




INTRODUCCIÓN
Cronológicamente, el primer trastorno de la función renal que aparece en los pacientes cirróticos es una disminución de la capacidad renal de excretar sodio. Inicialmente, cuando la enfermedad está compensada, esta alteración sólo puede ponerse de manifiesto mediante pruebas de sobrecarga (administración de solución salina, tratamiento con mineralcorticoides). Sin embargo, cuando la enfermedad progresa, la retención renal de sodio se hace más intensa y los enfermos son incapaces de excretar el sodio ingerido con la dieta. Éste se retiene junto con agua en proporciones isotónicas y se acumula en la cavidad peritoneal en forma de ascitis. La retención renal de sodio en la cirrosis es debida fundamentalmente a un aumento de la reabsorción tubular renal de este ion. Al inicio del desarrollo de la ascitis, cuando los pacientes tienen una retención renal de sodio moderada, las concentraciones plasmáticas de aldosterona y noradrenalina son normales. En esta fase de la enfermedad, por tanto, la retención de sodio es independiente de la actividad nerviosa simpática renal y del sistema renina-angiotensina-aldosterona, los 2 sistemas antinatriuréticos más conocidos. La concentración de péptidos natriuréticos atriales es elevada, lo que indica que la retención renal de sodio tampoco está en relación con una disminución de factores natriuréticos endógenos. Se ha planteado que mecanismos antinatriuréticos aún desconocidos podrían ser los responsables de este hecho. También se ha postulado la existencia de un aumento de la sensibilidad renal al efecto de la aldosterona. Cuando la retención renal de sodio es intensa, los pacientes cirróticos con ascitis presentan invariablemente hiperaldosteronismo y un aumento de la actividad nerviosa simpática.
Tiempo después del inicio de la retención renal de sodio y ascitis, los pacientes desarrollan una segunda alteración de la función renal, consistente en la disminución de la capacidad renal de excretar agua libre. Como ocurre en el caso del sodio, inicialmente esta alteración sólo puede ponerse de manifiesto mediante pruebas de sobrecarga acuosa. Sin embargo, en fases avanzadas de la enfermedad el trastorno se hace tan intenso que los pacientes son incapaces de excretar el agua ingerida con la dieta. El agua retenida diluye el medio interno y produce hiponatremia e hipoosmolaridad. La aparición tardía de este trastorno justifica su importante valor pronóstico. La hiponatremia dilucional (concentración plasmática de sodio < 130 mEq/l) es uno de los parámetros pronósticos más importantes en los pacientes cirróticos.
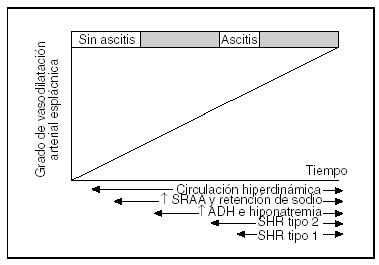
Finalmente, cuando la enfermedad está en sus etapas más avanzadas, los pacientes cirróticos desarrollan insuficiencia renal (síndrome hepatorrenal). Su aparición indica un pronóstico infausto en semanas o meses. Consiste en una insuficiencia renal funcional secundaria a una intensa vasoconstricción renal. Existen 2 tipos de síndrome hepatorrenal. El síndrome hepatorrenal tipo 1 consiste en una insuficiencia renal rápidamente progresiva. Generalmente se asocia a una complicación que actúa como factor precipitante, con frecuencia una infección bacteriana. El síndrome hepatorrenal tipo 2 consiste en una insuficiencia renal moderada que se mantiene estable durante meses. El principal problema de los pacientes con síndrome hepatorrenal tipo 1 es una insuficiencia hepática y renal rápidamente progresiva que determina la muerte del paciente en días. En los pacientes con síndrome hepatorrenal tipo 2 la principal manifestación clínica es una ascitis que no responde a los diuréticos (ascitis refractaria) (fig. 1).

Fig. 1. Esquema que muestra la evolución clínica en el tiempo de los parámetros hemodinámicos y hormonales de los pacientes cirróticos. SRAA: sistema renina-angiotensina-aldosterona; ADH: hormona antidiurética; SHR: síndrome hepatorrenal.
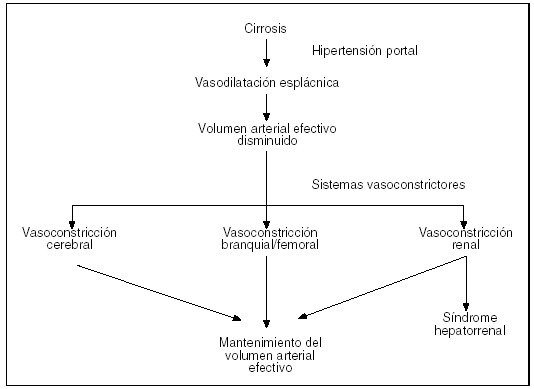
La disfunción renal en la cirrosis es consecuencia de un trastorno circulatorio secundario a una vasodilatación arterial esplácnica. Es bien conocido que la hipertensión portal, por mecanismos no aclarados, determina una masiva liberación de vasodilatadores locales, especialmente óxido nítrico, en este compartimiento vascular. Inicialmente el trastorno circulatorio queda compensado por un aumento del gasto cardíaco, lo que determina la característica circulación hiperdinámica de la cirrosis. Sin embargo, cuando la enfermedad hepática y la vasodilatación arterial esplácnica progresan, la circulación hiperdinámica es insuficiente para mantener la homeostasis circulatoria, los pacientes desarrollan hipotensión arterial y se activan mecanismos homeostáticos que tratan de mantener la presión arterial en límites normales o casi normales. Los 3 sistemas reguladores de la presión arterial más conocidos son el sistema nervioso simpático, el sistema renina-angiotensina y la hormona antidiurética. Se activan a través de receptores de presión existentes en el seno carotídeo y arco aórtico. El sistema nervioso simpático y el sistema renina-angiotensina son más sensibles a cambios de la presión arterial que la hormona antidiurética, lo cual explica el desarrollo de retención renal de sodio con anterioridad al trastorno del metabolismo renal del agua en la cirrosis. Es posible que existan mecanismos aún más sensibles, todavía desconocidos, que se activarían en fases muy iniciales de la enfermedad y que explicarían la retención renal de sodio y el desarrollo de ascitis en ausencia de activación de los sistemas renina-angiotensina y nervioso simpático. El síndrome hepatorrenal es la manifestación más extrema de la disfunción circulatoria de la cirrosis. Ocurre cuando existe una estimulación muy intensa de los sistemas renina-angiotensina, nervioso simpático y hormona antidiurética, y es debido no sólo al efecto vasoconstrictor de estos sistemas sobre la circulación renal sino, probablemente, también a una deficiente síntesis renal de sustancias vasodilatadores (prostaglandinas, óxido nítrico). En los pacientes con síndrome hepatorrenal existe vasoconstricción intensa en otros territorios vasculares como la piel, el músculo, la circulación cerebral y el hígado (fig. 2).

Fig. 2. Hipótesis del infrallenado arterial y activación de sistema de vasoconstrictores de manera compensadora en diferentes regiones.
El objetivo del presente capítulo es revisar el tratamiento actual de los pacientes con ascitis complicada, definida como aquella que se asocia a hiponatremia dilucional, ascitis refractaria y síndrome hepatorrenal tipo 1.
HIPONATREMIA DILUCIONAL EN LA CIRROSIS
Fisiopatología
Disminución de la capacidad renal de excretar agua libre
La administración oral o intravenosa (solución glucosada al 5%) de 20 ml/kg de peso en 45 min a un individuo normal determina, tras un período de 30-45 min, la excreción de una orina diluida (60-110 mOsm/kg; la osmolalidad plasmática es de 290 mOsm/kg) a una tasa de 8-12 ml/min. El volumen de agua excretada por este individuo idealmente puede dividirse en 2 partes. La primera consiste en el volumen de agua que disolvería los solutos urinarios isoosmóticamente con respecto al plasma (aclaramiento osmolar). La segunda parte consiste en agua libre de solutos (aclaramiento de agua libre). Dado que el aclaramiento osmolar en sujetos sin enfermedad oscila entre 1,5 y 2,5 ml/min, el aclaramiento de agua libre normal oscila entre 6 y 12 ml/min. Esto significa que una persona sana es capaz de mantener el contenido corporal de agua en sus límites normales incluso ante ingestas acuosas de 10 l por día o más2.
