




Obesity is sharply rising worldwide and is increasingly recognized in patients with cirrhosis. This review summarizes the available data documenting a detrimental role of obesity and insulin-resistance on the risk of appearance of clinical events in patients with cirrhosis. Molecular pathways explaining the harmful effect of obesity and insulin resistance in the natural history of cirrhosis are largely unknown. Increasing knowledge of mechanisms leading to white adipose tissue dysfunction on one side, and to portal hypertension on the other side, allow hypothesizing that a link between the pathophysiology of obesity, insulin resistance and portal hypertension in cirrhosis exists. Mechanisms likely involved in this interplay are discussed in this article.
La obesidad muestra un aumento drástico en todo el mundo y se reconoce cada vez más en los pacientes con cirrosis. Esta revisión resume los datos disponibles que documentan el papel perjudicial de la obesidad y la resistencia a la insulina en el riesgo de aparición de sucesos clínicos en los pacientes con cirrosis. Se desconocen en gran parte las vías moleculares que conducen a que la obesidad y la resistencia a la insulina desempeñen un papel perjudicial en la historia natural de la cirrosis. No obstante, a medida que aumenta el conocimiento de los mecanismos que conducen a la disfunción del tejido adiposo blanco por un lado, y a hipertensión portal por el otro, es posible mantener la hipótesis de que en la cirrosis existe un vínculo entre la patofisiología de la obesidad, la resistencia a la insulina y la hipertensión portal. Este artículo describe los mecanismos probablemente implicados en esta interacción.
Obesity is a state of excessive storage of body fat leading to detrimental consequences on health. A percentage of body fat >25% for men and >33% for women defines obesity,1 but usually the simplified World Health Organization (WHO) definition based on body mass index is used; accordingly, those subjects having a body mass index (BMI) ≥30kg/m2 are obese.
Obesity prevalence is constantly increasing worldwide; overall, over 10% of the world's adults and 20–30% of inhabitants of North America and Europe are obese.2 As other medical conditions, primary obesity is a multifactor disease due to the interplay between genetic and environmental factors. Genetic factors are presumed to explain 40–70% of variance in obesity, but the main reason explaining obesity pandemics is the environmental increased availability of tasty, inexpensive, and safe foods and beverages. Their intake, associated with reduced calories expenditure due to sedentary lifestyle, results in a net positive calories balance, with subsequent fat storage in the adipose tissue.
