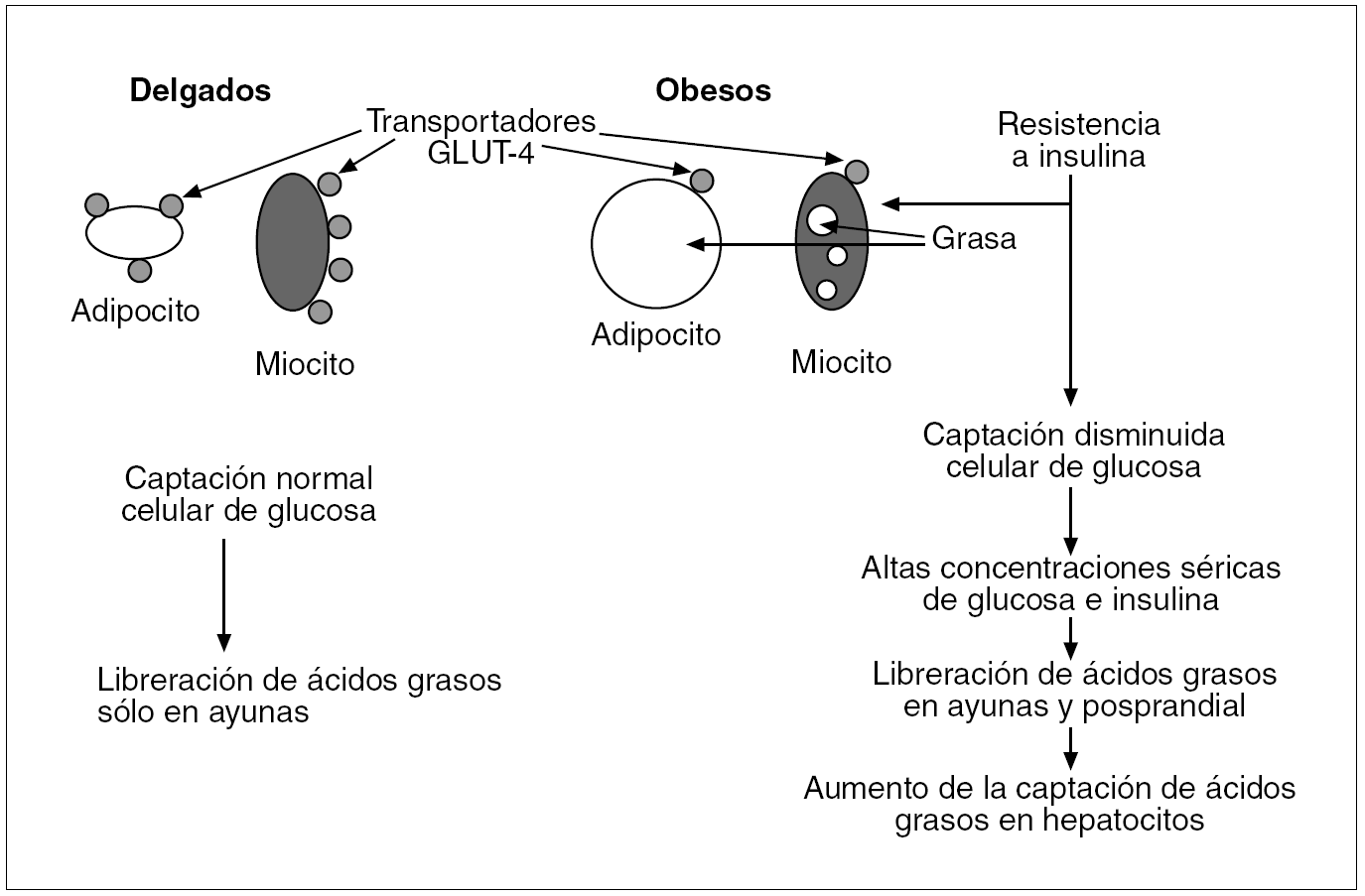
Fig. 2. En pacientes obesos los adipocitos agrandados y los miocitos cargados de grasa reflejan la resistencia a la insulina. La escasez de transportadores de la glucosa GLUT-4 en la membrana plasmática de estos pacientes ocasiona una captación insuficiente de glucosa y unas concentraciones séricas elevadas de glucosa/insulina.
Al menos 2 mecanismos contribuyen a una mayor resistencia a la insulina:
1. El estímulo crónico de la enzima IKK-β (inhibitor kappa beta kinase) promueve la activación del factor nuclear kappa-beta (NF-kβ), un factor de transcripción relacionado con la producción de citocinas inflamatorias, predominantemente el factor de necrosis tumoral alfa (TNF-α), mediante cambios en la fosforilación de la serina en lugar de tirosina en el ISR-1, lo que altera la señal intracelular ocasionada por el acoplamiento de la insulina a su receptor13; este NF-κβ se encuentra normalmente sintetizado y mantenido en forma inactiva en el citoplasma celular debido a que se halla unido a la proteína IKK14. Las concentraciones altas de ácidos grasos15 y la ulterior formación de radicales libres de oxígeno (RLO) tienen la capacidad, a concentraciones adecuadas, de activar la enzima IKK-β, que desacoplará al NF-κβ de la IKK y, por tanto, el factor quedará libre para migrar al núcleo e iniciar procesos de transcripción, lo que dará lugar a la formación de TNF-α. Dado que el TNF-α también activa la IKK-β, se inicia entonces un proceso de retroalimentación que perpetúa la resistencia a la insulina y contribuye a la patogenia del HGNA (fig. 3).
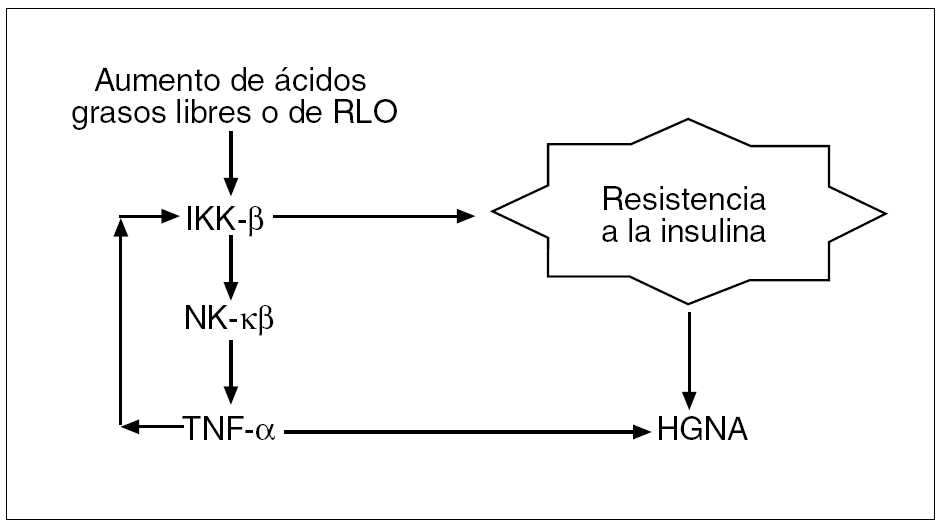
Fig. 3. La esteatosis hepática está vinculada con la resistencia a la insulina por la enzima IKK-β (inhibitor kappa beta kinase), que induce la activación del factor nuclear kappa-beta (NF-κβ). Esto da lugar a la transcripción de una citocina proinflamatoria, el factor de necrosis tumoral alfa (TNF-α), que al activar a su vez a la IKK-β inicia un proceso de retroalimentación que perpetúa la resistencia a la insulina y la producción de TNF-α.RLO: radicales libres de oxígeno; HGNA: hígado graso no alcohólico.
2. Aunque se ha postulado una disminución del aclaramiento hepático de insulina en el HGNA, recientemente se ha comprobado que la hiperinsulinemia es primordialmente secundaria a un aumento de su producción pancreática por la sensibilidad reducida a esta hormona16.
Actualmente se acepta que los adipocitos pueden tener un papel central en el desarrollo tanto de la resistencia a la insulina como del HGNA. El adipocito parece ser un importante órgano endocrino que puede desencadenar un proceso inflamatorio que facilite la evolución a EHNA, al ser capaz de secretar un conjunto de sustancias potencialmente tóxicas, tales como el TNF-α, la leptina y la resistina, además de los ácidos grasos, cuya concentración se relaciona con la resistencia a la insulina. Por tanto, la obesidad debe considerarse una entidad proinflamatoria, y el hígado de estos pacientes se halla expuesto a las citocinas producidas en el tejido adiposo.
Además se ha observado que la grasa visceral (no la masa de grasa corporal total) es un factor predictor de esteatosis hepática y de resistencia a la insulina, y que puede deberse a un acceso más directo de los ácidos grasos a través de la porta al hígado desde el tejido adiposo visceral y central, así como a una menor secreción de leptina17.
Mientras que los adipocitos de las personas delgadas liberan ácidos grasos durante el ayuno y los almacenan tras la ingesta, los adipocitos de los obesos siguen liberando ácidos grasos tras la ingesta, lo cual causa una liberación sostenida de dichos ácidos en el plasma18 (fig. 2).