La generación de agua libre se produce en la rama ascendente del asa de Henle, donde se reabsorben cloro y sodio sin reabsorción concomitante de agua (este segmento es impermeable al agua)3. Por tanto, el agua libre se produce por sustracción de solutos de la orina tubular. Este proceso ocurre constantemente y, así, el asa de Henle produce constantemente una orina hipotónica (diluida en relación con el plasma) que alcanza el túbulo distal y colector.
El aclaramiento de agua libre dependerá de la reabsorción de agua en el túbulo distal (que atraviesa la corteza renal, isotónica con el plasma) y, sobre todo, en el túbulo colector (que atraviesa la médula renal, progresivamente más hipertónica hasta la papila renal). La hormona antidiurética (ADH) es el principal factor que determina la reabsorción de agua en el segmento distal de la nefrona. Cuando se administra una sobrecarga acuosa, se suprime la secreción de ADH, el túbulo distal y colector se vuelven impermeables al agua, y la orina diluida generada en el asa de Henle se excreta prácticamente inmodificada. Por el contrario, en condiciones de deshidratación, las concentraciones de ADH son muy altas, el túbulo distal y colector son muy permeables al agua y el líquido tubular se concentra progresivamente a lo largo de la nefrona distal por reabsorción de agua, en virtud del gradiente osmótico existente entre el líquido tubular y el intersticio medular hipertónico.
El aclaramiento de agua libre tras sobrecarga acuosa es normal en pacientes con cirrosis hepática compensada y reducido en la mayoría de los pacientes con ascitis4. El grado de deterioro de la excreción de agua libre, sin embargo, varía considerablemente de un paciente a otro. Mientras que en algunos está sólo ligeramente reducido, en otros es muy bajo, y algunos pacientes son incapaces de diluir la orina en condiciones de sobrecarga acuosa (aclaramiento de agua libre negativo). Estos pacientes con aclaramiento de agua libre muy bajo (< 1 ml/min) son los que desarrollan hiponatremia dilucional5. Este concepto es importante desde el punto de vista terapéutico. La hiponatremia en la cirrosis con ascitis no se debe a un déficit de sodio, sino a un exceso de agua, y el tratamiento debe ir dirigido a corregir este aspecto. La incidencia de hiponatremia dilucional en pacientes cirróticos ingresados para el tratamiento de un episodio de ascitis es de aproximadamente el 35%5.
Hormona antidiurética
La ADH es producida por neuronas de los núcleos supraóptico y paraventricular del hipotálamo, se transporta en vesículas a través de los axones neuronales y, por último, se almacena en gránulos secretores en las terminaciones neuronales de la neurohipófisis. La secreción de ADH se inicia con la propagación de un estímulo neuronal que causa despolarización de la membrana celular, entrada de calcio, fusión de los gránulos con la membrana celular y liberación de su contenido al espacio extracelular y a la sangre6. Los factores más importantes que influyen en la secreción de ADH son la presión osmótica del plasma y el estado circulatorio2,3. Pequeños cambios en la osmolaridad plasmática, actuando a través de un grupo de neuronas concentradas en el hipotálamo anterior (osmorreceptor), producen cambios drásticos en la secreción de ADH. Las alteraciones hemodinámicas modifican la secreción de ADH mediante estímulos neurogénicos que, partiendo de receptores de presión y de volumen intravasculares (barorreceptores), alcanzan los núcleos supraópticos y paraventricular a través de los nervios glosofaríngeo y neumogástrico. Al contrario de lo que sucede con los estímulos osmóticos, los descensos pequeños del volumen circulatorio (< 10%) o de la presión arterial tienen poco efecto en las concentraciones plasmáticas de ADH. Sin embargo, descensos más intensos se acompañan de un aumento progresivo de la ADH circulante. Cuando la osmolaridad plasmática y la volemia envían estímulos contradictorios a los núcleos supraóptico y paraventricular (p. ej., coexistencia de hipovolemia e hiponatremia), predomina el estímulo hemodinámico2.
Los 2 efectos biológicos más importantes de la ADH son el aumento de la permeabilidad al agua en el túbulo distal y colector, lo cual facilita la reabsorción pasiva de agua desde la luz tubular al intersticio medular hipertónico, y la contracción de la musculatura lisa vascular (vasoconstricción). El efecto hidroosmótico de la ADH se inicia a través de su interacción con receptores V2 en la membrana basocelular de los túbulos colectores7. El resultado de esta interacción es una secuencia de eventos consistente en la activación de la enzima adenilciclasa, formación de adenosinmonofosfato cíclico, activación de una cinasa proteica citosólica e inserción de canales de agua (acuaporina 2) en la membrana luminal de las células epiteliales del túbulo colector. En ausencia de ADH, la membrana luminal del túbulo colector es impermeable al agua debido a la ausencia de canales de agua. Por el contrario, la membrana basocelular es muy rica en acuaporina 3 (un canal de agua diferente que también transporta urea) y altamente permeable al agua aun en ausencia de ADH. El efecto vasoconstrictor de la ADH se inicia con la interacción de la hormona con receptores V1 situados en la musculatura vascular lisa7. El efecto final es un aumento de la concentración intracelular de calcio que determina contracción celular. El efecto vascular de la ADH es particularmente intenso en las circulaciones esplácnica, cutánea y muscular, mientras que la circulación renal mucho menos sensible al efecto vasoconstrictor de esta hormona.
Existen muchas evidencias que indican que la ADH desempeña un papel muy importante en la disminución de la capacidad renal de excretar agua libre en la cirrosis4,8. Sus concentraciones plasmáticas están elevadas en la mayoría de los pacientes con cirrosis hepática y ascitis, y se correlacionan inversamente con la capacidad renal de excretar agua libre. Estudios longitudinales en ratas con cirrosis experimental y ascitis han demostrado que la disminución de la capacidad renal de excretar agua libre aparece en estrecha relación cronológica con el inicio de una hipersecreción de ADH. Ratas Brattleboro (ratas con deficiencia congénita de ADH) con cirrosis y ascitis no desarrollan alteración de la capacidad renal de excretar agua libre. Los riñones de las ratas con cirrosis y ascitis muestran una expresión génica de acuaporina 2 aumentada. Finalmente, el bloqueo farmacológico de los receptores V2 con antagonistas específicos peptídicos y no peptídicos de la ADH corrige completamente la disminución de la capacidad renal de excretar agua libre en ratas con cirrosis hepática y ascitis9-12. Este efecto también se ha observado tras la administración de niravolina (RU 51-599), un agonista kappa opioide que inhibe la liberación y el efecto tubular de la ADH, en ratas con cirrosis y a pacientes con cirrosis hepática y ascitis12,13.
Los elevados valores circulantes de ADH en la cirrosis con ascitis son debidos a un aumento de la síntesis hipotalámica, no a una disminución del aclaramiento hepático de la hormona. Existen evidencias de que la hipersecreción de ADH se debe a un estímulo hemodinámico4,6,8. La mayoría de los pacientes con hipersecreción de ADH tienen un grado de hiponatremia que suprimiría totalmente las concentraciones circulantes de la hormona en individuos normales. Por otra parte, la ADH en plasma de los pacientes cirróticos con ascitis se correlaciona directamente con la actividad de la renina plasmática y la concentración de noradrenalina, y sus valores se suprimen por maniobras que aumentan el volumen arterial efectivo, como la inmersión en agua hasta el cuello o la inserción de un shunt peritoneovenoso. Finalmente, el bloqueo de los receptores V1 con antagonistas específicos de la ADH en modelos experimentales de cirrosis hepática se asocia a un descenso significativo de la presión arterial, efecto que no se observa en los animales sanos. Esto indica que la hipersecreción de ADH en la cirrosis hepática constituye un mecanismo homeostático para mantener la presión arterial en límites normales o casi normales, y que el estímulo más probable de la ADH en esta enfermedad es la hipotensión arterial.
