




La histiocitosis es una enfermedad que se origina por la proliferación de células del sistema fagocítico mononuclear en diferentes tejidos. La histiocitosis de células de Langerhans (HCL) es un tipo de histiocitosis poco prevalente en edad adulta y más frecuente en niños entre 1-3 años de edad.
La infiltración de estas células en un 55% de los casos se limita a un solo órgano, siendo menos frecuente su diseminación sistémica a otras localizaciones como el hueso, la piel, los vasos linfáticos, los pulmones, el hígado, el bazo o el sistema nervioso central1.
El pronóstico y tratamiento dependen sobre todo de la edad del paciente y del número y disfunción de los órganos afectados.
Presentamos el caso de una paciente de edad avanzada con diagnóstico final de histiocitosis de células de Langerhans y afectación exclusivamente colónica.
Mujer de 81 años con múltiples antecedentes personales entre los que destaca DM2, HTA, esteatosis hepática e isquemia crónica grado IV en miembros inferiores. En tratamiento con insulina mixta, ramipril, gemfibrozilo y aspirina1.
Ingresa en el servicio de digestivo por cuadro de 2-3 meses de evolución2 de dolor abdominal periumbilical e hipogástrico, posprandial junto con vómitos ocasionales. No refería alteración del hábito intestinal ni productos patológicos en las heces. Además, presentaba astenia y mayor anorexia en los últimos meses.
La paciente mostraba buen estado general, delgadez evidente y ligera palidez mucocutánea. A nivel abdominal presentaba dolor abdominal difuso a la palpación profunda, sin palparse masas ni visceromegalias. En miembros inferiores presentaba signos de insuficiencia venosa crónica.