Durante la digestión los enterocitos convierten a los triglicéridos de la dieta en quilomicrones, que serán transportados por la linfa y posteriormente hidrolizados en ácidos grasos por la lipoproteinlipasa, localizada en el endotelio capilar de los tejidos adiposo y hepático. Los ácidos grasos libres así generados son muy miscibles con las membranas celulares, de modo que se dirigen de inmediato a los adipocitos, donde se esterifican para convertirse en triglicéridos, o bien penetran en el hígado para ser oxidados y generar adenosintrifosfato (ATP), ser esterificados y almacenados como triglicéridos en el citosol, o incorporarse a las lipoproteínas de muy baja densidad (VLDL) y ser exportados del hígado (fig. 4).
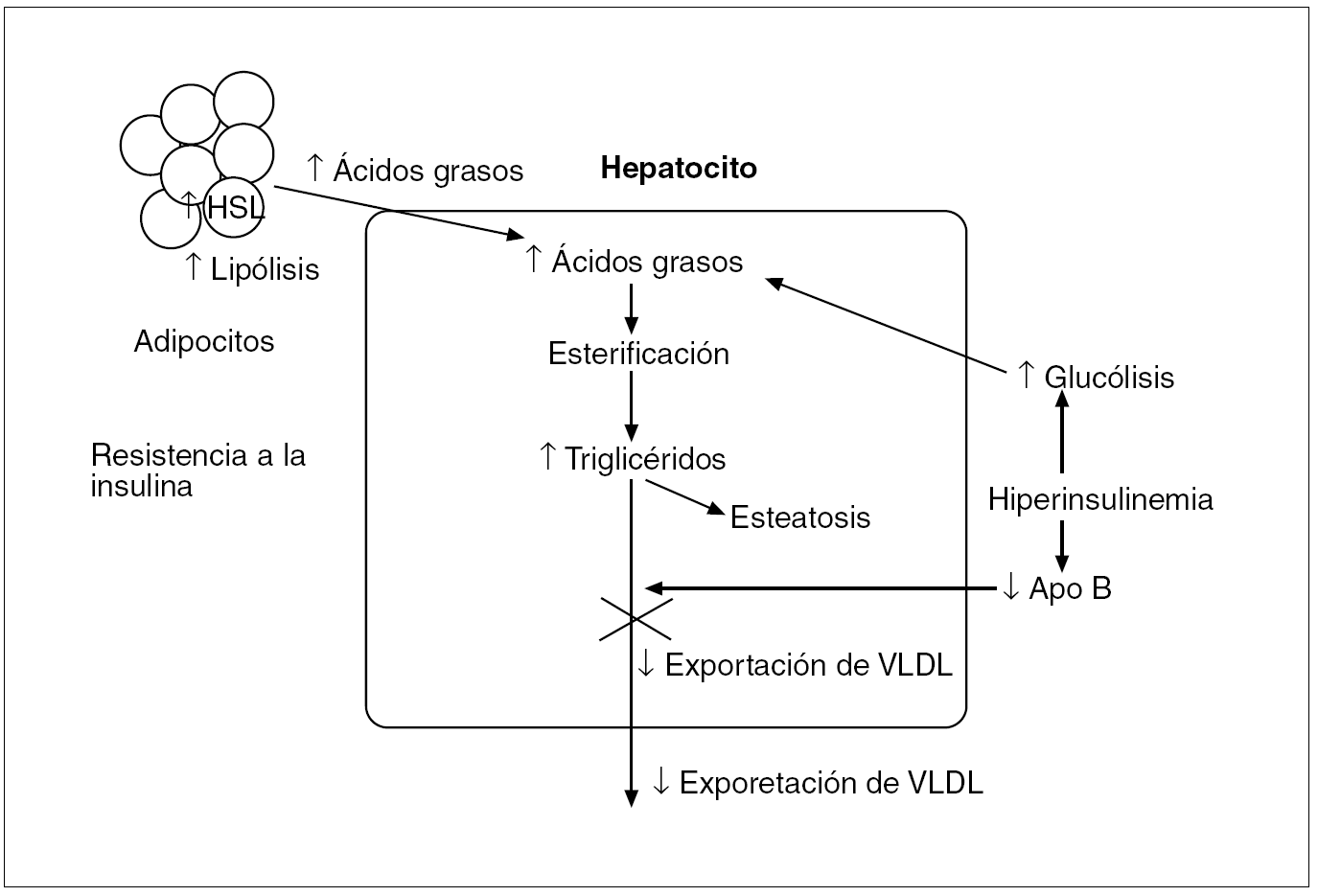
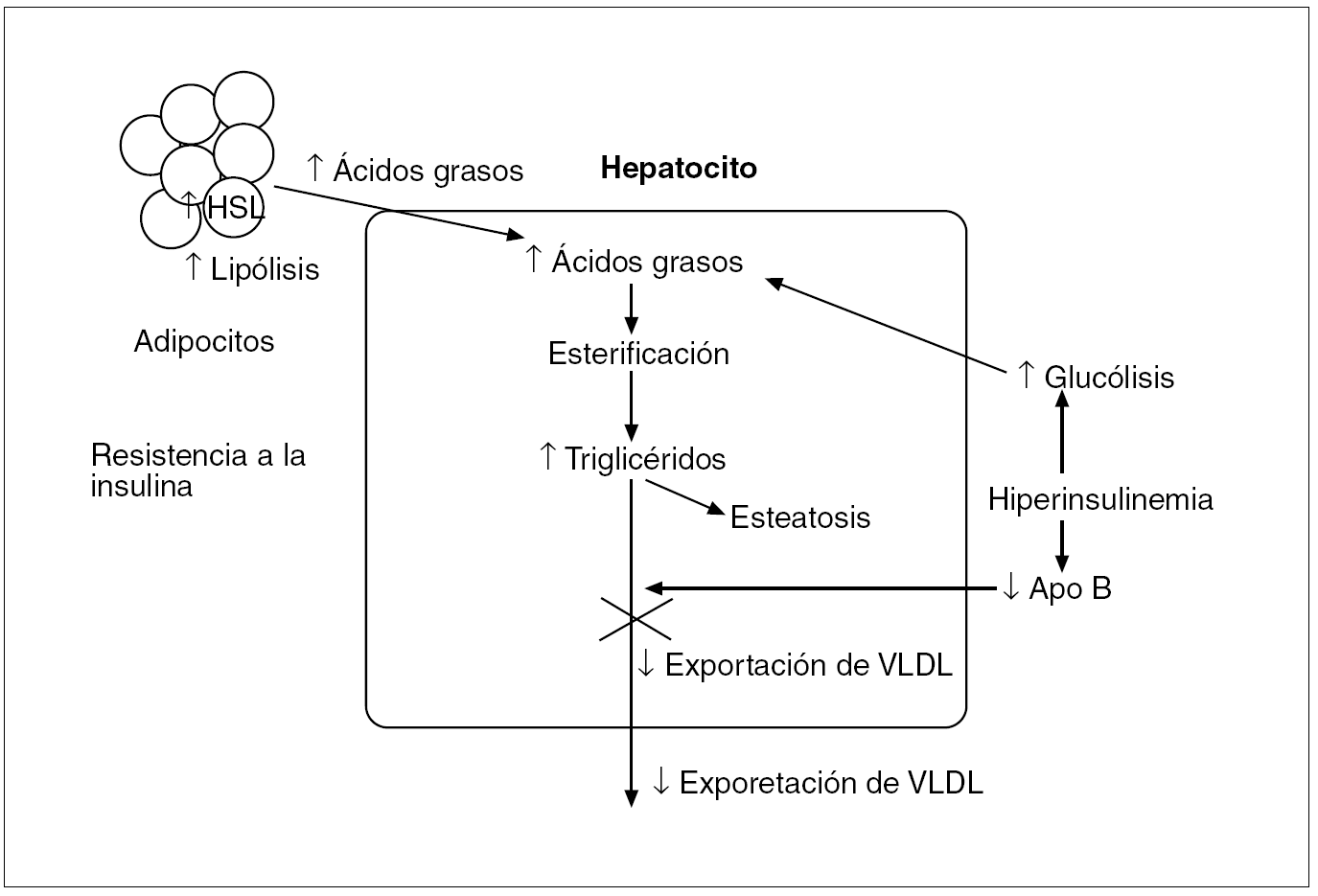
Fig. 4. En la resistencia a la insulina, a pesar de la elevada insulinemia, no se produce la supresión que ésta normalmente ejerce sobre la lipólisis, sino que, al contrario, hay una mayor liberación de ácidos grasos desde los adipocitos por un aumento de actividad de la HSL (hormone-sensitive lipase). Esto origina una mayor acumulación de los ácidos grasos almacenados en los hepatocitos, que tiende a compensarse mediante un aumento de la oxidación beta mitocondrial de estos ácidos con la formación de adenosintrifosfato y por la exportación de triglicéridos. No obstante, en la resistencia a la insulina la esterificación prevalece sobre la oxidación y estos ácidos grasos esterificados se almacenan como triglicéridos en el citoplasma o se dirigen a la síntesis de lipoproteínas de muy baja densidad (VLDL); la exportación disminuida de VLDL por una síntesis inhibida de apolipoproteína (Apo) B podría contribuir a la acumulación de triglicéridos y al desarrollo de la esteatosis hepática.