Existen muchos estudios que han demostrado que la hipertensión portal, por un mecanismo no bien conocido en el que intervendría una aumentada síntesis endotelial de óxido nítrico, se asocia a una marcada vasodilatación arterial esplácnica que compromete la presión arterial14. La activación del sistema nervioso simpático y la estimulación consiguiente del sistema renina-angiotensina-aldosterona y de la ADH serían hechos intermedios de este trastorno, mientras que la retención renal de sodio y la alteración de la capacidad renal de excretar agua libre serían las consecuencias finales.
Otros factores involucrados en la excreción de agua libre
En la cirrosis hepática con ascitis el aporte de solutos a la rama ascendente del asa de Henle disminuye como consecuencia de una disminución del filtrado glomerular y de un aumento de la reabsorción proximal de sodio8. Por tanto, la generación de agua libre en el segmento dilutor de la nefrona puede verse comprometida por una menor llegada de cloro y sodio a este segmento de la nefrona. El túbulo colector sintetiza prostaglandina E2 (PGE2) en respuesta a la acción de la ADH. Por otra parte, la PGE2 inhibe el efecto tubular renal de la ADH. Por tanto, en la nefrona distal existe un mecanismo de retroalimentación negativa mediante el cual la ADH estimula la síntesis de su inhibidor la PGE2, la cual modula su efecto hidroosmótico. La síntesis tubular renal de PGE2 está aumentada en pacientes con cirrosis hepática y ascitis. Por otra parte, la inhibición de la síntesis renal de prostaglandinas con antiinflamatorios no esteroideos en estos pacientes se asocia a un notable descenso de la capacidad renal de excretar agua libre. Estos resultados indican que la disminución del aclaramiento de agua libre en los pacientes cirróticos con ascitis sería aún más bajo si la PGE2 tubular no antagonizara el efecto de la ADH, y explican por qué muchos pacientes con cirrosis hepática y ascitis mantienen una capacidad renal de excretar agua libre relativamente preservada a pesar de tener concentraciones elevadas de ADH15. Es posible, por tanto, que el deterioro de la excreción renal de agua libre en la cirrosis apareciera cuando la síntesis renal de prostaglandinas fuera insuficiente para antagonizar el efecto de la ADH.
Tratamiento de la hiponatremia dilucional. Agentes acuaréticos
Tradicionalmente el tratamiento de la hiponatremia dilucional en la cirrosis se ha basado en la restricción de la ingesta de agua. Sin embargo, este tratamiento es difícil de aplicar y raramente efectivo. La administración de sodio en estos pacientes está contraindicada porque aumenta la velocidad de formación de ascitis8. El futuro del tratamiento de la hiponatremia dilucional en la cirrosis son los fármacos acuaréticos. Se trata de fármacos que interfieren con los efectos renales de la ADH y, por lo tanto, inhiben la reabsorción de agua en los túbulos colectores16-18. Incrementan el volumen urinario sin afectar la excreción de solutos, llevando a poliuria hipotónica, y por tanto son el tratamiento ideal de la hiponatremia en los pacientes con cirrosis u otras enfermedades asociadas con retención de agua tales como insuficiencia cardíaca congestiva o síndrome de secreción inapropiada de ADH (SIADH).
El primer agente acuarético se descubrió hace más de 25 años. La poliuria hipotónica se consideraba un efecto colateral de la demeclociclina, una tetraciclina usada para el tratamiento del acné. Este efecto se relaciona con una inhibición en el mensajero intracelular de la adenilatociclasa de la ADH en las células del túbulo colector. El papel de la demeclociclina en el tratamiento de la hiponatremia se estudió en pacientes con SIADH y cirróticos con ascitis. En ambos procesos se observaron corrección de la retención de agua y aumento de las concentraciones séricas de sodio. Desafortunadamente, la demeclociclina afectaba la perfusión renal y la filtración glomerular en la cirrosis, por lo que su uso se abandonó19. Durante los años setenta y principios de los ochenta se encontró que los péptidos opiodes con afinidad por los receptores μy κ tenían profundos efectos en el metabolismo renal de agua. Los agonistas μ típicamente causan antidiuresis por un incremento de la secreción de ADH. En contraste, los agonistas κ producen poliuria hipotónica por supresión de la ADH. A inicios de los noventa se desarrolló el agonista selectivo κ-opioide niravoline como un agente acuarético12,13,20 que producía un importante incremento de la excreción renal de agua y de la concentración sérica de sodio.
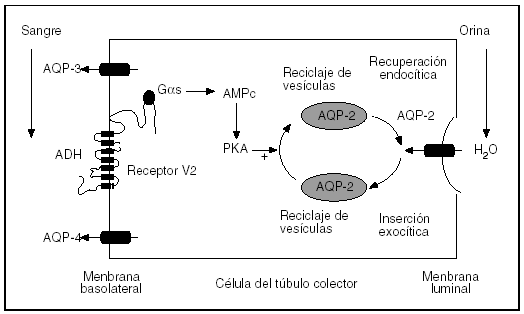
En los años setenta y ochenta los avances en la fisiología de la ADH fueron esenciales en el desarrollo de los actuales fármacos acuaréticos. El efecto hidroosmótico de la ADH está mediado por la inserción de canales de agua (acuaporina 2), los cuales se almacenan en las vesículas citoplasmáticas cerca de la luz tubular y en la membrana luminal de las células epiteliales del túbulo colector. En un estado no estimulado, la membrana es casi impermeable al agua por ausencia de estos canales. En contraste, la membrana basocelular, rica en acuaporina 3, es altamente permeable al agua. El efecto hidroosmótico de la ADH se inicia por la unión de la hormona al receptor V2 en la membrana basolateral de las células epiteliales del túbulo colector. Este receptor se une a la adenilatociclasa, y la estimulación da lugar al paso del adenosintrifosfato a adenosinmonofosfato cíclico. Éste activa una proteincinasa citosólica que promueve la inserción de las moléculas de acuaporina 2 en la membrana luminal. El agua se reabsorbe pasivamente del lumen tubular hipotónico al intersticio medular hipertónico (fig. 3). En contraste, el efecto vasoconstrictor de la ADH depende de la interacción de la hormona con los receptores V1 en la membrana de las células musculares lisas. El mecanismo intracelular es similar al de otros vasoconstrictores e involucra un incremento del calcio citosólico. Manning y Sawyer21,22 fueron los primeros investigadores que desarrollaron antagonistas específicos de los receptores V2. Ellos modificaron la molécula de desmopresina, un antagonista selectivo de V2, y obtuvieron numerosos péptidos con afinidad por los receptores V2, pero sin actividad antidiurética en animales experimentales. Clària et al9 usaron uno de estos péptidos en ratas con cirrosis, ascitis e hiponatremia y observaron la normalización del metabolismo renal de agua. Por desgracia, cuando estos péptidos se usaron en humanos, paradójicamente mostraban un débil agonismo V2.

Fig. 3. Esquema del receptor de acuaporina 2 (AQP): se muestran los mecanismos de inserción, procesos intracelulares de reciclaje y recuperación dependientes de la fosfocinasa. ADH: hormona antidiurética; AMPc: adenosinmonosfosfato cíclico; PKA: proteincinasa A.
La era moderna de los fármacos acuaréticos se inició en 1991, cuando se describió un antagonista V1 no peptídico activo oralmente. Un año más tarde, mediante modificaciones estructurales de la molécula, se obtuvo el primer antagonista selectivo no peptídico de los receptores V2 (OPC-31260) y se sentaron las bases para la síntesis de otros antagonistas V2 (VPA-985, SR.121463, OPC-41061 y YM-087)10,11,23-25. En animales y sujetos sanos, la administración de los antagonistas de los receptores V2 se asocia con un notable incremento del volumen urinario, sin cambios mayores en la excreción de solutos, lo que lleva a una poliuria hipotónica. Estudios posteriores han comunicado efectos similares en pacientes y animales de experimentación con SIADH, insuficiencia cardíaca congestiva y cirrosis.
