





INTRODUCCIÓN
El cáncer colorrectal (CCR) es la segunda neoplasia más frecuente en España. Cada año se diagnostican más de 25.000 casos y más de 11.000 personas fallecen a consecuencia de esta afección1,2. El cribado poblacional del CCR produce una disminución de la mortalidad por esta causa3, de acuerdo con diferentes estudios. Un porcentaje del cáncer colorrectal se debe a determinadas enfermedades hereditarias que confieren un riesgo muy elevado a las familias que lo presentan. Entre las enfermedades que se encuadran en la denominación cáncer colorrectal hereditario hay 2 bien definidas: la poliposis adenomatosa familiar4 y el cáncer colorrectal hereditario sin poliposis (CCRHSP). A diferencia de otros autores, utilizamos esta terminología de acuerdo con los argumentos indicados en otro artículo5.
En diferentes publicaciones se ha indicado que el porcentaje de CCRHSP varía entre el 1 y el 5% de todos los cánceres de colon que se diagnostican en una comunidad, según de los criterios diagnósticos utilizados y el área geográfica6-11. En un estudio reciente llevado a cabo en España en una muestra de casi 2.000 pacientes se ha observado un 2,5%12.
En el CCRHSP, la transmisión ocurre con un patrón autosómico dominante, por lo que la historia familiar es trascendental para identificar a las familias afectadas. La mejor manera de tratar esta enfermedad hereditaria es mediante un registro de cáncer colorrectal hereditario, ya que permite aplicar técnicas de cribado en estas familias para disminuir la incidencia de casos de cáncer y mejorar la supervivencia del CCR13-17. Los criterios clínicos más aceptados para la identificación del CCRHSP son los denominados criterios de Ámsterdam18 que posteriormente se modificaron (Ámsterdam II; tabla I)19, que son más específicos que sensibles y se diseñaron para llevar a cabo estudios multicéntricos con unos criterios diagnósticos unificados20,21. Se ha observado que esta enfermedad va ligada, en un porcentaje variable de casos, a la inestabilidad de microsatélites y a diferentes mutaciones de los ge nes reparadores de los errores de replicación del ADN22-24 . Los criterios de Bethesda revisados (tabla I) sirven para seleccionar a los pacientes en los que, a pesar de no cumplirse los criterios de Ámsterdam, presentan una alta sospecha de presentar un CCRHSP y, por tanto, está indicado un estudio para determinar la inestabilidad de microsatélites y el posible análisis genético de las muta ciones de los genes reparadores ligados a este síndrome25-27.

En una revisión realizada en que se utiliza la base de datos Medline, con el objetivo de identificar las publicaciones procedentes de España, hemos encontrado una serie de publicaciones referidas al CCRHSP sobre casos o familias aisladas, sin que hayamos podido identificar ningún registro que haya publicado sus datos12,28-39.
Por todo ello, nos ha parecido oportuno publicar la implantación de este registro de CCRHSP en nuestro hospital. Los resultados que presentamos hacen referencia únicamente al núcleo de las familias que cumplen los criterios de Ámsterdam I y II. Dejamos para otra ocasión los resultados ampliados con las familias que cumplen los criterios de Bethesda.
PACIENTES Y MÉTODO
En 2002 se puso en marcha, en nuestra unidad, un estudio para la identificación clínica de las familias afectadas de CCRHSP. En todos los casos de cáncer de colon de nuestro hospital con sospecha de CCRHSP (menos de 50 años de edad, localización derecha, neoformación sincrónica, antecedentes familiares) se realiza una historia familiar oncológica mediante entrevistas personales, por personal especialmente entrenado.
En cada familia se ha elaborado una historia familiar completa en que se han registrado los casos de cáncer correspondientes al espectro del CCRHSP (colon, endometrio, ovario, estómago, hepatobiliar, intestino delgado, uréter y pelvis renal), así como los miembros de cada familia en situación de riesgo. Mediante la entrevista personal se obtiene la máxima cantidad de información que los familiares puedan recordar y que pueda resultar de utilidad: estado actual del paciente (vivo o muerto), edad al diagnóstico, centro donde fue tratado, presencia de metástasis al diagnóstico, etc. Generalmente, en cada familia se localizó a 1 o 2 miembros, que funcionaban como "colaboradores especiales", y que recababan la mayoría de la información complementaria necesaria. Se elaboró, de esta forma, un árbol genealógico para cada familia.
Es importante tener en cuenta, además, que la historia familiar en un registro de estas características tiene un carácter dinámico y que en cada visita pueden y deben completarse o rectificarse datos anteriores de acuerdo con la información verbal o documentada que aportan los pacientes y sus familiares.