Durante el ayuno los ácidos grasos que se suministren al hígado provendrán de la hidrólisis de los triglicéridos almacenados en el tejido adiposo. En condiciones normales, las catecolaminas, el glucagón y la hormona de crecimiento estimulan esta lipólisis, y la insulina la inhibe. Sin embargo, en situaciones de resistencia a la insulina, a pesar de la hiperinsulinemia, no se produce la supresión que normalmente ésta ejerce sobre la lipólisis, sino que hay un aumento de la producción de ácidos grasos por parte de los adipocitos, secundaria a una hidrólisis incrementada de los triglicéridos por una mayor actividad de la HSL (hormone-sensitive lipase) en los adipocitos, lo que comporta un mayor aporte de ácidos grasos al hepatocito, donde prevalece la esterificación sobre la oxidación; esta reserva aumentada de ácidos grasos esterificados puede almacenarse en su citosol como triglicéridos o dirigirse a la síntesis de VLDL19 (fig. 4). Por otro lado, la hiperinsulinemia, al inhibir la síntesis de las apolipoproteínas B, disminuye la exportación de las VLDL, lo que provoca un acumulación de triglicéridos en el hepatocito que ocasiona la esteatosis.
Síntesis de novo de ácidos grasos
En el hígado hay una síntesis de novo de ácidos grasos, regulada independientemente por la insulina y por la glucosa20,21, que parece incrementada en la resistencia a la insulina.
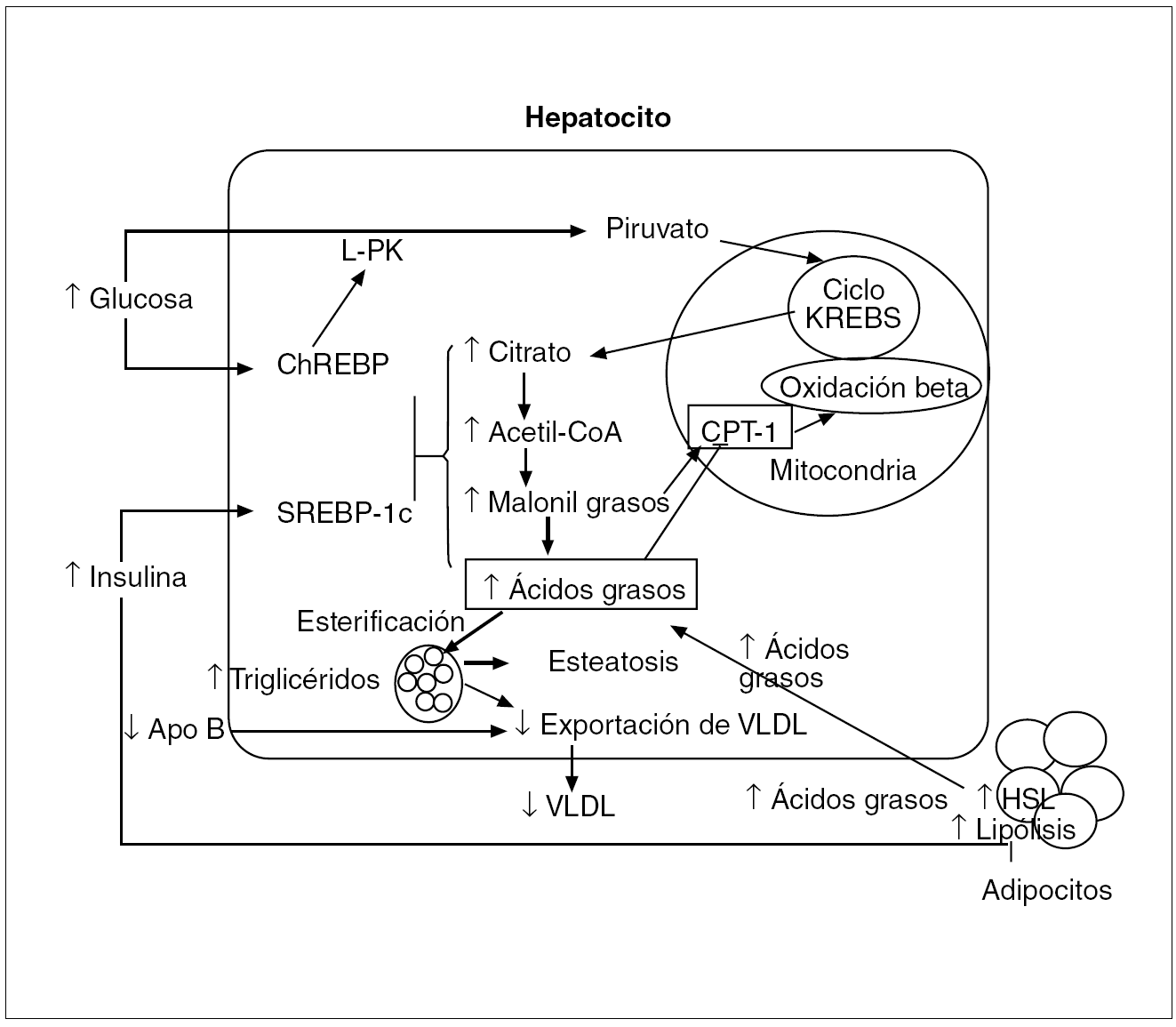
La hiperinsulinemia puede estimular la lipogénesis al inducir en el hígado la SREBP-1c (sterol regulatory element-binding protein-1c), una de las 3 formas isomorfas de las SREBP pertenecientes a la familia de los factores de transcripción. En el núcleo la SRBEP-1c activa transcripcionalmente todos los genes requeridos para la lipogénesis y facilita la aparición de esteatosis20. La SREBP-1c también activa la ACC-2 (acetil CoA carboxilase 2), que produce malonil-coenzima A (CoA) en la membrana mitocondrial, lo cual da lugar a una oxidación disminuida de ácidos grasos por la inhibición de la proteína CPT-1 (carnitine palmitoyl transferase-1), que transporta los ácidos grasos a la mitocondria.
Simultáneamente la hiperglucemia puede estimular la lipogénesis al activar la ChREBP (carbohidrate response element bindig protein), que transcripcionalmente activa la L-PK (liver-type pyruvate kinase) en todos los genes lipogénicos, catalizando la conversión de la glucosa a piruvato, que al incorporarse en la mitocondria al ciclo de Krebs da lugar a la formación de citrato, que en el citosol se convierte en acetil-CoA, el cual se metaboliza sucesivamente a malonil-CoA, ácido palmítico, ácido esteárico y, por último, a ácido oleico, que es el producto final de la síntesis de novo de ácidos grasos21 (fig. 5).
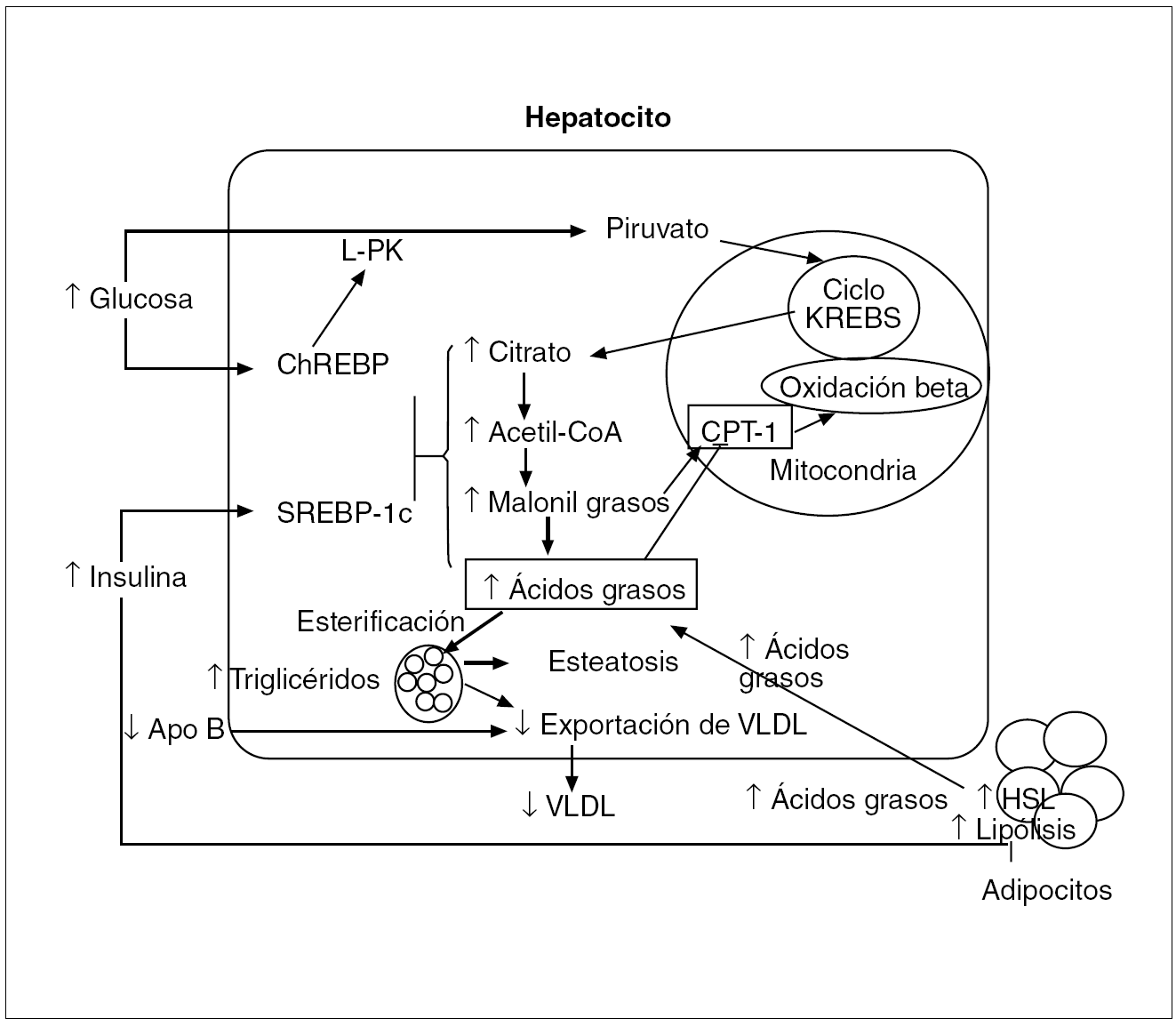
Fig. 5. En los adipocitos la resistencia a la insulina incrementa la actividad de la HSL (hormone-sensitive lipase), por lo que se produce una mayor lipólisis de los triglicéridos, con el consiguiente mayor aporte de ácidos grasos al hepatocito. La hiperinsulinemia induce la expresión de SREBP-1c (sterol regulatory element-binding protein-1c), que da lugar a la activación transcripcional de todos los genes lipogénicos. La hiperglucemia induce la expresión de la ChREBP (carbohidrate response element binding protein), que ocasiona la activación transcripcional de la L-PK (liver-type pyruvate kinase) y de todos los genes lipogénicos. La acción sinérgica de SREBP-1c y ChREBP activa las enzimas necesarias para convertir el exceso de glucosa en ácidos grasos. Como consecuencia de la síntesis aumentada de ácidos grasos hay una mayor producción de malonil-coenzima A (CoA), que inhibe la proteína CPT-1 (carnitine palmitoyl transferase-1), responsable del transporte de los ácidos grasos a la mitocondria. Por tanto, en la resistencia a la insulina tanto los ácidos grasos provenientes de la periferia como los de la lipogénesis de novo se esterifican preferentemente a triglicéridos, lo que facilita el desarrollo de la esteatosis hepática.Apo: apolipoproteína; VLDL: lipoproteínas de muy baja densidad.