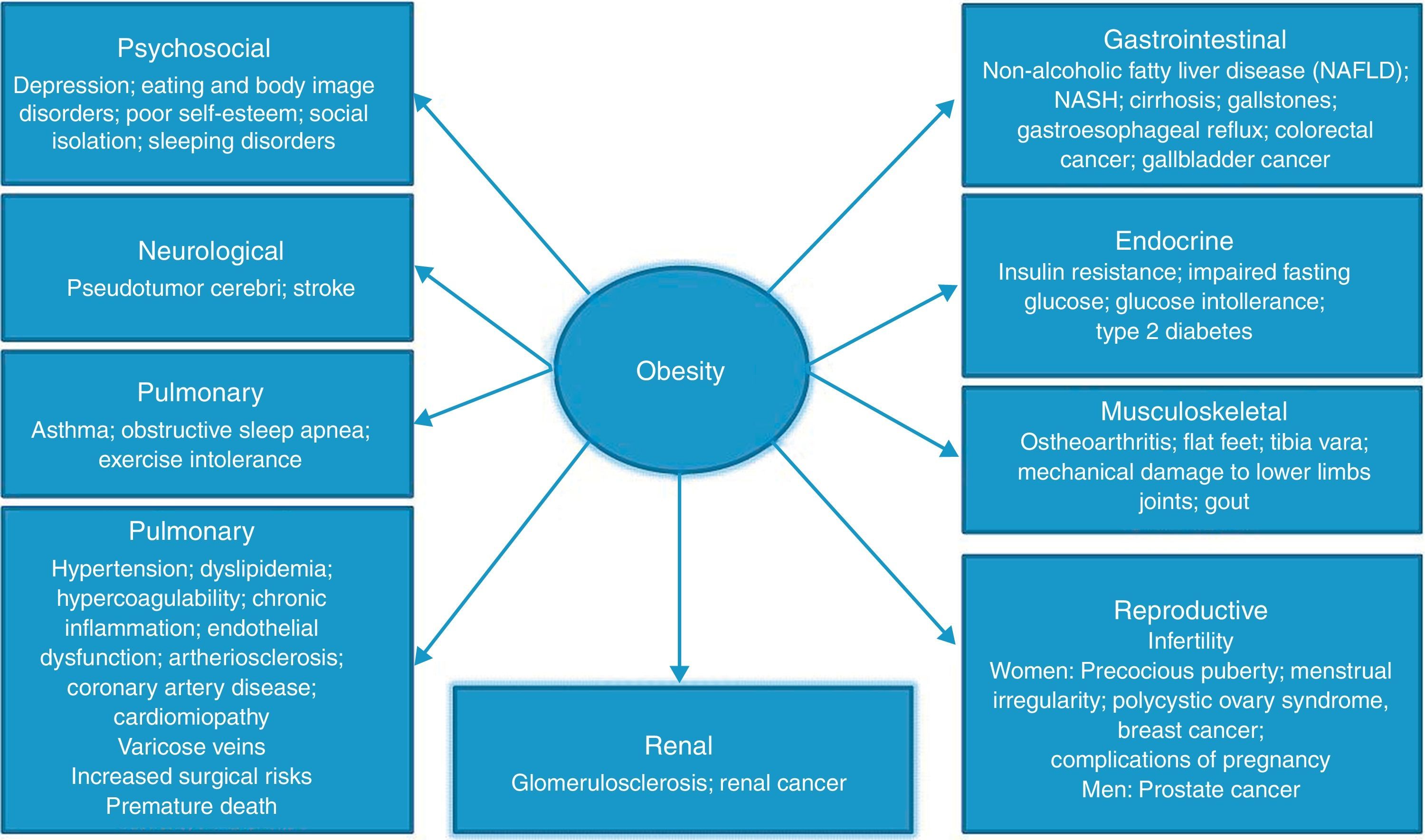
Large, prospective cohort studies, such as the Framingham and NHANES, clearly show that obesity is associated with an increase in morbidity and mortality rates, being their incidence proportional to the increase of BMI above normality. Type 2 diabetes, arterial hypertension, hyperlipidemia, cardiovascular diseases, and malignancies are the most common obesity-related disorders, but several other chronic diseases are favored by obesity (Fig. 1).

The negative effect of obesity on health has been linked to the onset of a chronic low grade inflammation originating in the excessive white adipose tissue, which is functionally an endocrine gland producing peptides (adipokines) and metabolites.
Indeed, the phenotype of adipose tissue (and particularly that of visceral and perivascular adipose tissue) in obesity is different from that of lean individuals. Adipocytes are enlarged in obese subjects; white adipose tissue becomes hypoxic as adipocyte size and tissue mass expand in obesity, and hypoxia promotes the development of inflammation and cellular dysfunction.3 Adipocytes shift their adipokines secretion from a protective to a damaging profile; in addition, macrophage infiltration and macrophage-derived cytokines secretion take place.4 Obesity-associated adipokines (such as IL-6, TNFa, monocyte chemoattracting protein 1and plasminogen activator inhibitor-1) and cytokines are involved in insulin sensitivity and appetite regulation, but also typically show pro-inflammatory, pro-fibrogenic, pro-angiogenic and pro-oxidant effects on several tissues and organs.3,4
Obesity in patients with cirrhosis and portal hypertensionTaking into account the above mentioned data, it is not surprising that obesity is per se a cause of chronic liver disease,5 as demonstrated by a 90% prevalence of non-alcoholic fatty liver disease (NAFLD) in obese subjects.6 As for the risk of cirrhosis, the results of the large prospective study “Million Women Study” performed in the United Kingdom7 in over a million women without known liver disease on inclusion showed that about 17% of incident cases of cirrhosis can be directly attributed to obesity.7
Obesity also plays a detrimental role in the progression of liver disease to cirrhosis. In patients with NAFLD obesity is independently associated with the presence of hepatic inflammation and fibrosis (non-alcoholic steato-hepatitis, NASH) and with an increased risk of progression to cirrhosis.8 Similarly, in patients with alcoholic liver disease9 and viral liver disease10 obesity is an independent risk factor for the presence of severe fibrosis, fibrosis progression and cirrhosis.