Se ha realizado una exhaustiva comprobación de los datos inicialmente registrados en la historia familiar; en el caso de los pacientes tratados en nuestro hospital, mediante revisión de la historia clínica.
Para la comprobación de los datos de los pacientes no tratados en nuestro centro se han seguido 2 mecanismos, según el área geográfica donde fueron tratados: a) en el caso de los pacientes tratados en la isla de Mallorca, se han revisado los datos del Registro Poblacional de Cáncer de Mallorca, y b) en el caso de los pacientes tratados fuera de Mallorca, se han requerido informes clínicos a los familiares o a los centros donde fueron tratados los pacientes.
Sólo se han aceptado como casos registrables aquéllos en los que se ha confirmado la presencia del cáncer reportado en la entrevista personal; aquéllos en los que no fue posible la comprobación se registraron como no definitivos, pendientes de una evaluación posterior y no se recogen en esta publicación.
En el Registre de Càncer Poblacional de Mallorca se registran todos los casos de cáncer que se producen en la isla, tanto de colon como del resto de localizaciones. Se recogen los datos referidos al tipo de tumor, histología, localización, estadio, fecha del diagnóstico y edad del paciente en el momento del diagnóstico, así como los datos epidemiológicos del paciente (edad, sexo, dirección, etc.).
Para realizar la consulta se cruzaron los datos de los pacientes de nuestro estudio con los del Registro de Cáncer Poblacional de Mallorca (con un total de 4.214 casos de CCR en el período de 1984-1998).
Toda la información de los pacientes se registra en una base de datos informática, en la que la identidad del paciente queda protegida por una codificación, que se actualiza periódicamente. Asimismo, se ha habilitado una consulta específica para el seguimiento de estas familias. Esta consulta de prevención del cáncer colorrectal, además de las familias con CCRHSP, atiende a las familias con poliposis adenomatosa familiar, a aquellas con riesgo elevado de presentar CCR (que no cumplen criterios de CCR hereditario), a los pacientes a quienes se ha realizado una polipectomía de uno o más pólipos colorrectales neoplásicos, y a los pacientes y familiares que son remitidos a esta consulta con sospecha de riesgo elevado de cáncer colorrectal.
Los familiares en situación de riesgo son identificados, se les informa sobre la posibilidad de desarrollar cáncer de colon y se les propone participar en un programa de seguimiento para su prevención. El seguimiento propuesto consiste en una colonoscopia completa anual, y se inicia el seguimiento a los 25 años o 5 años antes que la edad correspondiente al paciente más joven con cáncer de colon en esa misma familia.
En los casos de neoplasias diagnosticadas y tratadas anteriormente que siguen controles en las consultas externas, se informa a los pacientes acerca del riesgo elevado de recidiva neoplásica y se les propone un seguimiento mediante colonoscopia completa anual.
En los casos de neoplasia de nueva aparición durante el desarrollo del seguimiento se informa a los pacientes de la posibilidad de una colectomía subtotal como intervención ideal para evitar una recidiva tumoral, y se mantiene el seguimiento mediante endoscopias anuales del recto15,40.
Las endoscopias las lleva a cabo personal experimentado del servicio; caso de no poder completar la exploración hasta el ciego, el estudio se completa mediante un enema opaco. Todos los pólipos encontrados son resecados mediante asa de polipectomía. Se realizan biopsias endoscópicas de todas las masas o lesiones encontradas.
Las colonoscopias y las técnicas quirúrgicas, así como el examen histológico de las piezas quirúrgicas, siempre las realizada personal experimentado en nuestro propio centro. La no realización del seguimiento endoscópico en los tiempos estipulados no se consideró un criterio de exclusión del estudio, pero sí se registró el tiempo de retraso.
En todos los casos de CCR detectados durante este período de funcionamiento del registro se han obtenido los siguientes datos: edad, sexo, fecha intervención, histopatología del cáncer, estadificación, edad en el momento del diagnóstico, localización de la neoplasia, historia de cáncer sincrónico o metacrónico, así como tiempo y tipo de seguimiento.
RESULTADOS
Hemos recopilado la información referente a 12 familias (90 individuos). Se han registrado 38 pacientes con CCR, de los cuales 19 están vivos y en seguimiento en el momento actual, y 19 han fallecido.