Por tanto, la acción sinérgica de la SREBP-1c y de la ChREBP activa los mecanismos enzimáticos necesarios para la conversión del exceso de glucosa en ácidos grasos. Como consecuencia de la mayor producción de malonil-CoA, al inhibirse la proteína CPT-1, en el caso de resistencia a la insulina los ácidos grasos tanto provenientes de la periferia como los derivados de la lipogénesis de novo, en vez de oxidarse en la mitocondria, se esterifican preferentemente a triglicéridos18 (fig. 5), lo que facilita el desarrollo de esteatosis.
Receptores del peroxisoma proliferante activado
Un tercer factor de transcripción implicado en la esteatosis serían los receptores del peroxisoma proliferante activado (PPAR), pertenecientes a la superfamilia de los receptores nucleares. Existen 3 isotipos, PPAR-α, PPAR-β y PPAR-γ, que controlan diversas funciones celulares como el metabolismo de los lípidos y lipoproteínas, la oxidación de los ácidos grasos, el metabolismo de la glucosa, la adipogénesis, la diferenciación celular y también la inflamación vascular asociada con la aterogénesis. Los PPAR-γ son necesarios para la diferenciación normal del adipocito22; se expresan sobre todo en el tejido adiposo, músculos, corazón, hígado y riñones, y parecen regular muchos genes involucrados en la oxidación beta de los ácidos grasos, varios citocromos P450, en particular el Cyp2E1 y el Cyp4A; aunque en el hígado los PPAR normalmente se expresan en concentraciones muy bajas, aumentan significativamente en modelos animales con resistencia a la insulina y esteatosis hepática. Los PPAR son activados por ácidos grasos libres, lo que facilita su entrada en el hepatocito y su catabolismo al aumentar la oxidación beta de estos, disminuyendo la reserva de ácidos grasos disponibles para la síntesis de triglicéridos y su incorporación a las VLDL.
Los SREBP-1c pueden transcripcionalmente activar los PPAR-γ estimulando la producción de ligandos activadores del receptor nuclear23. En pacientes con EHNA se han identificado algunas mutaciones en el gen codificador de estos receptores nucleares, por lo que podrían estar implicados en la génesis del HGNA. Las tiazolidinedionas actúan como ligandos de los PPAR-γ disminuyendo los ácidos grasos circulantes al aumentar la actividad de estos receptores en los adipocitos24. La deleción genética de PPAR-γ en hígados de ratones ob/ob atenúa notablemente el desarrollo de esteatosis, con independencia de la existencia de hiperinsulinemia o hiperglucemia25.
AMPK
La AMPK (adenosinmonofosfato-proteincinasa activada) es una proteína celular que actúa como sensor de los niveles de energía celular26 y es activada por las concentraciones celulares de adenosinmonofosfato (un marcador de bajo nivel de energía celular). La activación de la AMPK estimula las vías catabólicas de producción de ATP, tales como la oxidación beta de ácidos grasos, y la inhibición de procesos consumidores de ATP como la lipogénesis26. La AMPK disminuye la lipogénesis por 3 mecanismos independientes: a) fosforilar e inhibir la actividad de la acetil-CoA carboxilasa disminuyendo la formación de malonil-CoA27; b) fosforilar la ChREBP inhibiendo su entrada en el núcleo, disminuyendo la L-PK y la expresión de genes lipogénicos28, y c) disminuir la expresión de la SREBP-1c29. Por tanto, la activación de la AMPK origina, por un lado, una disminución de la síntesis de ácidos grasos y, por otro, un descenso de las concentraciones de malonil-CoA y un aumento de la actividad CPT-1, con el consiguiente incremento de la oxidación de ácidos grasos. Tanto la metformina10 como las tiazolidinedionas24 activan la AMPK hepática, por lo que pueden ser útiles en el tratamiento de la esteatosis.
Segundo impacto
Estrés oxidativo. La esteatosis no es siempre quiescente, pues las grandes concentraciones intrahepáticas de ácidos grasos libres son susceptibles de un «segundo impacto» en el que intervendrían factores adicionales que condicionarían estrés oxidativo y peroxidación lipídica, lo cual originaría un alto aflujo de electrones a la cadena respiratoria mitocondrial y un aumento de producción de RLO, que a su vez causaría las lesiones hepáticas de la EHNA.
El estrés oxidativo es consecuencia del desequilibrio entre los mecanismos pro y antioxidantes. Entre los prooxidantes se encuentran los RLO, tales como el radical superóxido (O2), el peróxido de hidrógeno (H2O2) y el radical hidroxilo (OH), provenientes mayoritariamente de las mitocondrias y de otras organelas como los microsomas y peroxisomas. Entre los antioxidantes se hallan las enzimas superóxido dismutasa, la catalasa y la glutatión peroxidasa, y sustancias exógenas como la vitamina E, los carotenos y la vitamina C. Los RLO, por contener un electrón impar en su órbita exterior, son átomos o moléculas muy inestables y reactivas, con gran avidez por los ácidos grasos no saturados de las membranas celulares, lo que comporta alteraciones en su estructura y función; este proceso se denomina peroxidación lipídica.
Aunque la oxidación beta mitocondrial es la vía oxidativa predominante de los ácidos grasos en condiciones fisiológicas, también puede ser una causa de formación de RLO. Así, en la EHNA se producen grandes concentraciones intrahepáticas de ácidos grasos libres y una tasa elevada de oxidación beta mitocondrial, y en consecuencia, un alto aflujo de electrones a la cadena respiratoria mitocondrial, con un aumento de los RLO generados.