Varias enfermedades podrían beneficiarse de los fármacos acuaréticos, y la cirrosis es probablemente la principal indicación para estos agentes por los siguientes motivos:
1. La frecuencia de retención de agua en la cirrosis es muy alta. Mientras que la prevalencia de hiponatremia importante (sodio sérico < 130 mEq/l) en los pacientes hospitalizados oscila entre el 1 y el 4%, en los pacientes con cirrosis y ascitis es del 30%.
2. De momento no hay tratamiento para la hiponatremia en la cirrosis descompensada. La restricción de líquidos no es práctica ni efectiva. Por otro lado, la administración de sodio quizá incremente las concentraciones séricas de sodio, pero a expensas de un incremento de la ascitis.
3. El principal componente del edema en la cirrosis es el agua isotónicamente reabsorbida con sodio en los túbulos renales por acción de la aldosterona y otros sistemas antinatriuréticos. El agua retenida por acción de la ADH en pacientes con hiponatremia dilucional es también un componente importante. Por lo tanto, los agentes acuaréticos pueden mejorar el tratamiento de la ascitis y el edema en estos pacientes.
4. Aproximadamente el 25% de los pacientes cirróticos requieren dosis elevadas de diuréticos y secundariamente desarrollan hiponatremia14,26-28. El mecanismo más probable es el empeoramiento de la hipovolemia arterial efectiva secundaria a la depleción de volumen. Los agentes acuaréticos podrían prevenir este efecto adverso del tratamiento diurético.
5. Los síntomas asociados con hiponatremia incluyen un amplio espectro de manifestaciones neurológicas, que van desde síntomas inespecíficos como cefalea, náuseas o calambres, hasta problemas mayores como desorientación, confusión, letargia, convulsiones, déficit neuronales focales e incluso coma. Esto refleja edema cerebral, resultado del paso osmótico del agua hacia el cerebro. No suele haber síntomas neurológicos significativos hasta que el sodio sérico es menor de 125 mEq/l, con una variabilidad individual marcada. Gran parte de esta variabilidad puede entenderse considerando el proceso de regulación del volumen cerebral. El cerebro y la osmolaridad del líquido extracelular están en equilibrio. Después de la inducción de hipoosmolaridad en el líquido extracelular, el movimiento de agua hacia el cerebro provoca edema cerebral. Sin embargo, en respuesta al edema cerebral, la pérdida rápida de osmólitos orgánicos y electrólitos del espacio extracelular lleva a la pérdida de agua intracelular y a la disminución del volumen cerebral, con tendencia a la normalidad. Esta respuesta homeostática, la cual ocurre en un lapso de 48 h, explica por qué la encefalopatía hiponatrémica es frecuente en pacientes con hiponatremia aguda y no crónica. La hiponatremia espontánea en la cirrosis es por lo general asintomática ya que se desarrolla lentamente. Sin embargo, no es infrecuente que los pacientes presenten una caída rápida de la concentración sérica de sodio (después de paracentesis o mediante tratamiento agresivo con diuréticos), lo que conduce a encefalopatía hiponatrémica. El tratamiento en estos pacientes se basa en la infusión intravenosa de soluciones salinas y, en estos casos, los fármacos acuaréticos pueden ser otra indicación.
6. Finalmente, los fármacos acuaréticos quizá sean de interés en los pacientes cirróticos con hiponatremia previa al trasplante hepático. Muchos donantes tienen hipernatremia e hiperosmolaridad, y hay evidencias de que las grandes diferencias en la concentración sérica de sodio entre el donante y el receptor predisponen a la disfunción temprana del injerto29. Sin embargo, la rápida normalización del sodio sérico con la infusión de soluciones salinas y plasma durante la cirugía quizá precipite síndromes osmóticos desmielinizantes30. El incremento prequirúrgico del sodio sérico con los fármacos acuaréticos quizá reduzca este problema.
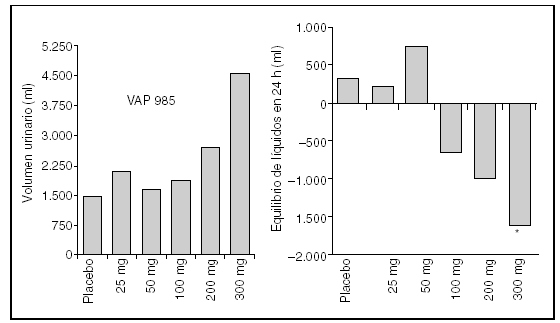
Varias investigaciones experimentales en fases 1 y 2 en pacientes han demostrado que los antagonistas V2 de la vasopresina son extremadamente efectivos a la hora de incrementar la aclaración de agua libre y normalizar las concentraciones séricas de sodio en pacientes cirróticos con ascitis e hiponatremia dilucional. El incremento del volumen urinario depende de la dosis y, cuando se administra la dosis apropiada, las concentraciones de sodio se normalizan en pocos días31 (fig. 4). Sin embargo, han de investigarse muchos aspectos concernientes al uso de los fármacos acuaréticos en los pacientes cirróticos. La manera en que estos agentes deben emplearse en dichos pacientes es un aspecto importante, ya que tienen múltiples interacciones con agentes natriuréticos usados en esta enferemedad. Por tanto, es esencial conocer cómo estos fármacos afectan la excreción de sodio en pacientes con cirrosis y ascitis. En condiciones normales los agentes acuaréticos incrementan el volumen urinario sin afectar la excreción de sodio. Esto no ha sido el caso en pacientes y en animales experimentales con cirrosis y ascitis, ya que, en adición a una poliuria hipotónica, estos agentes incrementan la excreción urinaria de sodio, por lo que las indicaciones de su uso fuera de la hiponatremia dilucional espontánea (p. ej., hiponatremia inducida por diuréticos, hiponatremia previa a trasplante) deben confirmarse. Finalmente, hay evidencia de que el 60% de los pacientes cirróticos con hiponatremia dilucional responden a los fármacos acuaréticos, por lo que deben investigarse los factores que influyen en dicha respuesta32.

Fig. 4. Efecto del VPA-985 (antagonista V2) sobre el volumen urinario y el balance de líquidos. El volumen urinario depende de la dosis y se obtiene de igual manera equilibrio negativo en el manejo de líquidos a partir de la dosis de 100 mg. (Tomada de Guyader et al27.)
ASCITIS REFRACTARIA
Definición y criterios diagnósticos
Con el objetivo de estandarizar los criterios diagnósticos de ascitis refractaria, el Club Internacional de Ascitis realizó una conferencia consenso en 1994 en la que propuso las siguientes definiciones y criterios diagnósticos33:
Ascitis refractaria. Es la ascitis que no puede eliminarse o cuya recurrencia (p. ej., tras paracentesis terapéutica) no puede evitarse mediante tratamiento médico. El término ascitis refractaria incluye 2 subtipos: la ascitis resistente a los diuréticos y la ascitis intratable con diuréticos.
Ascitis resistente a los diuréticos. Es la ascitis que no puede eliminarse o cuya recurrencia no puede evitarse debido a una falta de respuesta a la restricción de la ingesta de sodio y dosis máxima de diuréticos.
Ascitis intratable con diuréticos. Es la ascitis que no puede eliminarse o cuya recurrencia no puede evitarse debido al desarrollo de complicaciones inducidas por diuréticos que impiden el uso de dosis efectivas de estos fármacos.
La duración del tratamiento es necesaria para definir una ascitis como refractaria, ya que los pacientes deben estar tratados con dosis máximas de diuréticos durante, al menos, una semana.
Falta de respuesta a los diuréticos: pérdida de peso inferior a 200 g/día durante los últimos 4 días de tratamiento diurético máximo, asociada a una excreción urinaria de sodio inferior a 50 mEq/día.
Restricción de la ingesta de sodio: dieta con 50 mEq día de sodio.
Dosis máxima de diuréticos: 400 mg/día de espironolactona más 160 mg/día de furosemida (4 mg/día de bumetanida o dosis equivalentes de otros diuréticos de asa).
Recurrencia de la ascitis: reaparición de ascitis grado 2 (moderada) o 3 (masiva o a tensión) en 4 semanas después de su eliminación por paracentesis. La reacumulación de ascitis en los primeros 2-3 días tras la paracentesis no debe considerarse una recurrencia de la ascitis, dado que esta ascitis temprana es secundaria a una rápida translocación de líquido desde el espacio intersticial al intraperitoneal.