La información relativa a estos 38 pacientes se obtuvo de la siguiente forma: 23 mediante datos directos de nuestro hospital (60%), 8 mediante datos del Registro de Cáncer Poblacional de Mallorca (21%) y 7 mediante informes de otros hospitales (18%). Existen otros 9 casos de CCR referidos durante las historias familiares, pero que no han podido comprobarse mediante informes clínicos, por lo que no se han recogido en esta publicación. Únicamente en 4 pacientes la historia familiar refería la presencia de neoplasia, y la comprobación de los datos demostró que estos datos no eran correctos. Una familia inicialmente catalogada como cumplidora de criterios de Ámsterdam tuvo que ser retirada del registro, al realizar la comprobación de los datos.
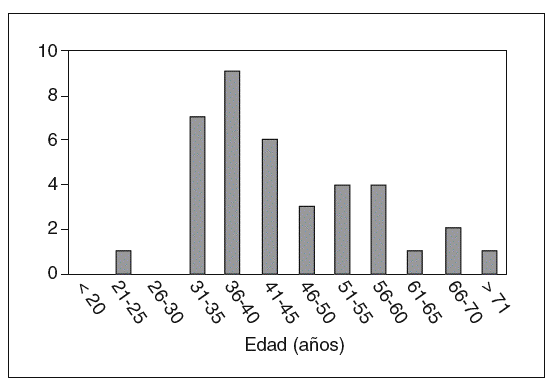
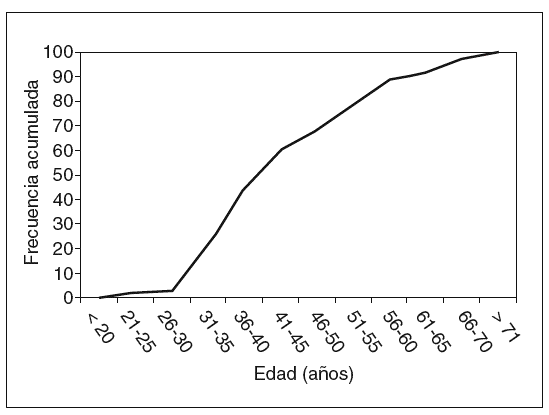
El total de casos de CCR registrados es de 46 en 38 pacientes (6 casos de tumoraciones metacrónicas y 2 de tumoración sincrónica). La edad media ± desviación estándar (DE) de presentación fue de 45,4 ± 12,7 años (rango, 25-73 años). La variabilidad, según los grupos de edad y la frecuencia acumulada, se muestran, respectivamente, en las figuras 1 y 2. Como puede apreciarse en la figura 2, en nuestro grupo el 59% de las neoplasias se diagnostican antes de los 45 años, y el 79%, antes de los 60. La distribución por sexos muestra 26 varones y 12 mujeres.
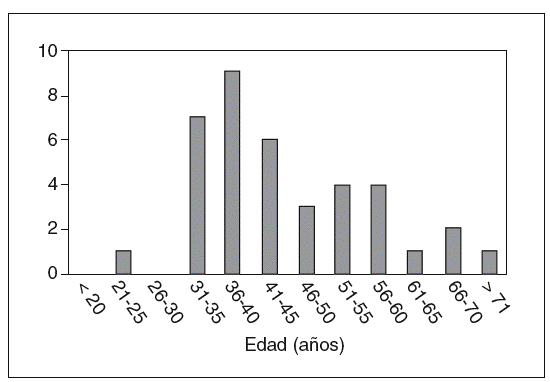
Fig. 1. Edades de distribución del diagnóstico del cáncer colorrectal.
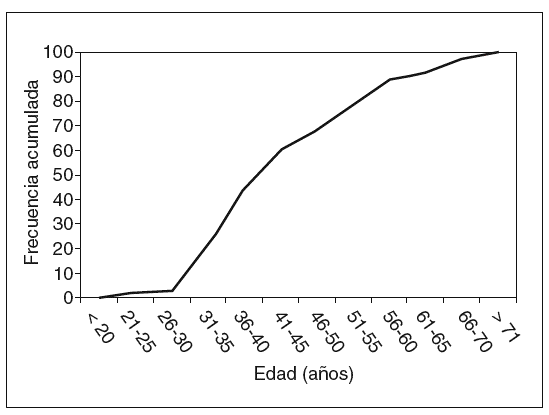
Fig. 2. Frecuencias acumuladas de cáncer colorrectal.
Disponemos de la localización de la neoplasia en 35 casos de los 46 registrados (77%). Del total, 20 se localizan proximales al ángulo esplénico del colon (ciego-colon ascendente-colon transverso) y 15 se localizan distalmente al ángulo esplénico del colon (colon descendente-sigma-recto).