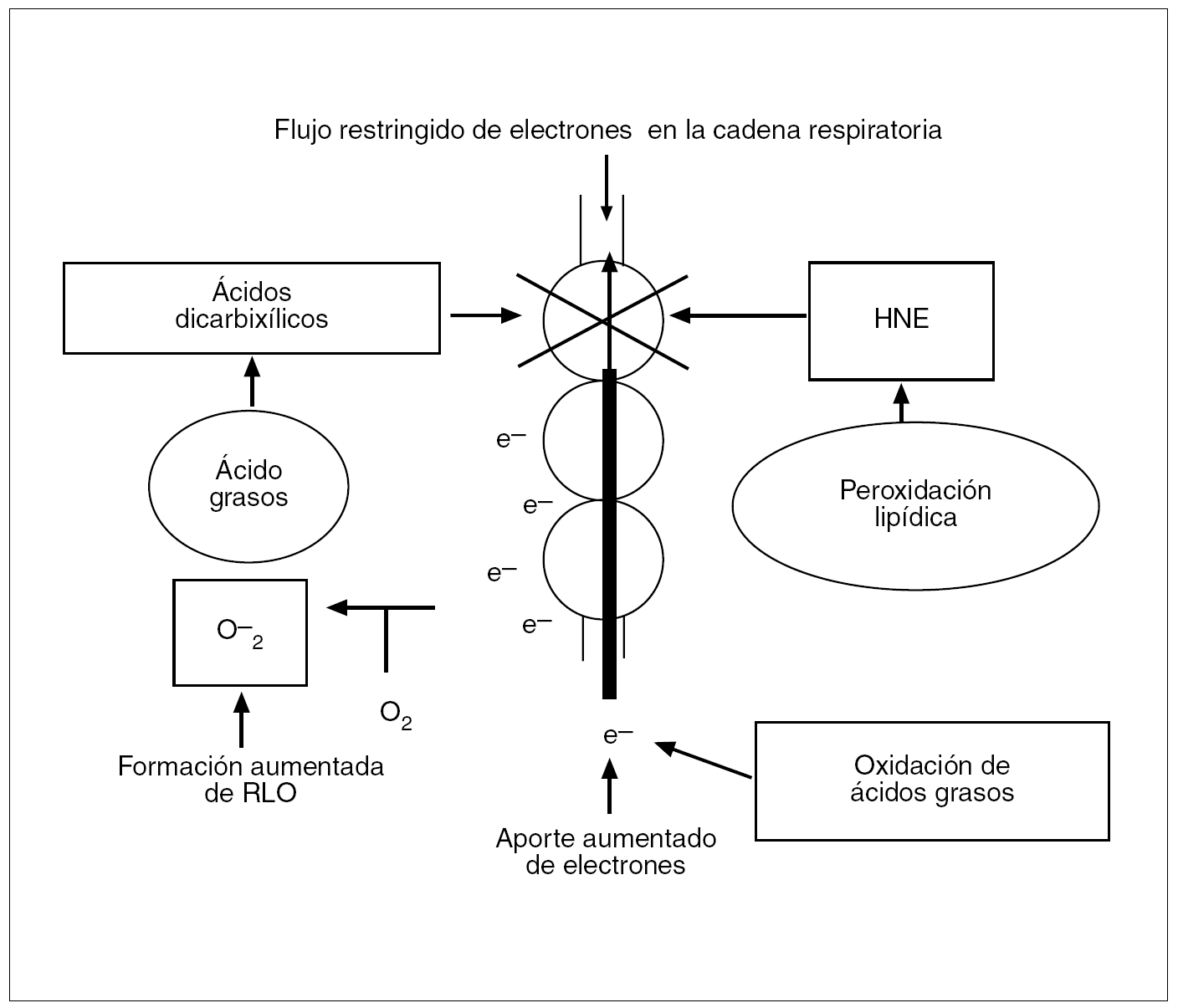
Además, los ácidos grasos libres pueden ser oxidados por vías alternativas dependientes, por un lado, de la oxidación beta peroxisómica catalizada por la acil-CoA oxidasa, con la formación H2O2 mediante la donación de electrones al oxígeno molecular, y por otro, de la oxidación omega microsómica por parte del citocromo P450 (CYP2E1, CYP4A)20 (fig. 6). Además, los ácidos dicarboxílicos, otro producto de la oxidación omega microsómica de los ácidos grasos, también pueden alterar la función mitocondrial al bloquear el flujo de electrones en la cadena respiratoria (fig. 7)30. En contraste con este bloqueo parcial del flujo de electrones, hay un aumento de la llegada de electrones a la cadena respiratoria por la incrementada oxidación beta mitocondrial de los ácidos grasos, con lo que se generan NADH (dinucleótido de nicotinamida adenina) y FADH2 (dinucleótido de flavina adenina) con transferencia de sus electrones para mantener la actividad de la cadena respiratoria mitocondrial11,31; este proceso se denomina fosforilación oxidativa. En pacientes diabéticos las glucemias elevadas y la aumentada oxidación de glucosa pueden incrementar aún más la llegada de electrones. El paso de electrones a lo largo de la cadena respiratoria mitocondrial está asociado a la síntesis de ATP, de modo que a partir de una molécula de glucosa se generan 36 moléculas de ATP, y a partir de un ácido graso libre, 146 moléculas de ATP. Por ello la mitocondria convierte las grasas y demás nutrientes en anhídrido carbónico y H2O y genera energía en forma de ATP. Si por cualquier motivo los electrones no fueran captados por el oxígeno en el complejo IV mitocondrial, se produciría una captación anómala de éstos por parte del oxígeno y entonces se generaría el O2 en lugar de H2O. Este hecho puede darse en condiciones basales, dado que suele producirse una fuga de electrones en los complejos mitocondriales I, II y III. A más actividad de la cadena respiratoria, mayor será la cantidad de O2 producido, desencadenando una reacción en cadena que origina más RLO, tales como H2O2 y OH. Sin embargo, en condiciones basales las células pueden afrontar tal formación de RLO, puesto que se mitigan sus efectos nocivos gracias a las sustancias antioxidantes.