Few data exist regarding the epidemiological and clinical impact of obesity in patients with pre-existent cirrhosis. This might be due to the fact that cirrhosis has long been diagnosed in very late stages characterized by ascites, infection and encephalopathy in which malnutrition, sarcopenia and cachexia are not unusual, while a preserved nutritional status was traditionally associated with a better prognosis.11
Contrary to these old observations, two large prospective studies recently demonstrated that obesity is frequent in patients with cirrhosis, and has a negative impact on the natural history of the disease and on portal hypertension.
In the HALT-C (Hepatitis C Antiviral Long-Term Treatment Against Cirrhosis) study from USA, enrolling 985 patients with chronic HCV-related liver disease and severe liver fibrosis or compensated cirrhosis, up to 43% had obesity, and 32% were overweight. Each quartile increase in BMI was associated with a 14% increase in the risk of clinical events in the follow-up (worsening of liver fibrosis or decompensation of cirrhosis)12; among obesity-related variables, the severity of insulin-resistance (assessed by HOMA-2 index) and histological grade of steatosis appeared as the factors more tightly associated with liver disease progression. A subanalysis focused on patients with cirrhosis was not performed.
The second study13 shows the results of a subanalysis of the population included in a multicentric randomized controlled trial comparing timolol vs placebo for the prevention of formation of varices in patients with Child A compensated cirrhosis.14 The study was conducted in Barcelona (Spain), London (UK), Boston and New Haven (USA). The BMI was available in 161 patients with compensated cirrhosis and no esophageal varices at inclusion, followed-up in mean for 59 months. In this population overweight and obesity were very frequent, accounting for 71% of the cases (40% overweight and 31% obese). The stratification by Center indicated that the prevalence of obesity in patients with cirrhosis reflected that of the referral general population, being much higher in American patients (54%) as compared to Europeans (18%; p<0.00001).
During follow-up clinical decompensation of cirrhosis (ascites, encephalopathy or jaundice) developed in 14% of patients with normal weight, in 31% of overweight patients and in 43% of patients with obesity. Multivariate analysis pointed out that the increase in BMI was a risk factor for the development of first clinical decompensation of cirrhosis, independent of liver function (albumin) and the severity of portal hypertension (HVPG). Each unit increase in BMI above its upper normal value (25kg/m2) was associated with a 6% increase in the risk of decompensation. Accordingly, obese patients with cirrhosis had a threefold risk of clinical decompensation as compared to normal weight patients. Of note, this increase in risk was independent of the etiology of cirrhosis.
Interestingly, the data showed that obesity also negatively impacts portal hypertension. Indeed, after 1 year of treatment with timolol (a non-selective beta-blocker which is able to decrease portal pressure) or placebo, only patients with normal weight or overweight showed a reduction of HVPG, whereas obese patients had a slight increase in HVPG. Moreover, patients who had an increase of BMI after 1 year of follow-up also had a significant increase in HVPG.
These observations hint about the existence of a complex interaction between obesity and portal hypertension. The pathophysiological mechanisms involved are still unknown, and represent a field for future research. From a speculative standpoint it is possible that the pro-inflammatory, pro-fibrogenic, pro-angiogenic and pro-oxidant effects of the above mentioned obesity-related cytokines might worsen the intrahepatic vascular tone and increase hepatic vascular resistance, resulting in an obesity-related component of the increased portal pressure.15
Experimental data permit to speculate that leptin might be one of the possible culprits of the adverse obesity-related outcomes in cirrhosis. Leptin is an adipose tissue-derived hormone that in healthy individuals contributes to the maintenance of a normal body weight by regulating energy intake (appetite/hunger) and energy expenditure (different metabolic pathways)16; in its absence, obesity takes place. Despite this, in obese individuals leptin is usually increased due to the resistance of target organs to its effects leading to compensatory increased release by the adipose tissue. When increased, leptin holds pro-oxidant, pro-fibrogenic and pro-angiogenic effects on several organs. Interestingly, leptin is also increased in cirrhosis and has been shown to induce fibrosis in the liver.17 Given the above mentioned evidence, a recent experimental study from our group tested the effects of the blockade of leptin receptor (ObR) in a murine model of cirrhosis,18 showing that the increase in portal pressure is attenuated by anti-ObR treatment with no changes in splanchnic blood flow. The simultaneous observation that ObR blockade increased intrahepatic nitric oxide bioavailability and decreased oxidative stress and protein nitrotyrosination suggests that leptin increases intrahepatic vascular resistance. Altogether, these data suggest that leptin receptor blockade might have potential for the treatment of portal hypertension18 in cirrhosis, in particular in cirrhosis associated with obesity.