En 30 de los 46 casos de CCR (65%), la histología se ha documentado mediante un informe anatomo-patológico, 21 procedentes de piezas quirúrgicas y 9 de biopsias endoscópicas; en este último grupo, la mayoría correspondió a tumores diagnosticados mediante endoscopia, pero que se encontraban en estadios avanzados y en los que no fue posible la intervención quirúrgica, por lo que se trataron de forma paliativa.
En 12 de los 30 casos de CCR con estudio histológico se han detectado pólipos sincrónicos, lo que supone un 40% de los casos.
El número total de neoplasias extracolónicas registradas en los familiares asciende a 11. De éstas, 9 corresponden a pacientes afectados exclusivamente de neoplasias extracolónicas y 2 a pacientes afectados también de CCR. La edad media de presentación fue de 48,8 ± 13,8 años (rango, 38-80 años). La distribución es la siguiente: 6 casos de cáncer de endometrio, 2 de estómago, 1 de ovario, 1 de vía urinaria y 1 de intestino delgado.
El número de familiares de riesgo registrados es de 43. De éstos, 14 han rechazado el seguimiento y 10 son seguidos correctamente en otros centros. Los 19 restantes se encuentran en seguimiento de forma activa en nuestro centro mediante colonoscopia anual y en consultas externas. La media de edad de los familiares de riesgo seguidos es de 29 ± 8 años. La distribución por sexos es de 12 varones y 7 mujeres.
Durante el tiempo de funcionamiento del registro, se han diagnosticado 4 casos de CCR: 2 correspondieron a familiares de riesgo que inicialmente rechazaron el seguimiento, el tercero correspondió a un paciente ya intervenido previamente que también había rechazado el seguimiento, y el cuarto, a un familiar de riesgo en seguimiento, en el que se detectó un pólipo plano en ángulo hepático del colon que fue sometido a cirugía por imposibilidad de resección endoscópica. En los 3 primeros, el estadio histológico fue avanzado (estadios III-IV), mientras que en el último caso se trató de un carcinoma in situ.
Por otro lado, se han realizado 10 polipectomías endoscópicas: 4 en familiares de riesgo en seguimiento y 6 en pacientes ya intervenidos previamente. En ninguno de los casos se observó displasia de alto grado.
DISCUSIÓN
Si bien existen estudios recientes en nuestro país referentes al CCRHSP12,28-32, de acuerdo con la revisión bibliográfica efectuada, éste es el primer artículo en el que se expone la puesta en marcha y los resultados de un registro hospitalario de CCRHSP en España.
Los objetivos del establecimiento de un registro de cáncer hereditario son claros: a) registrar los datos de pacientes afectados y de los familiares de riesgo; b) mejorar el pronóstico mediante el diagnóstico temprano y el seguimiento; c) educar a los pacientes y sus familias, incluyendo el consejo genético y, eventualmente, el estudio genético, y d) desarrollar la investigación básica y clínica41-42.
La utilidad del establecimiento de registros de cáncer de colon hereditario en cuanto a la disminución de la incidencia de casos de cáncer y la mejoría de la supervivencia se ha establecido tanto en el caso de la poliposis adenomatosa familiar43, como en el del CCRHSP13,15-17.
Los resultados obtenidos en nuestra serie, en cuanto a las características de los casos de cáncer de colon (comparados con los datos del Registre de Càncer Poblacional de Mallorca), son congruentes con los previamente publicados para el CCRHSP14,22-23. Está ampliamente descrito el inicio de la neoplasia en edades tempranas; así, en nuestra serie, la media de edad en el momento del diagnóstico del CCR es de 45 años frente a los 68,5 de media de la población general de Mallorca. También se ha descrito ampliamente la localización preferente de las lesiones en el colon derecho (un 57% en nuestra serie frente a un 20% en la población general). Estas diferencias se mantienen si la comparación se lleva a cabo con los resultados observados en otras áreas geográficas44.
El grado de aceptación del seguimiento endoscópico en los familiares de riesgo es relativamente aceptable 29/43 (67%), aunque posiblemente debamos incorporar estrategias de mejora para alcanzar un porcentaje ideal que se acerque al 100%. La edad media de estos familiares es muy baja (29 años). Estos datos son de gran importancia, ya que la potencialidad del registro para prevenir casos de cáncer se basa precisamente en lograr una buena tasa de seguimiento en los familiares de riesgo.