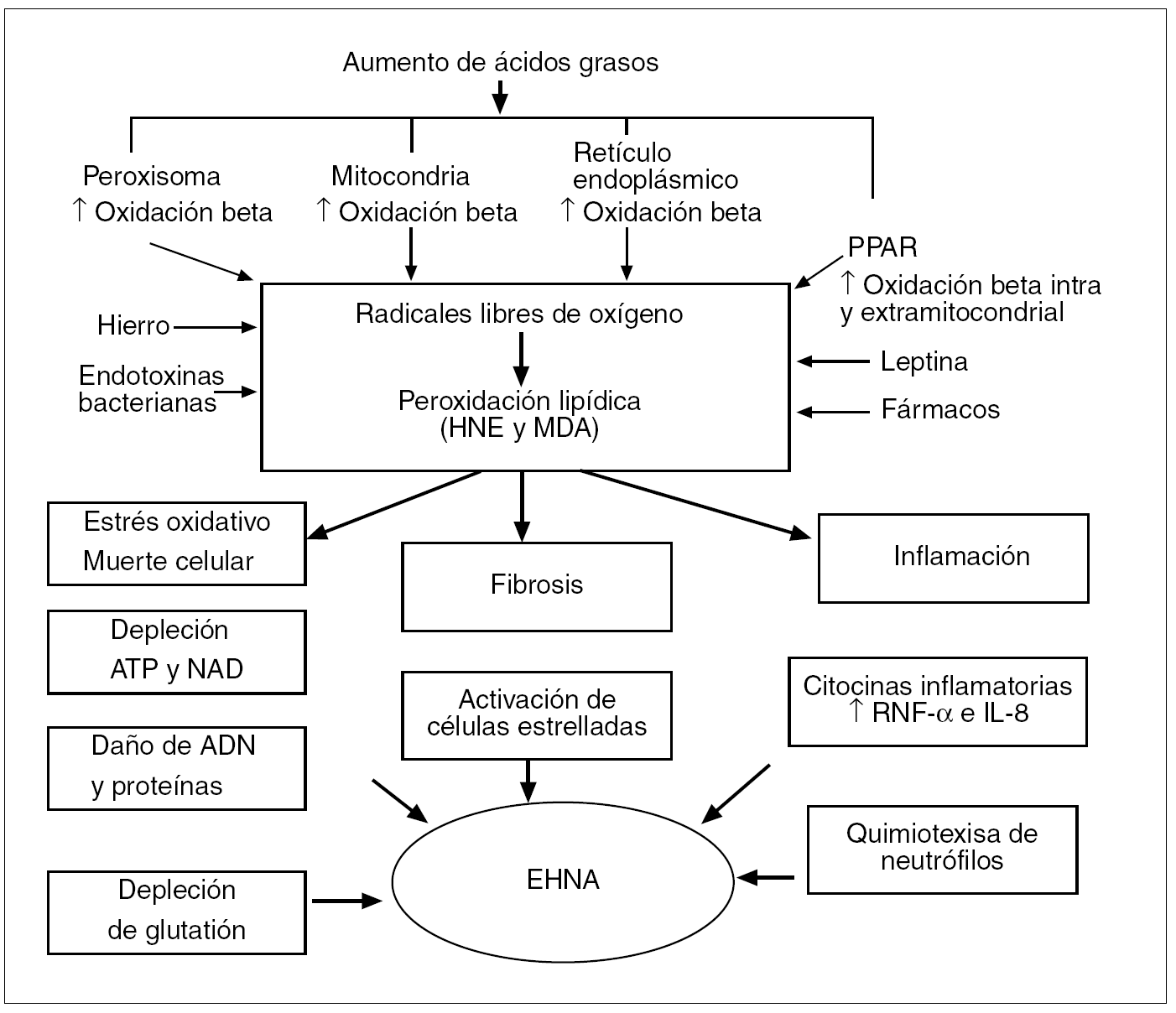
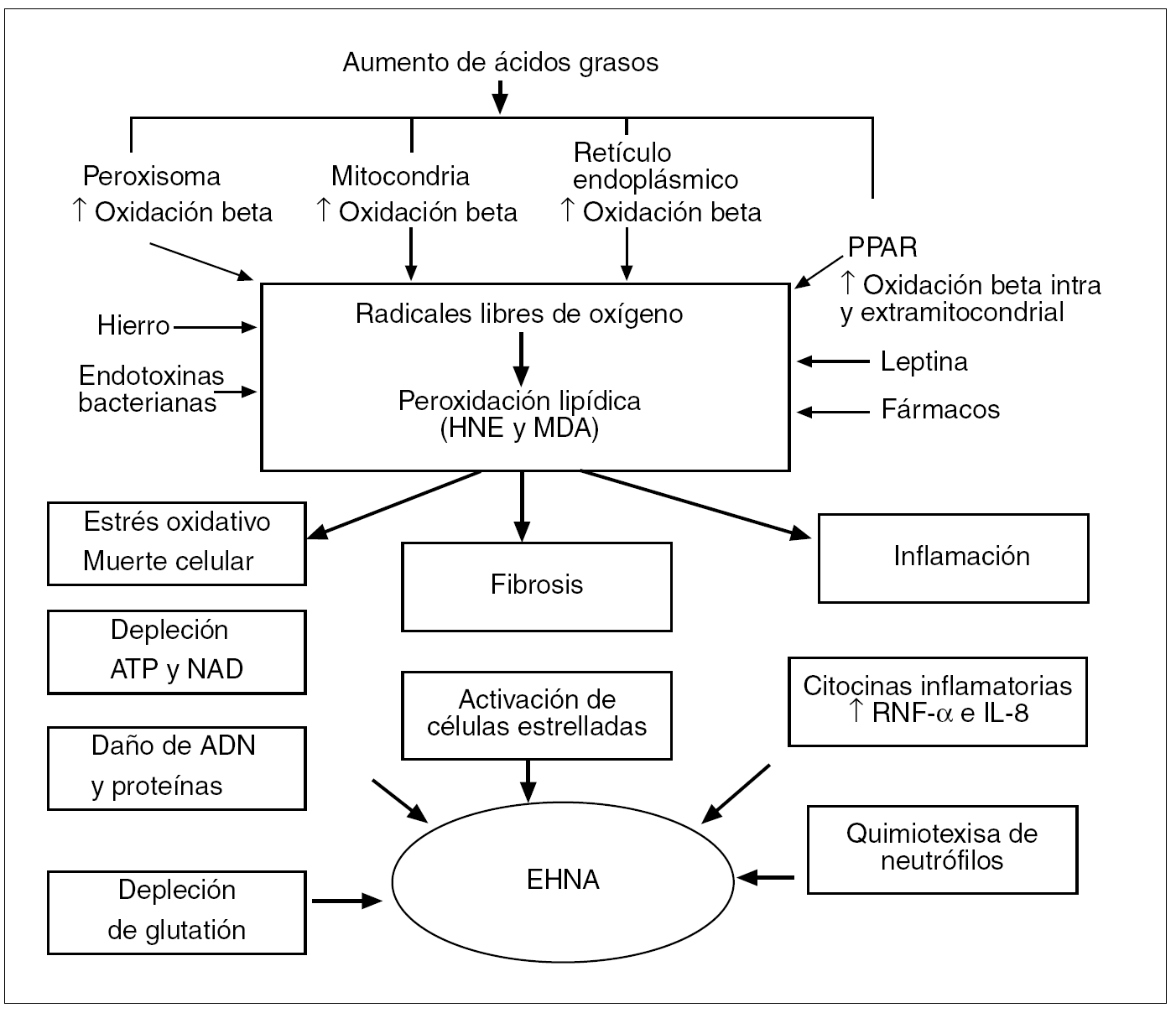
Fig. 6. Visión global de los mecanismos patogénicos de la esteatohepatitis no alcohólica (EHNA).ATP: adenosintrifosfato; dinucleótido de nicotinamida adenina; HNE: 4-hidroxinonenal; MDA: malondialdehído; PPAR: receptores del peroxisoma proliferante activado; TNF: factor de necrosis tumoral; IL: interleucina.
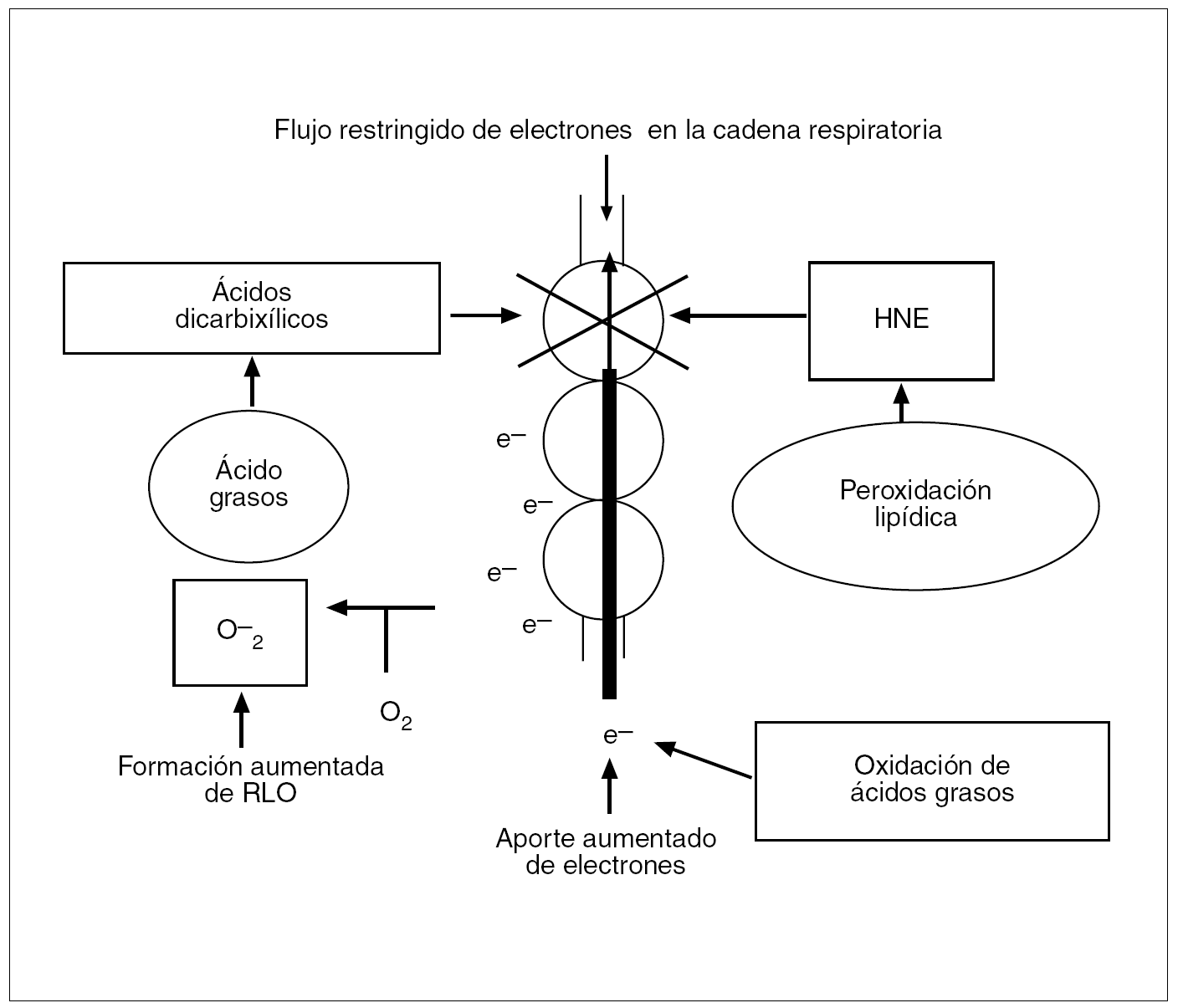
Fig. 7. En el hígado normal, la mayoría de los electrones aportados a la cadena respiratoria fluyen a lo largo de ésta hasta el citocromo C oxidasa (la oxidasa terminal), donde se combinan de forma segura con protones y oxígeno para formar agua. En el hígado graso de pacientes con resistencia a la insulina, la alta tasa de oxidación beta incrementa el aporte de electrones a la cadena respiratoria, mientras que los productos de la peroxidación lipídica y posiblemente el aumento de ácidos dicarboxílicos pueden bloquear parcialmente el flujo de electrones dentro de esta cadena. El desequilibrio entre un aporte elevado y un flujo disminuido de electrones puede hacer que la cadena respiratoria transfiera directamente sus electrones al oxígeno para formar el radical superóxido (O2).RLO: radicales libres de oxígeno; HNE: 4-hidroxinonenal.
Los pacientes con EHNA presentan lesiones mitocondriales ultraestructurales11,32 y una disminución de la actividad de los complejos mitocondriales11,33,34 que ocasionan una interrupción del flujo de electrones en algún punto de la cadena respiratoria mitocondrial, con transferencia de electrones al oxígeno molecular y producción de aniones superóxido y H2O2. El incremento de los RLO en el hígado graso ocasiona depleción de ATP35 y de nicotinamida, daño del ADN mitocondrial, alteración de la estabilidad proteica, desestructuración de las membranas lipídicas (peroxidación) y liberación de citocinas proinflamatorias.