From a clinical point of view, it can be hypothesized that the reduction of body weight in patients with cirrhosis and obesity might lead to a reduction in HVPG, thus reducing the risk of clinical decompensation.19 Data from the HALT-C study suggest that weight loss is indeed beneficial in patients with advanced chronic liver disease12 reducing the progression of fibrosis, but no data are available in the specific setting of patients with cirrhosis. In order to test the above mentioned hypothesis and gather data regarding the pathophysiological link between obesity and portal hypertension, we are conducting a pilot multicenter prospective study in patients with cirrhosis and obesity in 6 Spanish centers (SPORTDIET study, Clinical Trials.gov: NCT01409356). In this study, the degree of portal hypertension (assessed as the hepatic venous pressure gradient or HVPG20) is measured at inclusion and after completing a 4-month program of controlled diet and exercise to induce weight loss. The final results are expected by the end of 2013.
While obesity is increasingly seen in patients with decompensated cirrhosis,21 it is still unclear whether it might also worsen the prognosis in this stage of the disease. Recent data suggest that in patients awaiting liver transplantation obesity substantially and independently increases the risk of portal vein thrombosis.22 This needs to be confirmed in other series, but since it is well known that in non-cirrhotic patients obesity is a risk factor for venous thromboembolism and arterial thrombosis, it is possible that the proinflammatory, prothrombotic, and hypofibrinolitic milieu typical of obesity might explain this finding.
Finally, abundant data confirm that obesity and obesity-related variables (insulin resistance and type 2 diabetes) have negative impact on the development of hepatocellular carcinoma (HCC) (83% increase in risk as compared to the general population)23 and on HCC outcome,24,25 including post-transplantation recurrence and death.26
Insulin-resistance in obesity and cirrhosisInsulin signaling pathway is of striking importance in the maintenance of body metabolic homeostasis. In normal conditions, insulin binding to insulin receptor activates the phosphoinositide 3-kinase/serine/threonine kinase (PI3K/Akt), which accounts for most metabolic effects of insulin. In addition, at the vascular level, specifically in endothelial cells, insulin activates the endothelial nitric oxide synthase (e-NOS), leading to nitric oxide (NO) release and maintenance of a balanced vascular tone through vasodilatation.27,28
In subjects with obesity, the release of free fatty acids and adypokine/cytokine signaling mediate, directly and by inducing an inflammatory state, the impairment of insulin signaling pathway primarily through serine phosphorylation of insulin receptor substrate proteins.4 This blunts insulin action in the target cells leading to a condition known as “insulin resistance”. In insulin resistant individuals (nearly the totality of obese patients) besides a reduction of glucose intake in the skeletal muscle and the liver participating in the pathogenesis of hyperglycaemia, the insulin-induced synthesis of NO by e-NOS is impaired in the systemic vascular endothelium. Among other complex effects, this leads to a shift of vascular tone to vasoconstriction28 and lack of ability to vasodilate in response to physiological stimuli typical of endothelial dysfunction.
While the mechanisms and consequences of insulin resistance have been extensively studied in the systemic vascular endothelium, data regarding the effects of insulin resistance on the intrahepatic microvascular bed are scarce. Very recently, two studies showed in two different models of NAFLD that liver endothelial dysfunction develops in the absence of fibrosis and inflammation in the fatty liver, and this was associated with an increase in hepatic vascular tone and portal pressure.29,30 Further data showed that that insulin signaling at the liver endothelium is disrupted as early as after 3 days of a high fat administration,31 suggesting that this mechanism could probably be a major contributor to the development of liver endothelial dysfunction in NAFLD.
There are no specific studies addressing insulin resistance on intrahepatic vascular endothelial function in cirrhosis. However, it is well known that intrahepatic endothelial dysfunction is a characteristic feature of cirrhosis and participates in the onset and aggravation of portal hypertension.15 Hence, insulin resistance could contribute to further exaggerate intrahepatic endothelial dysfunction. Results obtained in clinical studies (summarized below) seem to support this hypothesis.