En el momento actual, coexisten 2 formas de identificar a las familias con CCRHSP: a) la identificación basada en criterios de carácter clínico (Ámsterdam I y II), y b) el diagnóstico basado en la detección de las mutaciones de los genes reparadores del ADN. Se está realizando un gran esfuerzo investigador para intentar determinar qué estrategia diagnóstica es más adecuada45-49.
La mayoría de los estudios desarrollados recientemente están dirigidos a establecer sistemas o criterios de selección (criterios de Bethesda, inestabilidad de microsatélites, inmunohistoquímica), que permitan determinar qué pacientes son tributarios de que se les realice el estudio genético para la detección de las mutaciones de los genes reparadores del ADN28,50.
De esta manera, se ha llegado a considerar que las familias que cumplían criterios de Ámsterdam, pero sin presencia de inestabilidad de microsatélites o mutación detectable, corresponderían a otra entidad con menor riesgo de CCR, para la que se ha propuesto el término de síndrome X51. En un artículo reciente se describen las características básicas de los pacientes correspondientes a este grupo29, que muestran una edad más avanzada en el diagnóstico, una mayor tendencia a localizarse en el colon izquierdo y un patrón histológico diferenciado.
Creemos importante puntualizar que una elevada proporción de los pacientes que cumple los criterios clínicos para el diagnóstico del CCRHSP (Ámsterdam I o II) no muestra mutaciones germinales en los genes reparadores del ADN o inestabilidad de microsatélites (un 50%, aproximadamente)52. Se ha observado, en estos casos, alteraciones en la vía común de la carcinogénesis colorrectal, lo que sugiere que otros genes, hasta ahora desconocidos, podrían ser causantes del CCR en estas familias53.
Consideramos que la aplicación de los criterios de Ámsterdam II para la identificación clínica de las familias (con independencia de que el estudio genético detecte luego la mutación) presenta ventajas: a) permite establecer, desde el primer momento y sin necesidad de esperar a los resultados genéticos, un programa de seguimiento eficaz para disminuir la morbimortalidad en estas familias13,15-17, y b) incluye a las familias con mutación negativa, lo cual es de gran importancia, al encontrarnos en un momento de grandes avances en el estudio del mecanismo de aparición de la enfermedad y de las mutaciones asociadas.
No es posible descartar que, en el futuro, el grupo de familias que cumplen los criterios de Ámsterdam II puedan desglosarse en varias entidades diferentes, con riesgos estratificados y diferentes mecanismos de predisposición al CCR. Sin embargo, en el momento actual, la conducta más prudente nos obliga a establecer una selección basada en criterios clínicos en estas familias, con el seguimiento apropiado54, sin por ello renunciar a la investigación que permita profundizar en el mejor conocimiento de este grupo de pacientes.
Este estudio permite resaltar la importancia de la historia familiar, basada en la entrevista personal y la revisión de las historias clínicas, en el estudio de las formas hereditarias de cáncer de colon55. La historia familiar representa, a pesar de sus limitaciones56, el elemento fundamental para la identificación de las familias afectadas de CCRHSP. Es importante destacar la precisión con que la historia familiar recoge los casos de cáncer en cada familia, ya que tan sólo hemos tenido 4 casos con información incorrecta y una familia que ha tenido que ser retirada del registro en la comprobación posterior.
Tal vez, estos resultados (bastante distintos de los descritos en los países anglosajones) se deban una diferencia cultural, al mantener las familias en el área mediterránea una relación mucho más estrecha, lo que mejora la eficacia de la historia familiar.
La importancia de la historia familiar y de los criterios clínicos radica también en la universalidad de su posible aplicación, que depende únicamente del interés y el tiempo que aplique el equipo médico responsable del paciente, sin depender de complejas técnicas de laboratorio con altos costes económicos asociados, que muchos centros no se pueden permitir. Se debe precisar, sin embargo, que la obtención de una historia familiar detallada y completa implica dedicar tiempo y disponer de personal bien entrenado para su obtención.
Los datos del seguimiento de las familias, aunque parciales, sugieren la utilidad del seguimiento endoscópico. En total, se han detectado 10 polipectomías sin complicaciones y 4 nuevos casos de cáncer, 3 con estadios avanzados en pacientes no seguidores del programa de cribado y 1 con un carcinoma in situ entre los cumplidores del seguimiento endoscópico.
Como conclusión final cabe afirmar que este estudio pone de manifiesto la utilidad de la puesta en marcha de un registro de familias con CCR hereditario basado en los criterios de Ámsterdam II para la identificación de un grupo de familias con alto riesgo de cáncer en edades juveniles que se beneficiarán del cribado endoscópico, así como del consejo y el estudio genéticos.