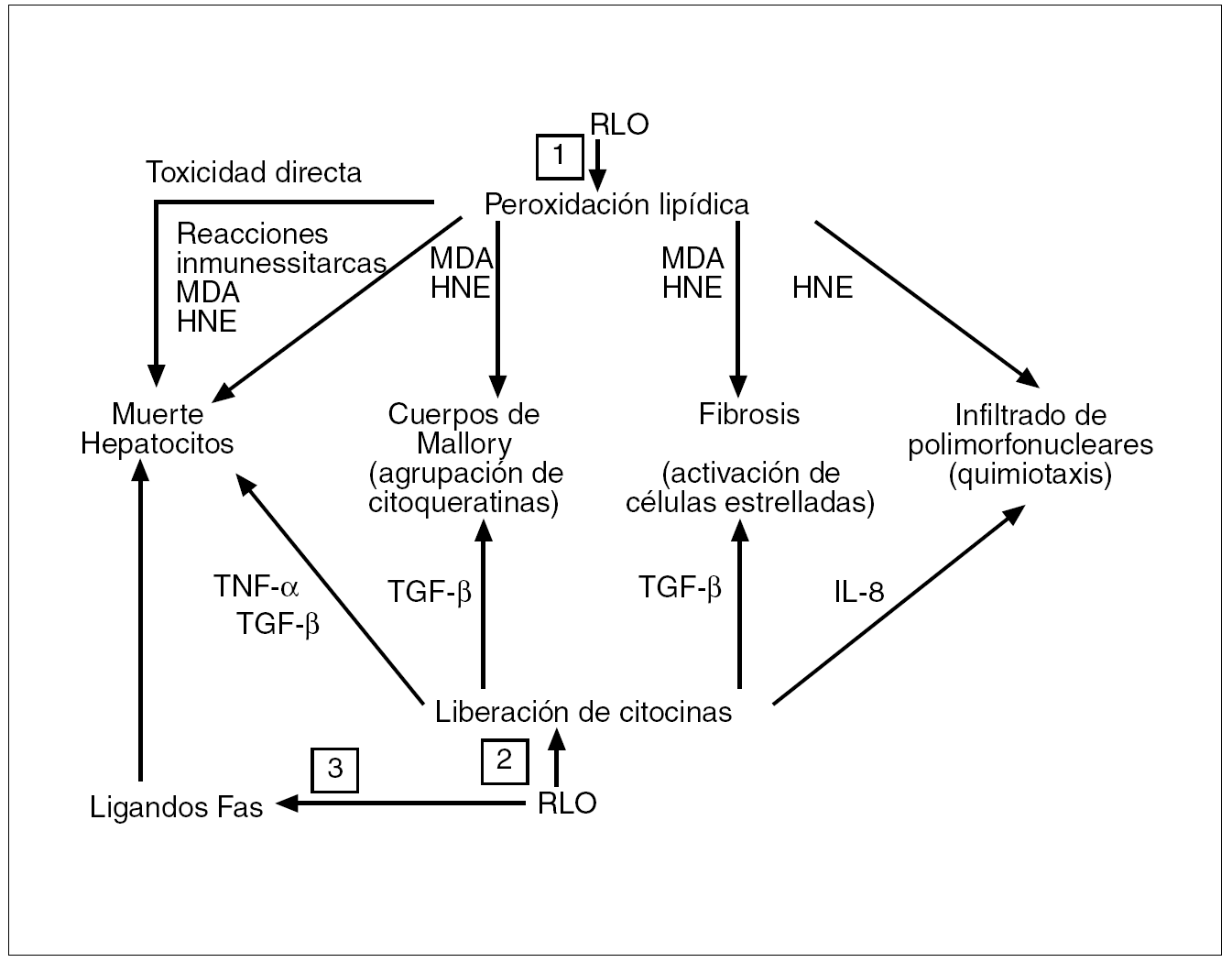
Los RLO determinan la generación de distintas citocinas desde distintos tipos celulares (hepatocitos, adipocitos y células de Kupffer). Este hecho depende de la capacidad de activación que tienen los RLO sobre factores de transcripción, fundamentalmente el NF-κβ, que ya vimos determinaba la formación de citocinas tales como el TNF-α, además del factor de crecimiento tumoral beta 1 (TGF-β1), de la interleucina 8 y de ligandos Fas. A su vez, cada una de estas citocinas puede estar implicada en la patogenia de las lesiones hepáticas, ya que el TNF-α, al activar la IKK-β, perpetuará la resistencia a la insulina y una mayor producción de TNF-α. El TNF-α y el TGF-β1 pueden activar la muerte celular programada o apoptosis al activar las caspasas, enzimas responsables de su ejecución. El TGF-β1 puede intervenir en la formación de cuerpos de Mallory y activar la síntesis de colágeno por parte de las células estrelladas, y la interleucina 8 es un potente activador de neutrófilos10,35 (fig. 8).
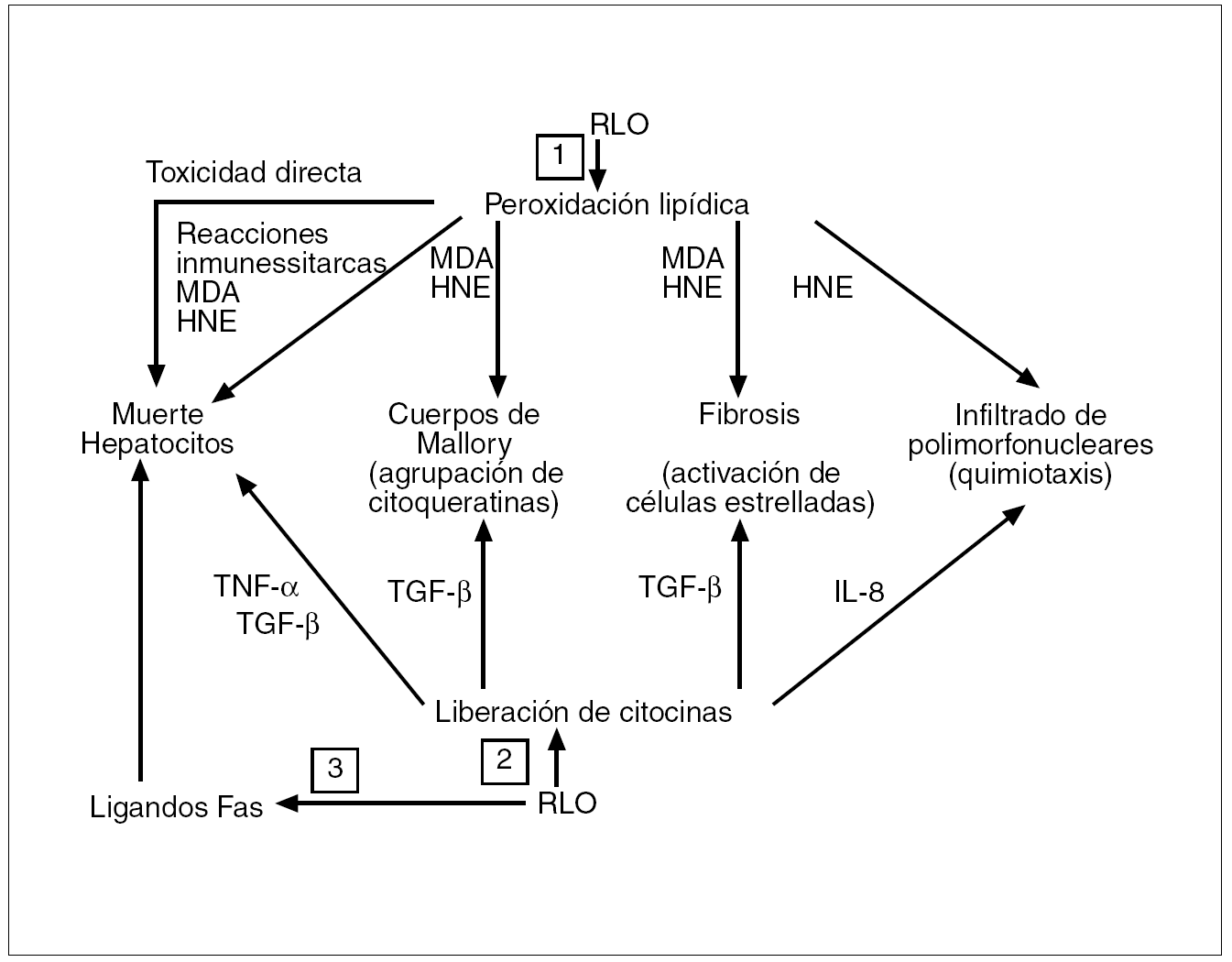
Fig. 8. 1) Al desencadenar los radicales libres del oxígeno (RLO) la peroxidación lipídica, la liberación de malondialdehído (MDA) y de 4-hidroxinonenal (HNE) causa toxicidad directa, pues ambos se acoplan covalentemente a proteínas, lo que ocasiona reacciones inmunitarias. MDA y HNE pueden intervenir en la formación de los cuerpos de Mallory, constituidos por la agregación de monómeros de citoqueratina. MDA y HNA también incrementan la síntesis de colágeno por las células estrelladas, y el HNA tiene actividad quimiotáctica por los neutrófilos. 2) Los RLO inducen la formación de citocinas por las células de Kupffer y por los hepatocitos. El factor de necrosis tumoral alfa (TNF-α) y el factor de crecimiento tumoral beta (TGF-β) activan las caspasas y la muerte de los hepatocitos. El TGF-β también activa la síntesis de colágeno por las células estrelladas, mientras que la interleucina 8 (IL-8) es un potente quimiotáctico sobre los neutrófilos. 3) Los RLO causan la expresión de ligandos Fas en los hepatocitos; los ligandos Fas de un hepatocito pueden interaccionar con los de otros hepatocitos causando «asesinatos fratricidas» entre ellos.