Insulin resistance holds specific features in cirrhosis. Definitely it is very frequent, as supported by several studies in the 1980s and 1990s32–36 using either an oral glucose load or the euglycemic–hyperinsulinemic clamp (gold-standard method for the measurement of hepatic insulin sensitivity37). However, its pathogenesis is different than that of obesity, being mainly due to portal hypertension-related porto-systemic shunting of insulin from the splanchnic circulation to systemic circulation,38 causing hyperinsulinaemia, desensitization and downregulation of insulin receptors leading to insulin resistance. Nonetheless, an impaired beta-cell secretion of insulin coexists.39 A minor role is played by insufficient clearance of insulin by the liver due to reduced hepatocellular function.38 The pivotal role of porto-systemic shunting on the onset of insulin resistance is confirmed by several data: (a) insulin resistance is also found in patients with non-cirrhotic portal hypertension, having a completely normal hepatic function but porto-collateral vessels40; (b) TIPS placement leads to a worsening of insulin resistance as a consequence of increased porto-systemic shunting,41 and (c) insulin resistance is largely corrected or reversed by abolishing porto-systemic shunting by liver transplantation.42
Independent of its cause, insulin resistance in cirrhosis appears to identify a subgroup of patients with worse prognosis. In a recent prospective study conducted in 249 patients with HCV-related compensated cirrhosis with or without diabetes (not on insulin treatment), Nkontchou et al. reported that that insulin resistance, assessed by the classical homoeostasis model assessment (HOMA-1 or HOMA-IR),43 was independently associated with an increased risk of liver-related death or liver transplantation (OLT),44 together with parameters reflecting liver function (prothrombin time) and portal hypertension (platelet count). The five-years risk of death or OLT in patients with the highest tertile HOMA was around 30% as compared to a <20% risk in the remaining groups.44 In the same study, both HOMA and BMI were independently associated with the risk of hepatocellular carcinoma.
The same group recently reported that the use of metformin, an insulin-sensitizing agent, is associated with a decreased risk of death or OLT, and of HCC in patients with HCV-related cirrhosis and type 2 diabetes.45 Unfortunately, the level of evidence provided by this observational, uncontrolled study is not strong enough to allow clear-cut conclusions, and should be confirmed by appropriately designed randomized controlled trials. Nonetheless, in 100 patients included in this study (26 of whom treated with metformin), BMI and metformin were independently associated with the risk of death or OLT,45 suggesting that obesity and insulin resistance possibly act through different pathways in the pathophysiology of cirrhosis.