Complicaciones inducidas por diuréticos. La encefalopatía inducida por diuréticos consiste en el desarrollo de encefalopatía hepática sin ningún otro factor precipitante. La insuficiencia renal inducida por diuréticos se define como un aumento de la concentración plasmática de creatinina superior al 100% por encima de 2 mg/dl en pacientes con ascitis que responden al tratamiento diurético. La hiponatremia inducida por diuréticos consiste en un descenso de la concentración plasmática de sodio superior a 10 mEq/l que alcance un valor inferior a 125 mEq/l. La hipo o hiperpotasemia inducidas por diuréticos se definen como un descenso de la concentración plasmática de potasio por debajo de 3 mEq/l, o un aumento hasta valores superiores a 6 mEq/l, a pesar de usar medidas apropiadas para normalizarla.
La prevalencia de ascitis refractaria en pacientes con cirrosis hepática que ingresan en un hospital general para el tratamiento de un episodio de ascitis se estima que oscila entre un 5 y un 10%. Sin embargo, no existe ningún estudio específicamente realizado para evaluar este punto. La mayoría de los datos de que disponemos son estimaciones indirectas a partir de estudios que analizan la efectividad e incidencia de complicaciones en pacientes con cirrosis hepática, y que se publicaron antes de los criterios definidos por el Club Internacional de Ascitis expuestos más arriba. En un estudio de Strauss et al34 en 100 pacientes consecutivos con cirrosis hepática y ascitis tratados con dieta de 40 mEq/día de sodio y dosis progresivamente crecientes de diuréticos, sólo el 2% tenía ascitis resistente a los diuréticos. Sin embargo, un 4% tenía ascitis intratable con diuréticos por encefalopatía hepática inducida por estos fármacos. Un 24% de los pacientes desarrolló azoemia, aunque no puede conocerse por sus datos cuántos de ellos entrarían en el grupo de ascitis intratable sobre la base del deterioro de la función renal. En un estudio similar de Gatta et al35 sólo el 4% de los pacientes presentaron ascitis resistente a los diuréticos y, sorprendentemente, ninguno presentó ascitis intratable por encefalopatía hepática o deterioro de la función renal. En un estudio en más de 300 pacientes estudiados por Stanley et al36, la tasa de ascitis resistente fue del 10%. Finalmente, Bernardi et al37 encontraron una incidencia de ascitis resistente del 5% y de ascitis intratable del 2%. Todos estos datos, sin embargo, probablemente infravaloran en cierta manera la prevalencia real del problema, dado que existe el sesgo de los pacientes con insuficiencia hepática y/o renal grave, que no suelen incluirse en este tipo de estudios.
Patogenia, pronóstico y diagnóstico
La mayoría de los pacientes con ascitis resistente a los diuréticos tiene síndrome hepatorrenal tipo 2, incluso en aquellos casos con creatinina sérica inferior a 1,5 mg/dl (valor de corte que define la existencia de dicho síndrome). Si se determinan el flujo sanguíneo renal y el filtrado glomerular por técnicas de aclaramiento (aclaramiento de paraaminohipúrico e inulina, respectivamente), se obtendrán valores muy bajos, lo cual refleja la poca sensibilidad de la creatinina sérica para detectar cambios importantes en el filtrado glomerular. La patogenia de la ascitis resistente está relacionada con este deterioro de la función renal33,38. Los diuréticos más comúnmente empleados en el tratamiento de la ascitis son la furosemida y la espironolactona. La furosemida, como otros diuréticos de asa (bumetanida, ácido etacrínico), inhibe la reabsorción de sodio en la rama ascendente del asa de Henle. La espironolactona inhibe la reabsorción de sodio en el túbulo distal y, sobre todo, en el túbulo colector. En cambio, no disponemos de ningún diurético que inhiba la reabsorción proximal de sodio. La retención de sodio en los pacientes cirróticos con filtrado glomerular normal o sólo moderadamente bajo ocurre de forma predominante en la nefrona distal. Por tanto, no es extraño que estos pacientes respondan satisfactoriamente a los diuréticos. Por el contrario, la retención de sodio en los pacientes con síndrome hepatorrenal se debe a un descenso del sodio filtrado y a un aumento de la reabsorción proximal de este ion. Muy poco sodio alcanza la nefrona distal, que es el lugar de acción de los diuréticos.
El componente farmacológicamente activo de la furosemida es aquel que alcanza la luz tubular por mecanismos de secreción activa en el túbulo proximal. La furosemida secretada viaja con el líquido tubular y, al alcanzar la rama ascendente del asa de Henle, inhibe un transportador de sodio específico en la membrana luminal de la célula tubular. La espironolactona penetra en la célula del túbulo colector a través de la membrana plasmática y compite con la aldosterona en los receptores específicos citosólicos. Ambos fármacos han de llegar en cantidades apropiadas a las células tubulares para poder actuar. En pacientes cirróticos con síndrome hepatorrenal el descenso del flujo sanguíneo renal, por tanto, puede contribuir a la resistencia a los diuréticos.
Antes de considerar que un paciente presenta ascitis refractaria y, por tanto, de indicar un tratamiento potencialmente invasivo, es importante evaluar si ingiere realmente una dieta hiposódica y conocer el tipo de medicamentos con los que está siendo tratado. La causa más frecuente de ascitis refractaria es la ingesta de una dieta con alto contenido en sodio. Los fármacos que descienden la presión arterial pueden agravar el deterioro circulatorio y renal que presentan los pacientes cirróticos y, por tanto, la respuesta a los diuréticos39. Finalmente, el efecto natriurético de los diuréticos de asa está parcialmente mediado por prostaglandinas. Por tanto, los antiinflamatorios no esteroideos reducen de forma muy intensa el efecto diurético de la furosemida40. Existen estudios que demuestran que estos fármacos también reducen la respuesta a la espironolactona.
La probabilidad de supervivencia de los pacientes con ascitis refractaria es muy corta. Aproximadamente la mitad fallece antes de 6 meses. Por consiguiente, cualquier paciente joven con ascitis refractaria debe ser incluido sin dilación en un programa de trasplante hepático.
Tratamiento
En 1975 LeVeen et al diseñaron una prótesis que comunica la cavidad peritoneal con el torrente circulatorio. Posee una válvula unidireccional que permite el paso de ascitis en sentido ascendente pero impide el reflujo de sangre al interior del segmento intravenoso de la prótesis. El objetivo de esta anastomosis peritoneovenosa es revertir las consecuencias de la formación de ascitis. Si la ascitis es líquido que escapa del compartimiento intravascular, lo que pretende la prótesis es retornarlo al torrente intravascular y, con ello, mejorar el estado circulatorio y corregir la hipovolemia arterial. Existen numerosos estudios que demuestran que la anastomosis peritoneovenosa de LeVeen realmente consigue mejorar el estado circulatorio y reducir de forma marcada las concentraciones plasmáticas de renina, aldosterona, noradrenalina y ADH41. En pacientes con síndrome hepatorrenal tipo 2 mejora la función renal y aumenta la eficacia del tratamiento diurético. Por ello fue el primer tratamiento realmente útil en pacientes con ascitis refractaria. El principal inconveniente del shunt peritoneovenoso es la frecuencia con la que se obstruye. La obstrucción puede ocurrir en la válvula por acumulación de restos celulares y fibrina, o bien en el segmento intravenoso por reflujo de sangre y obstrucción trombótica. La probabilidad de obstrucción de un shunt peritoneovenoso es del 40% durante los primeros 6-9 meses tras su colocación42,43. Ello obliga a frecuentes reintervenciones quirúrgicas para extraer el shunt obstruido y reemplazarlo por uno nuevo. No es extraño que un paciente tenga que ser reintervenido en varias ocasiones si se desea mantener comunicada la cavidad peritoneal con el torrente circulatorio. Aunque se ha señalado que la colocación de una punta de titanio (sustancia muy tromborresistente) en el segmento intravenoso de la prótesis podría reducir la incidencia del obstrucción, esto no se ha confirmado en un estudio controlado efectuado en una serie amplia de pacientes43. Otras complicaciones relacionadas con el shunt de LeVeen son la obstrucción trombótica de la vena cava superior por lesión del endotelio vascular y una peritonitis plástica por reacción desmoplástica al segmento intraperitoneal de la prótesis. Las consecuencias de estas complicaciones son el síndrome de la vena cava superior, generalmente mortal, y la obstrucción intestinal, en muchas ocasiones inoperable. Sin embargo, estas complicaciones, por fortuna, son muy infrecuentes.