Sería erróneo pensar que todos los efectos derivados de la formación de RLO y de la activación del NF-κβ son nocivos, o se encaminan exclusivamente a desarrollar muerte celular; de hecho, también se han descrito efectos antiapoptóticos, tanto de los RLO como del NF-κβ. Parece pues que un ligero incremento celular de RLO no produce mayor muerte celular, e incluso puede ejercer efectos inflamatorios defensivos, pero un aumento intenso y prolongado de RLO sí puede desencadenar daño hepático.
Peroxidación lipídica
Conviene señalar que los productos derivados de la peroxidación lipídica como el malondialdehído y el 4-hidroxinonenal36 también parecen implicados en la génesis de las lesiones hepáticas de la EHNA. Mientras que los RLO son moléculas de vida corta y su efecto es primordialmente local, al actuar sobre los ácidos grasos no saturados de las membranas celulares a las que lesionan (lo que constituye la peroxidación lipídica), el malondialdehído y el 4-hidroxinonenal son moléculas de vida más larga que los RLO y con efectos más distantes intra y extracelulares, por lo que amplían los efectos del estrés oxidativo. La peroxidación de los ácidos grasos no saturados estimula la proteólisis en el retículo endoplásmico de las apolipoproteínas B y, en consecuencia, disminuye la exportación de las VLDL, con acumulación de triglicéridos en el hepatocito. Tanto el malondialdehído como el 4-hidroxinonenal causan toxicidad directa y, al acoplarse covalentemente a las proteínas, pueden desencadenar reacciones inmunitarias con aumento de la producción de TNF-α, promueven el aflujo de células inflamatorias al hígado, deplecionan los antioxidantes naturales tipo glutatión e impiden la síntesis de nucleótidos y proteínas (fig. 8); pueden inducir la formación de cuerpos de Mallory al promover la agrupación de citoqueratinas y también incrementan la síntesis colágena al activar las células estrelladas hepáticas. El 4-hidroxinonenal además tiene actividad quimiotáctica sobre los neutrófilos (fig. 8). Todo ello induce la necrosis de los hepatocitos, con inflamación y fibrosis (los marcadores histológicos de la EHNA).
Otros mecanismos potencialmente implicados
Ligandos Fas. Normalmente los hepatocitos expresan en sus membranas receptores Fas, pero no producen ligandos Fas37. Sin embargo, en situaciones de activación del NF-κβ los hepatocitos pueden sintetizar ligandos Fas, secretarlos y, consecuentemente, actuar sobre los receptores Fas de hepatocitos vecinos, en los que causan muertes fratricidas entre ellos38 debido a que aumentan la permebilidad de la membrana externa mitocondrial, lo que permite la salida desde el espacio intermembranoso mitocondrial del citocromo C, procaspasas y otros factores proapoptóticos que, al activar la caspasa 9 en el citosol, desencadenan la muerte celular por apoptosis.
Hierro. La mayoría de los pacientes con sobrecarga primaria de hierro no debida a hemocromatosis presentan resistencia a la insulina39-42, la cual puede mejorar con la realización de flebotomías43-47. La resistencia a la insulina provoca una mayor expresión de los receptores de la transferrina (fig. 1) en la superficie celular y un incremento de la exocitosis de los receptores intracelulares preexistentes, se asocia a concentraciones elevadas de ferritina sérica12,39,42,47 y a aumento de hierro hepático en algunos pacientes. Una concentración elevada de ferritina no refleja necesariamente un incremento del hierro hepático, sino que puede deberse a la propia EHNA (como un reactante de fase aguda). El hierro ferroso es un poderoso generador de OH, y se este modo contribuye a la acumulación de RLO y a la fibrogénesis. Queda por aclarar si una sobrecarga moderada de hierro en la EHNA interviene en la patogenia de esta entidad, está relacionada con anomalías metabólicas asociadas o se debe a factores ambientales o genéticos no identificados. Por ejemplo, mutaciones heterocigotas del gen HFE podrían incrementar la acumulación de hierro hepático en estos pacientes48. Las flebotomías terapéuticas pueden retrasar e incluso revertir el daño hepático en pacientes con sobrecarga de hierro y EHNA. Por ello, la biopsia hepática, además de confirmar el grado de EHNA, permite valorar el hierro hepático y la indicación de distintas opciones terapéuticas, entre ellas las flebotomías. Sin embargo, diversos trabajos no han evidenciado que la concentración hepática de hierro sea un factor predictivo de fibrosis en los pacientes con EHNA.
Adipocinas. El adipocito segrega varias proteínas bioactivas o adipocinas49, que regulan el metabolismo de los lípidos y de la glucosa tanto en el hígado como en la periferia. Estas adipocinas incluyen la leptina, el TNF-α, la resistina y la adiponectina.
Aunque la resistina inhibe la acción de la insulina en modelos animales, su expresión no está aumentada en humanos con resistencia a la insulina50.
El TNF-α, como ya hemos comentado, aumenta la resistencia a la insulina, lo cual favorece la esteatosis, y desempeña también un papel proinflamatorio en la EHNA51.
La adiponectina es una hormona producida en el tejido adiposo periférico y tiene efectos antilipogénicos, pues puede proteger al hepatocito de la acumulación de lípidos al aumentar la oxidación beta de los ácidos grasos y disminuir el contenido de triglicéridos en el hígado. En la dislipemia, la obesidad y la diabetes se han encontrado concentraciones descendidas de adiponectina52. Por tanto, la adiponectina y el TNF-α tienen efectos contrarios respecto a la sensibilidad a la insulina y la inflamación, y el equilibrio entre estos 2 sistemas de adipocinas puede ser importante en la patogenia de la EHNA. El hígado y el músculo tienen receptores para la adiponectina53; la estimulación de los receptores 2 de la adiponectina en el hígado activa la AMPK y los PPAR-α.
Se ha señalado que la leptina podría clasificarse como una citocina, ya que, además de regular la ingesta alimentaria y el gasto energético, modula las respuestas inmunitaria e inflamatoria54. Al parecer, las concentraciones séricas elevadas de leptina se relacionan con la gravedad de la esteatosis hepática, por lo que se ha planteado que podría tener un papel patogénico en la resistencia a la insulina hepática y/o a un fallo de su actividad antiesteatósica. También su producción por las células estrelladas puede desempeñar un papel importante en la fibrosis hepática55-58. Puede contribuir a la progresión de la esteatosis a EHNA y ulteriormente a cirrosis, dada su actividad profibrogénica y moduladora de la respuesta inflamatoria en el hígado59. Por otro lado, se ha objetivado que el tratamiento con leptina en ratones con déficit congénito de ésta y lipodistrofia generalizada induce una reducción de la grasa corporal y una acusada disminución de la resistencia a la insulina60. Otros autores no encuentran correlación entre sus concentraciones y el desarrollo de EHNA.
Endotoxinas bacterianas. Otro factor con probable implicación en la patogenia de la EHNA podría ser la liberación de productos bacterianos desde el intestino hacia la sangre que irriga la grasa mesentérica y el hígado, tales como los lipopolisacáridos, que son un potente factor activador de macrófagos. Éstos podrían inducir la expresión de TNF-α en el tejido adiposo y en el hígado, y el TNF-α podría activar en el hígado la IKK-β, lo cual ocasionaría resistencia a la insulina y HGNA. En ratas ob/ob con tendencia al sobrecrecimiento bacteriano por estasis intestinal, los hidratos de carbono de la dieta fermentan produciendo etanol, el cual aumenta la permeabilidad de la barrera intestinal; el etanol y otros productos bacterianos tales como los lipopolisacáridos podrían estimular excesivamente la producción de TNF-α, y se ha objetivado en estas ratas una disminución de la producción endógena de etanol tras ser tratadas con neomicina oral61. Por ello cabe plantearse si la pequeña cantidad de etanol producido por la flora intestinal detectada en algunos pacientes podría intervenir en la génesis de esta entidad.
Esteatopatitis no alcohólica inducida por fármacos. Fármacos cardiovasculares como la amiodarona, el maleato de perhexilina y más raramente los bloqueadores de los canales del calcio como la nifedipina y el diltiazem; los glucocorticoides a dosis altas, los estrógenos sintéticos, el tamoxifeno, la cloroquina, entre otros, también pueden asociarse a la aparición de EHNA. La amiodarona, el maleato de perhexilina, el dietilaminoetoxihexestrol y el tamoxifeno32, por sus propiedades lipofílicas, atraviesan fácilmente la membrana externa de la mitocondria y desde el espacio intermembranoso son «empujados» dentro de la mitocondria por el alto potencial electroquímico existente en la membrana interna; así alcanzarían altas concentraciones intramitocondriales, inhibirían la oxidación beta (lo que causaría esteatosis) y bloquearían la transferencia de electrones a lo largo de la cadena respiratoria; de este modo se incrementaría la transferencia de sus electrones al oxígeno y se formaría el anión O2; así pues, estos fármacos generarían RLO.