Portal hypertension is the most important factor driving the progression of cirrhosis from a compensated to a decompensated stage,19 and its relationship with insulin resistance has been evaluated in two studies. In the first, including cross-sectionally 104 patients with compensated HCV-related cirrhosis irrespective of body weight and presence of diabetes (obesity in 15%, diabetes in 26%), insulin-resistance, assessed by the HOMA-IR model, was associated to the presence of esophageal varices independent of liver function and non-invasive markers of portal hypertension (platelet count and spleen size).46 This association was expected, since hyperinsulinemia and insulin resistance are a consequence of porto-systemic shunting occurring in the presence of porto-systemic collaterals such as esophageal varices. The authors concluded that a HOMA >3.5 suggests the presence of varices; on the other hand, the study did not include HVPG measurement to analyze more in detail the clinical relationship of insulin resistance and portal hypertension.
The second study was conducted in 49 patients with compensated cirrhosis of different etiologies, free of overt diabetes or metabolic syndrome.47 In this population insulin resistance was evaluated by the HOMA-2 model that accounts for variations in hepatic and peripheral glucose resistance48 at the time of hepatic vein catheterization and HVPG measurement. Insulin resistant patients (61% of the sample) were characterized by a higher HVPG, more accentuated hyperdynamic syndrome, worse liver function and higher BMI. Interestingly, even in the absence of a clear metabolic syndrome, both the degree of liver failure and BMI were independently associated with insulin resistance, suggesting a dual component of insulin resistance in cirrhosis (liver disease and overweight/obesity). HOMA-2 and HVPG showed a significant but weak correlation; hence, while values of HOMA-2 above 2 are globally associated with CSPH,47 HOMA-2 could not predict accurately the value of HVPG or acute HVPG response to i.v. propranolol.47
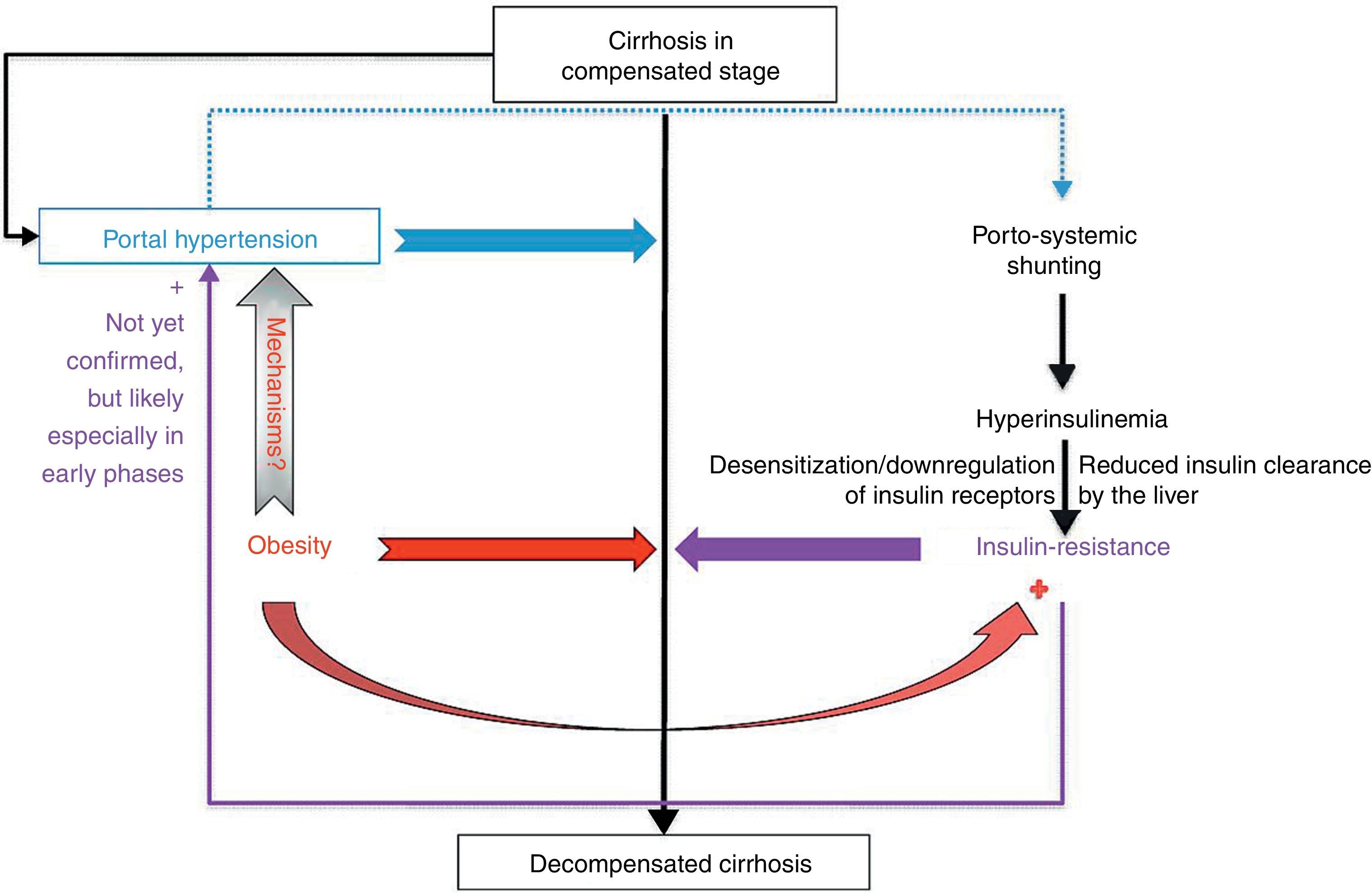
Concluding remarksThe complex interplay of obesity, insulin resistance and cirrhosis is far from being elucidated (Fig. 2). Even if it seems clear that obesity and insulin resistance increase the risk of decompensation in patients with compensated cirrhosis, several issues need to be clarified regarding the exact mechanisms mediating the negative effects of these conditions on pre-existing cirrhosis. Furthermore, high-quality evidence is needed to support a benefit of weight loss and of correction of insulin resistance on the clinical course of cirrhosis and on the risk of hepatocellular carcinoma in this intriguingly multifaceted scenario.

Interplay of obesity and insulin resistance on compensated cirrhosis. Obesity and insulin resistance increase the risk of decompensation of cirrhosis; the detrimental role of obesity seems to be only partially due to a further induction of insulin resistance. A negative role of different obesity-related adipokines and cytokines on hepatic resistance is postulated, but needs to be proved by future studies.
The authors have no conflict of interest to declare.