La reintroducción de la paracentesis como tratamiento de la ascitis a tensión ha cambiado radicalmente la actitud terapéutica en pacientes con ascitis refractaria. Existen 2 estudios controlados en series amplias de pacientes con ascitis refractaria que comparan la paracentesis con el shunt de LeVeen y demuestran que la primera es un tratamiento muy eficaz y seguro en estos pacientes42,43. Aunque el número de rehospitalizaciones por ascitis, como era de esperar, es más alto en los pacientes tratados con paracentesis, el tiempo total en el hospital durante el seguimiento es similar con ambos tratamientos. Este hecho se debe a las frecuentes hospitalizaciones en el grupo tratado con shunt peritoneovenoso para reoperaciones por obstrucción de la prótesis. No existen diferencias entre ambos grupos terapéuticos en relación con la supervivencia. La introducción de la paracentesis terapéutica determinó una disminución progresiva del uso de la derivación peritoneovenosa hasta que prácticamente se abandonó.
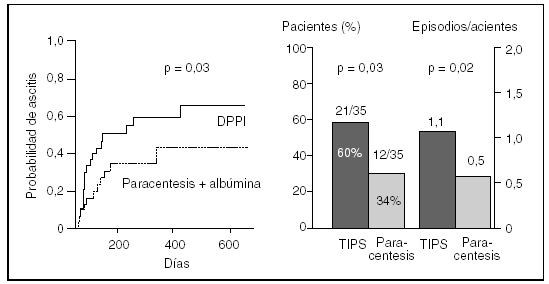
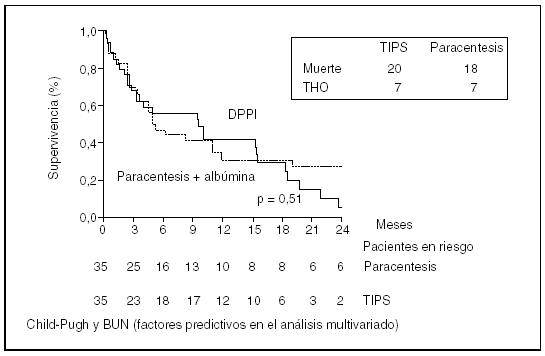
La derivación portocava percutánea intrahepática (DPPI), al corregir la hipertensión portal, mejora el deterioro circulatorio y la función renal en los pacientes con ascitis refractaria44. Muchos de estos pacientes permanecen sin ascitis a pesar de ingerir una dieta normosódica y recibir dosis bajas de diuréticos45. La DPPI tiene 2 problemas importantes. El primero es la frecuencia de disfunción u obstrucción del shunt, comparable a la de la prótesis de LeVeen, que obliga a frecuentes reintervenciones para angioplastia o colocación de un nuevo stent. El segundo es que se comporta como un shunt portocava laterolateral, con importante sustracción de sangre arterial desde el hígado a la vena porta y a la vena cava. Ello determina un empeoramiento de la función del hígado y una alta incidencia de encefalopatía hepática. Existen 2 estudios controlados en pacientes con ascitis refractaria que comparan la paracentesis terapéutica con la DPPI46,47. Aunque el control de la ascitis fue mejor en los tratados con DPPI, la incidencia de encefalopatía hepática fue más elevada en estos pacientes, sin que hubiera diferencias en relación con la supervivencia (figs. 5 y 6).

Fig. 5. En la gráfica de la izquierda se observa la probabilidad de recidiva de la ascitis al comparar el tratamiento con derivación portocava percutánea intrahepática (DPPI) con la paracentesis más albúmina en un seguimiento de 600 días. En la gráfica de la derecha se muestran el porcentaje de pacientes y el número de episodios por paciente de encefalopatía al comparar nuevamente estos grupos. (Tomada de Ginès et al47.)

Fig. 6. Gráfica que muestra la supervivencia a los 2 años en el tratamiento de la ascitis refractaria cuando se compara la derivación portocava percutánea intrahepática (DPPI) con la paracentesis más albúmina. BUN: nitrógeno ureico sanguíneo; THO: trasplante hepático ortotópico. (Tomada de Ginès et al47.)
Actualmente, la anastomosis peritoneovenosa prácticamente no se utiliza en el tratamiento de la ascitis refractaria de la cirrosis. El Club Internacional de Ascitis recomienda interrumpir el tratamiento diurético en los pacientes que no alcanzan una excreción urinaria de sodio superior a 30 mEq/l, al objeto de evitar complicaciones relacionadas con estos fármacos (hiperpotasemia, encefalopatía hepática). El tratamiento de elección sería la paracentesis terapéutica asociada a la administración de albúmina. La DPPI estaría indicada en los pacientes sin encefalopatía hepática y con una puntuación de Child-Pugh igual o inferior a 12 en quienes la reacumulación de ascitis fuera muy rápida y que no toleraran paracentesis frecuentes48.
PATOGENIA DEL SÍNDROME HEPATORRENAL EN LA CIRROSIS
Además de la retención renal de sodio, que es el principal factor causante de la formación de ascitis y edemas, los pacientes con cirrosis muestran una disminución de la capacidad renal de eliminar agua libre y vasoconstricción de las arterias renales, que son responsables del desarrollo de hiponatremia dilucional y síndrome hepatorrenal, respectivamente8,33,49. Los resultados de las investigaciones realizadas en las 2 últimas décadas han establecido de forma convincente que estas anormalidadas de la función renal y la formación de ascitis en la cirrosis están relacionadas patogénicamente con las importantes alteraciones en la función del sistema circulatorio14,50,51. Las principales anormalidades en la función del sistema circulatorio son el aumento del volumen sanguíneo total, el incremento en la compliancia vascular, la reducción de las resistencias vasculares sistémicas y el aumento del gasto cardíaco. El volumen sanguíneo total en los pacientes con cirrosis no está reducido, tal como proponía la teoría clásica de la formación de ascitis, sino que está aumentado en comparación con los sujetos sanos52. El incremento del volumen sanguíneo total es una manifestación temprana de la cirrosis que ocurre antes de la formación de la ascitis y que es la primera consecuencia de la retención renal de sodio. La compliancia vascular total está incrementada en los pacientes con cirrosis respecto a los sanos53,54. Este aumento de la compliancia vascular ocurre no sólo en la circulación arterial, sino probablemente también en la circulación venosa. Los mecanismos que contribuyen a este incremento en la compliancia vascular son la vasodilatación tanto de arterias como de venas, la disminución de la respuesta de vasoconstrictores endógenos y los cambios estructurales en la pared vascular. La reducción de las resistencias vasculares sistémicas totales se debe a una acentuada vasodilatación en la circulación esplácnica, ya que la resistencia al flujo en los lechos vasculares no esplácnicos (p. ej., miembros superiores e inferiores, riñones y cerebro) es normal o incluso elevada14,50,51. Esta vasodilatación esplácnica es consecuencia de la hipertensión portal, la cual no sólo causa un incremento de presión en el lado venoso de la circulación esplácnica, sino también vasodilatación de las arterias esplácnicas. El mecanismo exacto que lleva a la vasodilatación esplácnica no se conoce, pero probablemente se deba a un aumento de la síntesis/actividad de factores vasodilatadores, incluidos oxído nítrico, péptidos vasodilatadores y/o monóxido de carbono55-57. La acentuada vasodilatación arterial puede ser la responsable no sólo de la reducción de las resistencias vasculares sistémicas, sino también de la distribución anormal del volumen sanguíneo, con la reducción del volumen arterial efectivo (p. ej., volumen sanguíneo en el corazón, pulmones y lecho arterial central, que es percibido por los receptores arteriales), y una subsiguiente activación de factores vasoconstrictores y antinatriuréticos mediada a través de barorrecepto-res14,50,51. En la actualidad se cree que esta reducción del volumen arterial efectivo es fundamental en el desarrollo de la retención de sodio y formación de ascitis en la cirrosis. La acumulación predominante de líquido en la cavidad peritoneal a manera de ascitis es consecuencia de una elevada filtración en los capilares esplácnicos, resultado de un incremento anterógrado y retrógrado en la presión hidrostática secundario a la hipertensión portal y la vasodilatación arterial esplácnica, respectivamente, y a un incremento del coeficiente de filtración41,58. La hiponatremia dilucional espontánea y el síndrome hepatorrenal están también patogénicamente relacionados con la disfunción circulatoria y son debidos, al menos en parte, a la hipersecreción no osmótica de vasopresina ADH y a la acción de factores vasoconstrictores en la circulación renal, respectivamente. La disfunción renal en la cirrosis es de gran importancia clínica porque el desarrollo de insuficiencia renal reduce la supervivencia de los pacientes49.