En la analítica solo destacaba una PCR mínimamente elevada y leucocitosis sin neutrofilia.
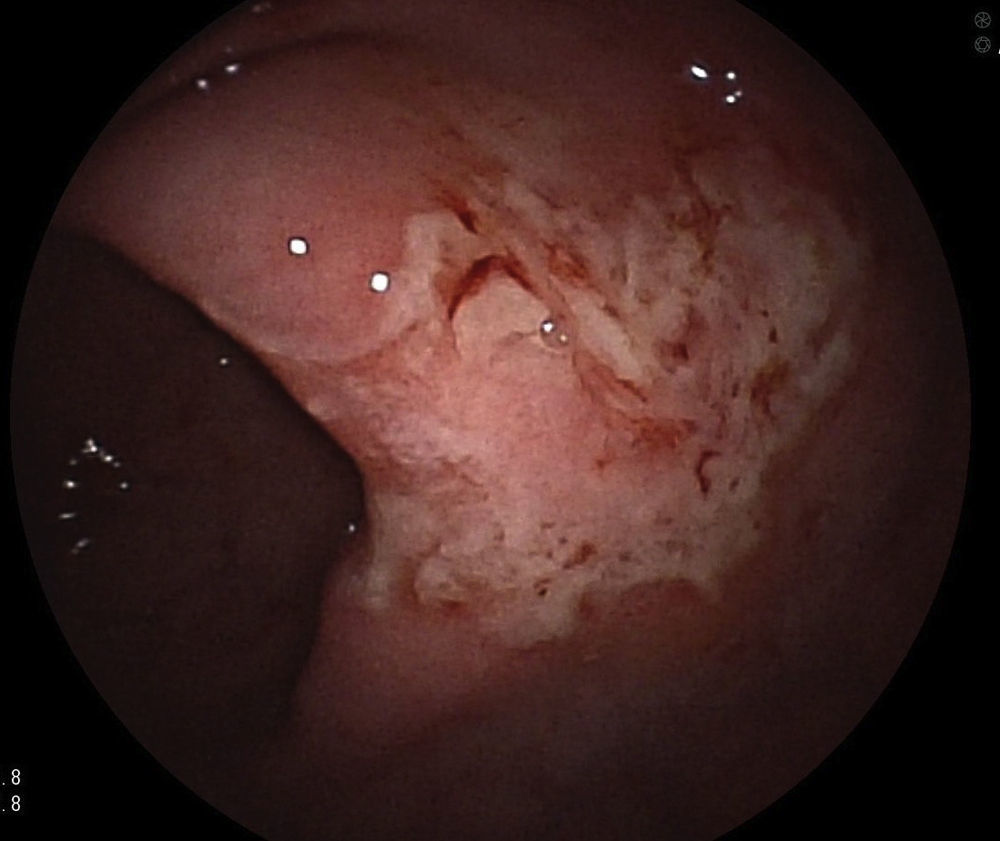
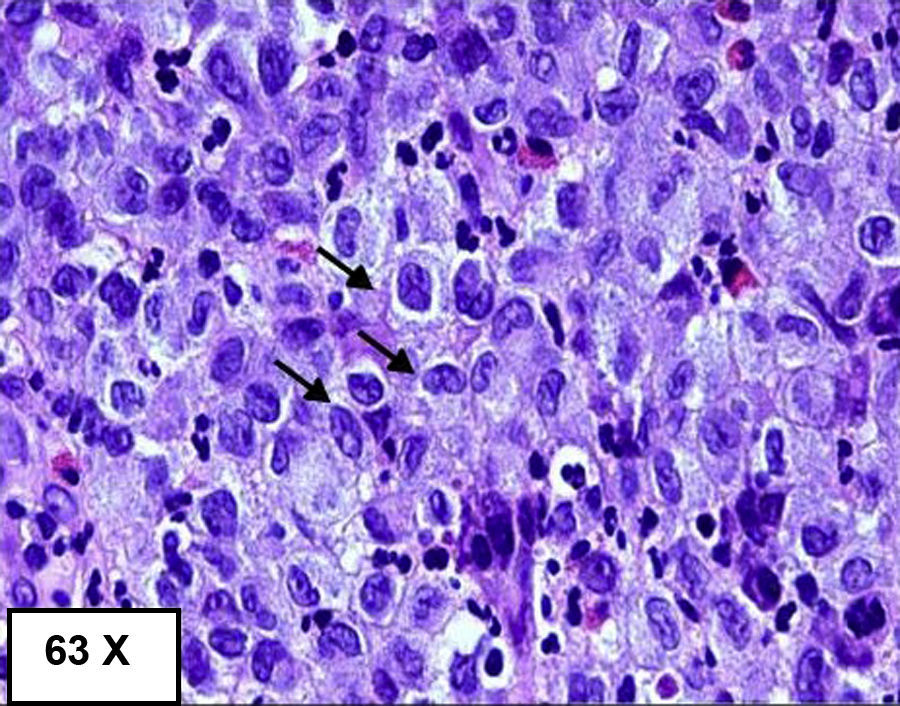
Se le realizó una colonoscopia apreciándose a 55cm del margen anal (colon descendente), mucosa con ulceraciones planas y superficiales, con algunas sufusiones hemorrágicas, que se extendían unos 10cm de longitud3 cuyo diagnóstico inicial era compatible con una colitis isquémica, sin embargo, la biopsia fue diagnóstica de HCL (se adjunta imagen de la colonoscopia inicial: fig. 1)4. En la biopsia se halló proliferación celular densa y difusa de naturaleza neoplásica, con células de hábito histiocitario, núcleos lobulados y citoplasma eosinófilo. La inmunohistoquímica fue positiva para proteína S100, vimentina, CD68 y CD1a5. En una segunda colonoscopia realizada al mes del ingreso se comprobó la evolución hacia la curación mucosa de las lesiones previamente descritas, persistiendo varias ulceraciones pequeñas y superficiales3.

En el TAC abdominal existía un aumento de la grasa pericólica con engrosamiento mural del colon a nivel de ángulo esplénico. Se le realizó como estudio de extensión un TAC de tórax, un rastreo óseo con tecnecio 99 siendo estos negativos para afectación sugerente de histiocitosis en otros territorios.
El diagnóstico final fue de neoplasia de células de Langerhans de afectación únicamente en colon descendente, por lo que la paciente fue remitida a consultas de hematología donde recibió tratamiento con corticoides, vinblastina durante 6 semanas y 6-mercaptopurina durante 12 meses.