Tratamiento
Vasoconstrictores y expansión de volumen
El fundamento de este tratamiento es la fisiopatogenia actual del síndrome hepatorrenal tipo 1. Los vasoconstrictores se administran para revertir la vasodilatación arterial esplácnica y la expansión de volumen, mejorar el retorno venoso y el gasto cardíaco. Los análogos de vasopresina (ornipresina y terlipresina) fueron los fármacos inicialmente usados para el tratamiento del síndrome hepatorrenal tipo 1 por su efecto predominante sobre la circulación esplácnica. La terlipresina, aprobada en varios países para el tratamiento de la hemorragia variceal aguda, ha sido el fármaco más frecuentemente usado. La ornipresina también es efectiva, pero su infusion constante produce con frecuencia complicaciones isquémicas.
La administración de terlipresina (0,5-2 mg/4-6 h por vía intravenosa) puede lograr una respuesta terapéutica completa, definida por la reducción de la creatinina sérica por debajo de 1,5 mg/dl, en el 50-75% de los pacientes tratados59-63. Esto se asocia con una marcada supresión de renina y noradrenalina y un incremento importante de la presión arterial media. No hay una relación directa entre la supresión de los sistemas neurohumorales endógenos, lo cual ocurre entres los primeros 3 días de tratamiento, y la disminución de la creatinina sérica, que se inicia entre 2 y 4 después de iniciado el medicamento. Pese a la normalización de la creatinina sérica, la función renal no alcanza valores normales y persiste una filtración glomerular baja, con rangos entre 30 y 50 ml/min en la mayoría de los casos (normal: 120 ml/min). En la mayor parte de los estudios, el tratamiento con terlipresina se mantiene hasta el descenso de la creatinina sérica por debajo de 1,5 mg/dl o durante un máximo de 15 días. Se desconoce si la administración continua de la terlipresina después de lograr un valor de creatinina inferior a 1,5 g/dl causa un mayor incremento en la filtración glomerular. En los pacientes que responden el volumen urinario mejora en las primeras 24 h. En algunos pero no en todos los pacientes, el tratamiento causa un incremento en la excreción de sodio59-63. Este efecto tardío lleva a considerar a la terlipresina como un agonista V2 de la vasopresina. Se ha señalado que la mejoría en la excreción de agua libre en el síndrome hepatorrenal tipo 1 se relaciona más con anormalidades en el manejo intrarrenal del sodio que con la secreción no osmótica de la ADH. En todos los estudios la albúmina intravenosa se administra a dosis variables durante el tratamiento con terlipresina. Estos datos indican que la respuesta terapéutica a la terlipresina es muy pobre si no se administra conjuntamente albúmina por vía intravenosa61,63. Un esquema recomendado para la administración de albúmina es 1 g/kg de peso durante el primer día, seguido de 20-40 g/día63. La administración de albúmina se interrumpe si la presión venosa central es mayor de 18 cmH2O. La edad superior a 65 años, el fallo hepático grave (Child-Pugh > 13) y la administración concomitante de albúmina se han identificado como predictores de la respuesta a la terlipresina61,63. La probabilidad de supervivencia en los pacientes con síndrome hepatorrenal tipo 1 que responden a la terlipresina se ha estimado en un 50% a los 3 meses y un 30% al año61. Estas cifras son comparables a las comunicadas en los pacientes con síndrome hepatorrenal tipo 2 y considerablemente superior a las de los pacientes con síndrome hepatorrenal tipo 1 no tratados. La recurrencia es poco frecuente después de la interrupción del tratamiento (aproximadamente el 15% de los pacientes), y el retratamiento es por lo general efectivo. La incidencia de efectos isquémicos que requieren interrumpir el tratamiento con terlipresina es baja (5-10%), y en la mayoría de los estudios se plantea excluir a los pacientes considerados de riesgo (enfermedad cardíaca isquémica o arterial periférica)59-63.
Las catecolaminas son efectivas en el tratamiento del síndrome hepatorrenal. Angeli et al64 administraron midrodrina (un agonista alfaadrenérgico) en asociación con albúmina por vía intravenosa y octreótido subcutáneo (suprime el glucagón) a 5 pacientes con síndrome hepatorrenal tipo 1. La dosis de midrodrina (7,5-12,5 mg cada 8 h) se ajustó a los incrementos de la presión arterial media de 15 mmHg o más. Los pacientes recibieron tratamiento durante al menos 20 días en el hospital y lo continuaron en casa. Hubo una notable mejoría en la perfusión renal y el filtrado glomerular, con supresión de renina, noradrenalina y ADH a valores normales o cercanos a la normalidad en todos los casos. Se trasplantó a pacientes a los 20 y 64 días, respectivamente, después de iniciado el tratamiento. Un paciente que no fue candidato a trasplante hepático estaba vivo sin tratamiento a los 472 días de salir del hospital. Los restantes 2 pacientes murieron a los 29 y 75 días, respectivamente. Un estudio reciente ha valorado los efectos del octreótido en los sistemas hemodinámico, neurohumoral y renal de los pacientes con síndrome hepatorrenal, sin observar cambios significativos. La eficacia terapéutica en el estudio de Angeli et al estuvo, por lo tanto, exclusivamente relacionada con el efecto vasoconstrictor de la midrodrina y con la expansión del volumen sanguíneo central por la albúmina.
Duvoux et al65 trataron a 12 pacientes con síndrome hepatorrenal tipo1 con noradrenalina (0,5-3,0 mg/h) y albúmina intravenosa durante un mínimo de 5 días. La reversión del síndrome se observó en 10 pacientes en asociación con un incremento de la presión arterial media y una notable reducción de la renina y aldosterona. Hubo un episodio de hipocinesia miocárdica reversible. Se practicó trasplante a 3 pacientes y otros 4 tuvieron una supervivencia superior a los 6 meses.
Derivación portocava percutánea intrahepática (DPPI)
Dado que la hipertensión portal es el acontecimiento inicial de la disfunción circulatoria en la cirrosis, la disminución de la presión portal a través de anastomosis portocava es un método racional en el tratamiento del síndrome hepatorrenal. Se han comunicado varios casos que muestran reversión del síndrome hepatorrenal después de un shunt quirúrgico portocava66,67. Sin embargo, la aplicabilidad de procedimientos quirúrgicos mayores en los pacientes con este síndrome es pobre. El desarrollo de los DPPI ha reintroducido la idea de tratar el síndrome hepatorrenal disminuyendo la presión portal.
Cuatro estudios validan el DPPI en el manejo del síndrome hepatorrenal68-71. Se trató en total a 30 pacientes. En 2 series no se realizó trasplante hepático, mientras que en las otras 2 se practicó un trasplante a 3 de los 9 pacientes a los 7, 13 y 35 días después del DPPI. La colocación del DPPI fue exitosa en todos ellos. Sólo uno murió a consecuencia del procedimiento. La filtración glomerular mejoró notablemente entre las semanas 1 y 4 después del DPPI y posteriormente se estabilizó. En un estudio donde se valoraron específicamente los sistemas neurohumorales, la mejora en la filtración glomerular y creatinina sérica se correlacionó con una marcada supresión de las concentraciones de renina y ADH69. La supresión de la noradrenalina plasmática fue menor que la de renina, situación observada en los casos de ascitis refractaria tratadas con DPPI. El seguimiento de la función hepática se llevó a cabo en 21 pacientes. La encefalopatía de novo o el deterioro en la encefalopatía preexistente ocurrió en 9 pacientes, en 5 de los cuales se controló con lactulosa. La supervivencia en los 27 pacientes sin trasplante hepático al mes, 3 y 6 meses fue del 81, el 58 y el 44%, respectivamente. Estos estudios apuntan fuertemente a que el DPPI tiene éxito en el tratamiento del síndrome hepatorrenal tipo 1. Hacen falta estudios que comparen el DPPI con el tratamiento farmacológico.
Como se indica arriba, uno de los puntos de interés en el tratamiento del síndrome hepatorrenal tipo 1 con vasoconstrictores y albúmina intravenosa es la observación de que, pese a la notable supresión de la renina y noradrenalina, indicativa de una mejoría importante en la función circulatoria, hay una filtración glomerular persistentemente baja. La razón de este retraso en la normalización es desconocida, pero podría deberse a la existencia de un componente del fallo renal que no responde a los cambios en la función circulatoria, o al hecho de que el volumen arterial efectivo no se normaliza con el tratamiento farmacológico. Un estudio reciente de Wong et al72 estudia esta hipótesis. El tratamiento con DPPI en pacientes que respondían a la farmacoterapia (midrodina, octreótido y albúmina) se asoció a la normalización de la filtración glomerular en la mayoría de los casos. Por lo tanto, el efecto del DPPI en la normalización de la filtración glomerular se debió a la corrección de la vasodilatación arterial, a un incremento de la precarga cardíaca y función ventricular o a ambos, situaciones que deben investigarse.
Trasplante hepático
Es el tratamiento de elección en el síndrome hepatorrenal73-77. Inmediatamente después del trasplante se observan alteraciones en la filtración glomerular y muchos pacientes requieren hemodiálisis (el 35% de los pacientes con síndrome hepatorrenal frente al 5% de aquellos que no lo presentan)73. La ciclosporina y el tacrolimus quizá contribuyen a esta afección en la función renal. Se ha planteado que el retraso en la administración de estos fármacos hasta la recuperación de la función renal, usualmente a las 48-72 h del trasplante. Después de este compromiso inicial en la función renal, la filtración glomerular comienza a mejorar un media de 30-40 ml/min cada 2 meses. Este fallo renal moderado persiste durante el seguimiento y es más acusado que el observado en los pacientes trasplantados sin síndrome hepatorrenal, probablemente debido a la nefrotoxicidad mayor de la ciclosporina y el tacrolimus en los pacientes con fallo renal previo al trasplante73. Las anormalidades hemodinámicas y neurohumorales asociadas con el síndrome hepatorrenal desaparecen al primer mes de la cirugía y los pacientes recuperan la excreción de sodio y agua libre78.
Los pacientes con síndrome hepatorrenal sometidos a trasplante tienen más complicaciones, permanecen más días en la unidad de cuidados intensivos y presentan mayor mortalidad intrahospitalaria que los trasplantados sin dicho síndrome73-77,79. La supervivencia, sin embargo, es buena (entre el 70 y el 80% al año)73-79.
El principal problema del trasplante hepático en el síndrome hepatorrenal tipo 1 es su aplicabilidad. Debido a la extremadamente corta supervivencia, la mayoría de los pacientes muere antes del trasplante. La introducción de la escala MELD, que incluye los valores de creatinina sérica, bilirrubina y razón normalizada internacional, para los pacientes en lista de espera ha resuelto parcialmente este problema al colocar a los pacientes con síndrome hepatorrenal en los primeros lugares de la lista. Debe realizarse tratamiento del síndrome hepatorrenal antes del trasplante porque la recuperación de este síndrome disminuye tanto la morbilidad temprana como la mortalidad después del trasplante y prolonga la supervivencia a largo plazo80.
Otros métodos terapéuticos
La hemodiálisis se usa con frecuencia en el tratamiento del síndrome hepatorrenal tipo 1 en muchos centros, particularmente en pacientes que son candidatos a trasplante hepático, con la finalidad de prevenir las complicaciones asociadas al fallo renal y mantener a los pacientes vivos hasta el trasplante. Sin embargo, los efectos benéficos de este procedimiento en el síndrome hepatorrenal no se han demostrado convincentemente81. Las complicaciones durante la hemodiálisis en estos pacientes son comunes e incluyen hipotensión arterial, hemorragia e infecciones. Por otro lado, los hallazgos clínicos o bioquímicos indicadores de terapia de reemplazamiento renal, tales como fallo cardíaco o respiratorio, acidosis o hipercalemia graves, son infrecuentes en el síndrome hepatorrenal tipo 1.
Durante muchos años, para el tratamiento del síndrome hepatorrenal se han usado otros fármacos diferentes de los vasoconstrictores sin eficacia comprobada, caso de los vasodilatadores renales, como la dopamina o las prostaglandinas82. Comunicaciones aisladas apuntan al efecto benéfico del octreótido solo, un fármaco que inhibe varios péptidos vasodilatadores de origen esplácnico, especialmente el glucagón. Sin embargo, un reciente estudio aleatorizado y controlado en que se administró a dosis de 50 μg/h en infusión no mostró efecto positivo83. Finalmente, la N-acetilcisteína mostró eficacia en una serie pequeña de pacientes a dosis de 300 mg cada 12 h, pero estos resultados requieren confirmación en series con mayor número de casos84.
Sistema molecular recirculante absorbente (MARS)
La diálisis extracorpórea de albúmina, un sistema que usa un dializado con albúmina que recircula y perfunde a través de columnas de carbón e intercambiadoras de aniones, mejora la función renal y la supervivencia, según se ha comunicado en pequeñas series de pacientes con síndrome hepatorrenal. Mitzner et al85 realizaron un estudio prospectivo y controlado para comparar el MARS con el tratamiento de soporte estándar (líquidos por vía intravenosa, dopamina y, si era necesario, vasoconstrictores). El objetivo era valorar la supervivencia a los 30 días en pacientes con síndrome hepatorrenal tipo 1 e insuficiencia hepática grave (bilirrubina > 15 mg/dl). Se incluyó a 13 pacientes, todos con Child-Pugh C, con una puntuación de 12,4 ± 1, situación pretrasplante 2A según el sistema UNOS y valores de bilirrubina de 25,7 ± 14 mg/dl. En el grupo MARS se observó una disminución significativa de los valores de bilirrubina y creatinina, así como un incremento del sodio sérico y de la actividad de protrombina (p < 0,01). La mortalidad fue del 100% en el grupo control al día 7, mientras que en el grupo MARS fue del 62,5% al día 7 y del 75% al día 30 (p < 0,01). Se necesitan futuros estudios para validar esta modalidad terapéutica.
Prevención del síndrome hepatorrenal
Dos estudios aleatorizados y controlados en series grandes de pacientes han demostrado que el síndrome hepatorrenal puede prevenirse con medidas específicas. En el primer estudio85, la administración de albúmina (1,5 g/kg por vía intravenosa en el momento del diagnóstico de la infección y 1 g/kg por vía intravenosa 48 h después) a pacientes con cirrosis y peritonitis bacteriana espontánea reduce notablemente la incidencia de disfunción circulatoria y síndrome hepatorrenal tipo 1 (un 10% de incidencia de síndrome hepatorrenal tipo 1 en pacientes que reciben albúmina frente al 33% en el grupo control). La mortalidad hospitalaria (un 10 frente a un 29%, respectivamente) y a los 3 meses (un 22 frente al 41%, respectivamente) fue a favor del grupo que recibió albúmina. En un segundo estudio86, la administración de pentoxifilina (un inhibidor del factor de necrosis tumoral), a dosis de 400 mg 3 veces al día, a pacientes con hepatitis alcohólica aguda grave redujo la aparición de síndrome hepatorrenal (un 8% en el grupo de pentoxifilina frente al 35%) y la mortalidad hospitalaria (el 24 frente al 46%, respectivamente). Dado que las infecciones bacterianas y la hepatitis alcohólica aguda son factores precipitantes importantes de síndrome hepatorrenal tipo 1, las medidas profilácticas quizá disminuyan la incidencia de esta complicación.