En el seguimiento posterior la paciente fue mejorando clínicamente, aunque dado que se trasladó a otra ciudad no disponemos de colonoscopia de control. Debido al mal control de la glucemia se decidió tras 6 dosis de vinblastina y prednisona, suspender la corticoterapia.
A los 7 meses de seguimiento ambulatorio la paciente había mejorado considerablemente con el tratamiento.
La HCL se presenta en la mayoría de los casos con afectación aislada de un solo órgano, siendo menos frecuente su diseminación sistémica. La afectación gastrointestinal es poco frecuente, generalmente son un hallazgo casual de pólipos solitarios colorrectales en pacientes asintomáticos. Tan solo un 2% de los pacientes presentan como sintomatología diarrea o malabsorción2,3. Existen pocos casos publicados sobre afectación exclusivamente colónica de la HCL, siendo la mayoría de ellos en forma de pólipos colónicos solitarios.
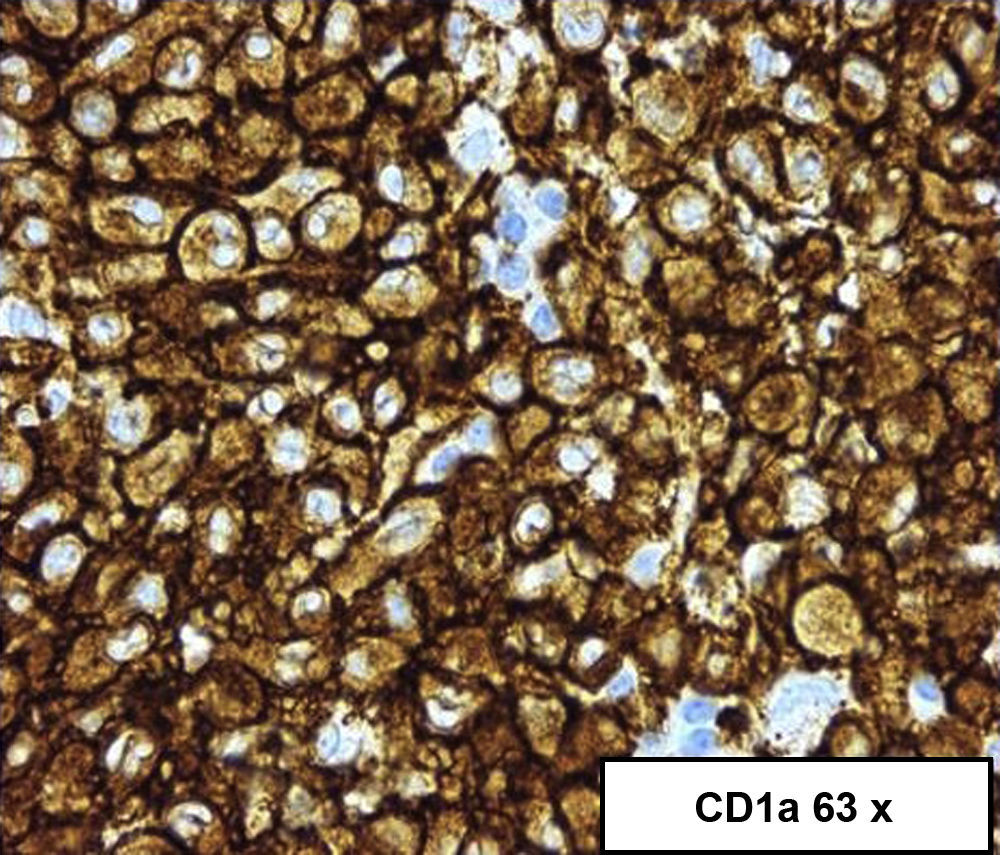
Es preciso el estudio endoscópico y la toma de biopsias para confirmar la presencia de4,5 infiltrado histiocítico en las células de la mucosa, además de realizar tinciones específicas para CD1a y S100 o demostrar la presencia de gránulos de Birbeck en el citoplasia con el microscopio electrónico (figs. 2 y 3)4,5.

.")
Con el aumento del número de colonoscopias realizadas por otro motivo se debería tener en cuenta la HCL como causa rara de enfermedad mucosa en colon dado sus implicaciones terapéuticas y pronósticas.

